
Abstract
Malus baccata (L.) Borkh. and Malus toringo (Siebold) Siebold ex de Vriese of the genus Malus Mill. (Rosaceae) are wild crabapples occurring in temperate East Asia. Despite their horticultural importance as ornamental trees and the natural resources in apple breeding, their phylogenetic relationships have never been determined clearly owing to lack of resolution in previous studies. We characterized four complete chloroplast genomes of these two species and conducted various phylogenomic analyses comparatively to the previously reported plastomes of other wild Malus species. They were highly conserved in genomic structures and gene contents, containing 129 genes including 84 protein-coding genes, eight rRNA genes, and 37 tRNA genes. Phylogenetic analysis of 23 representative Malus plastomes did not support the current classification of the major sections in Malus, revealing non-monophylies. The plastomes of M. toringo revealed two chloroplast types corresponding to their geographic distribution; M. toringo from China was more closely related to other sympatric species, while two conspecific M. toringo from Japan and Korea were in a sister relationship with M. baccata from Korea. We identified one positively selected gene (ndhD) and seven mutation hotspots (trnK-rps16, trnR-atpA, petN-psbM, trnT-psbD, psbZ-trnG, ndhC-trnV, and ycf1) and variable SSRs as potential useful plastid markers.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The genus Malus Mill. (Rosaceae) includes economically important apple species with cultivated apple fruits and wild crabapples. The domesticated apple, M. domestica Borkh., is one of the most widely cultivated fruit crops in temperate regions worldwide, and wide varieties of apple cultivars are bred for various tastes and use (Korban and Skirvin 1984; Morgan and Richards 2003). Its wild relatives, known as crabapples, usually having fruits less than 5 cm in diameter, offer a useful source of genetic diversity for apple breeding (Brown 2012), and are widely planted as ornamental and landscaping trees. The use of these natural resources in breeding and the development of effective conservation programs for apples require a good understanding of the genetic relationships as well as the genetic polymorphisms within and between cultivated apples and related wild crabapples (Cornille et al. 2014). Wild crabapples also provide habitats for wildlife and serve as a direct source of food for both human and wildlife or as components of hedges in agricultural landscapes.
The genus Malus comprises approximately 25 to 55 species, divided into five sections including six series (Harris et al. 2002; Phipps et al. 1990; Rehder 1974; Robinson et al. 2001), although recent phylogenetic studies have suggested elevating several series to sections as specified in Table 1 (Langenfelds 1991; Jiang et al. 1996; Qian et al. 2006, 2008). This study follows Phipps et al.’s treatment (1990) of infrageneric Malus classification (Table 1), adapted from Rehder (1974), Huckins (1972), and Williams (1982). The section Malus is traditionally sub-divided into two series, Malus and Baccatae (Rehder) Rehder, based on differences in fruit size and deciduous or persistent calyces. In the nomenclature of the taxa in the series, Malus of section Malus accommodating the cultivated apple, M. domestica, is complex. With few discrete characters to differentiate species, the morphological characters used to delimit the species are continuous and overlapping. Moreover, the intimate association that humans have with apples has blurred the distinction between wild and cultivated species, which has been further complicated by hybridization. The origin of the cultivated apple from its wild progenitors is relevant; however, difficulty in the delimitation of progenitor species has hampered investigations of parental contribution to its origin (Harris et al. 2002; Robinson et al. 2001). Additionally, the genetic identification of cultivars of artificial cross hybridization is difficult to determine their phylogenetic relationships or population genetic inference. The artificial selection that has been repeated through vegetative reproduction and re-crossing for a long period must have yielded a phylogeny like a network or disturbed the genetic structure of populations (Forte et al. 2002). Recently, several studies have provided insight into the origin of the cultivated apple and pointed out that several different wild species could have contributed organelle and nuclear genomes to the domesticated apple. The current most widely accepted theory based on morphological (Forte et al. 2002), phylogenetic (Forte et al. 2002; Harris et al. 2002; Robinson et al. 2001), population genetic (Cornille et al. 2012; Coart et al. 2006), and genome-wide (Nikiforova et al. 2013; Velasco et al. 2010) evidence suggests that M. sieversii (Ldb.) Roem in the Tian Shan Mountains of Central Asia was initially domesticated, and subsequently dispersed to West Europe along the great trade route known as the Silk Route, allowing hybridization and introgression of other wild crabapples from Siberia (M. baccata), Caucasus (M. orientalis Uglitz.), and Europe (M. sylvestris Mill) (Cornille et al. 2014). In addition to the initial progenitor, M. sieversii, the wild European crabapple, M. sylvestris, was, specifically, identified by population genetic study using microsatellite markers (Cornille et al. 2012), to be a major secondary contributor to the gene pool of current varieties of cultivated apple.
Malus baccata (L.) Borkh. in the series Baccatae of section Malus is a 10–14 m tall tree commonly called Siberian crabapple. It is native to Bhutan, China, India, Kashmir, Korea, Mongolia, Nepal, and Russia (Siberia) in northern Asia (Gu and Spongberg 2003), and has been introduced to Japan, Europe, northeastern USA, and Canada (USDA Natural Resources Conservation Service n.d.). It is one of the wild relatives of M. domestica that can be readily hybridized with varieties of cultivated apples (Cornille et al. 2014). Therefore, it is widely used as a rootstock and breeding resource in high-latitude apple-producing areas because of its disease resistance, and cold tolerance (Chen et al. 2019). Malus toringo (Siebold) Siebold ex de Vriese (Toringo or Siebold crabapple) is a 2–6 m tall shrub distributed naturally in China, Japan, and Korea (Iketani and Ohashi 2001; Lee 2007), and introduced to the USA and Europe as high horticultural value ornamental trees with semi-weeping branches (Dickson 2015); it is sometimes referred to by its illegitimate name, Malus sieboldii (Regel) Rehd. (Akiyama et al. 2014). M. toringo was traditionally classified in the series Sieboldianae of section Sorbomalus based on its lobed young leaves (among older entire leaves) and conduplicate bud vernation; however, M. baccata was placed in the series Baccatae of section Malus owing to its entire leaves and convolute or involute vernation (Fig. 1) (Phipps et al. 1990; Rehder 1974). The necessity for additional studies to clarify the systematic position of series Sieboldianae has been recognized, especially concerning its relationship to the series Baccatae. Despite different sectional assignments in Malus and Sorbomalus, the series of Baccatae and Sieboldianae displayed genetic similarity from molecular evidence as well as in morphology. Morphologically, they share several characters in common: flowers with deciduous calyces, umbellate inflorescences, and no or relatively few sclereids in fruits (Phipps et al. 1990; Rehder 1974; Forte et al. 2002). In nuclear DNA internal transcribed spacer (ITS) phylogeny, section Sorbomalus was polyphyletic, and the species belonging to series Sieboldianae (M. sieboldii and M. sargentii) were nested in the clade comprising the species of series Baccatae and Sieboldianae, suggesting their common origin (Forte et al. 2002; Harris et al. 2002; Robinson et al. 2001). Such phylogenetic similarity has also been revealed by amplified fragment length polymorphism (AFLP) and retrotransposon-based polymorphism analyses (Savelyeva and Kudryavtsev 2015; Savelyeva et al. 2017). However, chloroplast DNA (cpDNA) matK phylogeny and randomly amplified polymorphic DNA (RAPD) analyses did not provide sufficient resolution to infer robust species relationships (Forte et al. 2002; Harris et al. 2002; Robinson et al. 2001). Haplotype analyses of expanded cpDNA regions (trnH-psbA, trnS-trnG, trnL-trnF, and trnT-trnL) were also inconclusive owing to low resolution (Volk et al. 2015). Simple sequence repeat (SSR) markers have turned out to be inadequate in resolving interspecific relationships concordant with taxonomic classification or geographic origin, or both, although they have proven to be quite robust for many germplasm management applications (Hokanson et al. 2001). Disentangling the relationships between these two series has been exceptionally problematic owing to the high degree of hybridization between them and the application of the name “wild apple” to these hybrids, which blurs the boundaries between the two series. Robinson et al. (2001) claimed that the series Sieboldianae could be of hybrid origin, formed by hybridization between a series Baccatae taxon and a section Sorbomalus taxon.

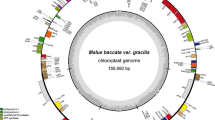
a Gene map of the chloroplast genomes of Malus baccata and Malus toringo collected from China, Japan, and Korea and sequenced in this study. b Flowers and leaves of Malus baccata. c Flowers and leaves of Malus toringo. Photo credit: Min Sung Cho, Sungkyunkwan University, Republic of Korea
To investigate their phylogenetic relationships, we sequenced and assembled four whole chloroplast genomes of two species representing the series Baccatae and Sieboldianae: one wild accession of M. baccata (series Baccatae; section Malus) from Korea and three accessions of M. toringo (series Sieboldianae; section Sorbomalus) from China, Japan, and Korea. The genetic and morphological similarity between M. baccata and M. toringo has been reported in previous studies despite taxonomic assignments into different sections of Malus and Sorbomalus, respectively, but their phylogenetic relationship has never been determined clearly yet. Specifically, we sampled M. toringo from natural environments in three countries of Korea, China, and Japan to examine the plastome variation among allopatric populations of M. toringo. With the advent of high-throughput sequencing technologies for next-generation sequencing (NGS), massive amounts of data are now available which improves the poor resolution in previous cpDNA phylogenies, to reveal considerable genome-wide variation in the sequences and structures of entire plastomes. Comparative genomic analysis of whole plastomes is now available as one of the effective markers to infer the phylogenetic relationships and evolutionary histories of numerous plant groups, including Rosaceae (Cheng et al. 2017; Daniell et al. 2016; Jansen et al. 2011; Jeon and Kim 2019; Njuguna et al. 2013; Parks et al. 2009; Terakami et al. 2012; Yang et al. 2018). Genome-wide data of Malus plastomes could provide vital information regarding genetic variation among wild crabapple species, not only increasing phylogenetic resolutions but also enhancing our understanding of organelle genome evolution. Based on the complete plastome sequences, we tested the previous phylogenetic hypotheses, focusing particularly on the relationship between Baccatae and Sieboldianae. Comparative plastome analyses allowed us to determine the structure, gene content, and rearrangements in the chloroplast genomes of wild Malus crabapples. Furthermore, highly variable chloroplast regions were identified as potentially useful markers for crabapples. Lastly, this study provided a glimpse into the infraspecific plastome variation of one of East Asian crabapple species, M. toringo.
Materials and methods
Plant sampling, DNA isolation, and plastome sequencing/annotation
The silica-gel dried leaves of four Malus wild crabapples were collected in the field; we collected one sample of M. baccata from a forest in Gangwon-do, Korea (37° 49′ 29.9″ N, 128° 21′ 46.5″ E, altitude 535 m), and three samples of M. toringo from three countries, i.e., Zhejiang, China (30° 18′ 01.2″ N, 119° 07′ 02.0″ E, 1117 m), Nagano-ken, Japan (35° 54′ 14.4″ N, 138° 09′ 43.0″ E, 1724 m), and Jeollanam-do, Korea (34° 57′ 50.3″ N, 127° 25′ 59.4″ E, 441 m). The voucher specimens were collected and deposited in the Ha Eun Herbarium (SKK) at Sungkyunkwan University, Republic of Korea. The total genomic DNA was isolated using a DNeasy Plant Mini Kit (Qiagen, Carlsbad, CA, USA) following the manufacturer’s protocol. An Illumina paired-end (PE) genomic library was constructed and sequenced using the Illumina HiSeq platform (Illumina, Inc., San Diego, CA, USA) at Macrogen Corporation (Seoul, Korea). By the de novo genomic assembler, Velvet 1.2.10 (Zerbino and Birney 2008), four Malus plastid genomes were assembled from the produced paired-end sequence reads (i.e., total 42,661,706 reads for M. baccata; 113,097,408, M. toringo China; 123,360,708, M. toringo Japan; 87,614,680, M. toringo Korea) with coverages of 364x (M. baccata), 289x (M. toringo China), 921x (M. toringo Japan), and 1054x (M. toringo Korea), respectively. The programs of Velveth and velvetg in Velvet were run using the optimized parameters of various hash length (k-mer) and coverage values to assemble each plastome. Annotation was performed using the Dual Organellar GenoMe Annotator (Wyman et al. 2004), ARAGORN v1.2.36 (Laslett and Canback 2004), and RNAmmer 1.2 Server (Lagesen et al. 2007). Using Geneious R10 (Biomatters, Auckland, New Zealand) (Kearse et al. 2012), annotation was inspected and corrected manually by comparison with other Malus plastomes. The annotated plastome sequences were deposited in the GenBank databank under the accession numbers MK571561 for M. baccata, and MK571562, MK571563, and MK571564 for M. toringo from China, Japan, and Korea, respectively. The annotated GenBank (NCBI, Bethesda, MD, USA) format sequence file was used to draw a circular map (Fig. 1) using the OGDRAW software v1.2 (CHLOROBOX, Postdam-Golm, Germany) (Lohse et al. 2009).
Comparative plastome analysis
We performed several comparative plastome analyses among the eight Malus plastomes including the previously reported four Malus species representing its major sections, i.e., M. angustifolia from section Chloromeles (NC_045410); M. sieversii from section Malus (NC_045390); M. tschonoskii from section Docyniopsis (NC_035672); and M. trilobata from Eriolobus (NC_035671); and those of M. baccata and M. toringo assembled in this study. The codon usage frequency was calculated using MEGA7 (Kumar et al. 2016) with relative synonymous codon usage (RSCU) value, which is the relative frequency of occurrence of the synonymous codon for a specific amino acid. The online program predictive RNA editor for plants (PREP) suite (Mower 2009) was used to predict the potential RNA editing sites for annotated protein-coding genes with 35 reference genes available with known edit sites, based on a cutoff value of 0.8 (suggested as optimal for PREP-Cp). Overall sequence divergence was estimated using the LAGAN alignment mode (Brudno et al. 2003) in mVISTA (Frazer et al. 2004). The nucleotide diversity (Pi) was calculated using sliding window analysis (window length = 1000 bp and step size = 200 bp, excluding sites with alignment gaps) to detect the most divergent regions (i.e., mutation hotspots) in DnaSP (Librado and Rozas 2009). Two types of repeat sequences were identified and compared among eight plastid genomes. REPuter (Kurtz et al. 2001) was used to detect the various types of repetitive sequences with search parameters of maximum computed repeats = 50, minimum repeat size = 8 bp, and hamming distance=1. Simple sequence repeats (SSRs) were identified using MISA web (http://pgrc.ipk-gatersleben.de/misa/) with search parameters of 1–15 (unit size-minimum repeats, i.e., mono-nucleotide motifs with 15 minimum numbers of repetition), 2-5, 3-3, 4-3, 5-3, and 6-3 with 100 interruption (maximum difference for two SSRs). To evaluate natural selection pressure in the protein-coding genes of the eight plastomes, the rates of nonsynonymous to synonymous substitution (ω=dN/dS) were estimated using EasyCodeML (Gao et al. 2019) based on PAML (Phylogenetic Analysis by Maximum Likelihood) algorithms (Yang 1997). Seven codon substitution models with heterogeneous ω across sites (Yang et al. 2000) implemented in EasyCodeML were employed to investigate the aligned sequences of protein-coding genes of eight Malus plastomes, M0 (one ratio), M1a (nearly neutral), M2a (positive selection), M3 (discrete), M7 (beta), M8 (beta and ω>1), and M8a (beta and ω=1). The fit of these models to the sequence data was compared in preset running mode using likelihood ratio test (LRT), and the pairwise comparison of codon models, M7 vs. M8, was effective for identifying amino acid residues that have potentially evolved under selection among Malus plastomes. The site potentially evolving under positive selection was presented based on the posterior probability higher than the standard threshold (0.95) (Scheffler and Seoighe 2005) calculated by the Bayes empirical Bayes (BEB) method (Yang et al. 2005).
Phylogenetic analysis
Phylogenetic relationships of the newly sequenced accessions of M. baccata and M. toringo assembled in this study were investigated in the context of their relationships with other Malus species. We analyzed 23 plastome sequences of major sections of the genus Malus: eight accessions of section Malus including two accessions of M. baccata (MK571561 and NC045389), M. domestica cultivar M9 (MK434916), M. halliana (MT246302), M. hupehensis (NC040170), M. micromalus (NC036368), M. prunifolia (NC031163), and M. sieversii (NC045390); nine accessions of section Sorbomalus, i.e., five accessions of M. torigno (MT268884, NC050059, MK571562, MK571563, and MK571564), M. florentina (NC035625), M. toringoides (NC049113), M. transitoria (MK098838), and M. yunnanensis (NC039624); three species of section Chloromeles, i.e., M. coronaria (NC045308), M. ioensis (NC045393), and M. angustifolia (NC045410); two species of section Docyniopsis, M. doumeri (NC045343) and M. tschonoskii (NC035672); one species of section Eriolobus, M trilobata (NC035671). The analysis included Pyrus pyrifolia (NC015996) from the same tribe (Maleae) as an outgroup species. The sequences of concatenated sequences of 79 common protein-coding genes (excluding duplicated ones in IR regions) among the Malus species were aligned using MAFFT v. 7 (Katoh and Standley 2013), and the ML phylogenetic tree was constructed using IQ-TREE v. 1.4.2, with 1000 replicate bootstrap analysis (Nguyen et al. 2015). The best-fit evolutionary model was chosen as “TVM+F+I,” which was scored according to the Bayesian information criterion (BIC) scores and weights by testing 88 DNA models of ModelFinder (Kalyaanamoorthy et al. 2017) implemented in IQ-TREE v. 1.4.2.
Results and discussion
Genome features, content, order, and organization of wild Malus plastomes
The plastome of M. baccata contained 160,149 base pairs (bp), and consisted of four typical regions: a large single-copy (LSC) region of 88,260 bp, a small single-copy (SSC) region of 19,181 bp, and a pair of inverted repeat regions (IRs) of 26,354 bp. The total lengths of the three M. toringo plastomes were 160,105 (Japan), 160,138 (Korea), and 160,168 bp (China), and comprised of the same four regions of LSC, SSC, and a pair of IRs (Fig. 1, Table 2). The overall guanine-cytosine (GC) content of M. baccata was 36.5%, while that of M. toringo, 36.5% (China) or 36.6% (Japan and Korea), respectively (Table 2). Each of the four plastomes contained 129 genes, including 84 protein-coding (excluding pseudogenes), eight rRNA, and 37 tRNA genes. Eighteen genes contained introns, including seven tRNA genes. Three genes, clpP, rps12, and ycf3, exhibited two introns. The trnK-UUU gene harbored the largest intron that contained the matK gene within it (Table 3). In total, 16 genes were duplicated in the IR regions, including seven tRNAs, four rRNAs, and six protein-coding genes. The trans-splicing gene rps12, consisting of three exons, was located in the LSC region of exon 1, but exon 2 and exon 3 of the gene were embedded in the IR regions. The infA gene located in the LSC region became a pseudogene, and part of each ycf1 and rps19, duplicated in the IR region, also became pseudogenes.
A genomic comparison of eight wild Malus plastomes, including M. baccata and M. toringo (sequenced in this study), and four other crabapple species (M. angustifolia, M. sieversii, M. tschonoskii, and M. trilobata) revealed high conservation in their plastome organization. They shared the most common genomic features of sequences, and gene content and numbers, demonstrating a 99.2% pairwise sequence identity, despite their different sectional assignments. Generally, the length of the chloroplast genome and its quadripartite regions vary among plant lineages due to the contraction and expansion of inverted repeat regions. Evaluating their contraction and expansion by comparing the location of the boundaries among the four chloroplast regions (two IRs, LSC, and SSC) can provide some insights into plastome evolution (Menezes et al. 2018). All eight Malus plastomes shared exactly the same genes and similar gene contents at all boundaries among the four regions, with only slight changes in the length of intergenic regions. They all contained the functional protein-coding gene of ycf1 at SSC/IR with its pseudogene copy, ycf1Ψ at IR/SSC, and functional rps19 at LSC/IR with pseudogene copy rps19Ψ at IR/LSC endpoints (Fig. 2).

Comparison of the border positions of the large single-copy (LSC), small single-copy (SSC), and inverted repeat (IR) regions among eight wild Malus chloroplast genomes. Gene names are indicated in colored arrow boxes, and their lengths in the corresponding regions are displayed beside the boxes. Ψ indicates a pseudogene
Comparative phylogenomic analyses of wild Malus plastomes
The frequency of codon usage in the eight wild Malus plastomes was calculated based on the sequences of protein-coding genes (Fig. S1, Table S1). The average codon usage in all plastomes was identical at 26,527, except for M. trilobata (26,532), and the patterns of frequently used codons were also consistent among them. The genetic code encoding protein in a mode of triplet codon is said to be redundant in that the same amino acid residue can be encoded by more than one, so-called synonymous codons. Most amino acids are encoded by several synonymous codons, as 64 different codons are translated into 20 amino acids and termination of translation (three stop signals). Synonymous codons are not used in equal frequency, but specific codons are used more often than other synonymous codons during translation of genes. This feature of preferential use of codons is known as codon usage bias. The codon usage bias and variation in codon usage within and among species suggest some selective constraint on codon choice. The frequency of codon usage varies by factors in species-specific ways, showing different preferences for codons used to encode specific amino acids, probably as a result of evolution in the presence of mutational biases, selection for translation rate and accuracy, and possibly other factors (Orešič and Shalloway 1998). Codon usage values are described by the relative synonymous codon usage (RSCU), which is a reflection of how often a particular codon is used relative to the expected number of times that codon would be used in the absence of codon usage bias. In our analyses, all RSCU values for each amino acid considered were similar among the eight plastomes. The highest RSCU value was indicated in the usage of the UUA codon for leucine (1.94–1.95) followed by GCU for alanine (1.84) and AGA for arginine (1.83–1.84), while the lowest were AGC for serine (0.38) and GAC for aspartic acid (0.38). We found that codon usage was biased toward a high RSCU value of U and A at the third codon position as found in other Rosaceae species (Yang et al. 2020).
RNA editing alters the nucleotide sequence of transcribed RNA molecules from that of the DNA template encoding it, which usually results in a change in the amino acid sequence of the translated protein. In plants, RNA editing affects mitochondrial and plastid transcripts of all major lineages of land plant, and the site-specific modification of cytidines to uridines (C-to-U conversion) is prevalent in organellar genomes of all land plants (Chateigner-Boutin and Small 2011). Comparison of editing frequencies and editing patterns shows that RNA editing is a transcript- and species-specific process, but its frequencies and patterns are not correlated with the phylogenetic position of the species, sometimes revealing extensive species-specific divergence among closely related species. Species specificity of the editing frequencies and gene-specific editing patterns suggest multiple independent acquisitions and occasional losses of editing at a specific site (Freyer et al. 1997). This raises questions about the selection pressures acting to maintain editing in the evolution of angiosperms that are yet to be completely resolved. Editing tends to correct the effect of DNA mutations that would otherwise compromise the synthesis of functional proteins, and its additional function could be generating protein diversity or regulating gene expression (Chateigner-Boutin and Small 2011). The extent of variation in the number of RNA editing sites is currently less known at shallow taxonomic levels (i.e., among congeneric species or across multiple genera belonging the same family), although several studies demonstrated that the number of editing sites can vary widely among large taxonomic groups of land plants and also between the two organellar genomes (Corneille et al. 2000; Guo et al. 2015; Tsudzuki et al. 2001). We have found that the RNA editing patterns across the eight Malus plastomes were similar in gene location and codon conversion type of the predicted RNA editing sites; only slight changes were observed in the numbers of editing sites for several codon conversions. The total number of RNA editing sites identified among them ranged from 62 to 64 for 25 of the 35 protein-coding genes. These genes included photosynthesis-related genes (atpA, atpB, atpF, atpI, ndhA, ndhB, ndhD, ndhF, ndhG, petB, petG, psaI, psbE, psbF, and psbL), self-replication genes (rpoA, rpoB, rpoC1, rpoC2, rps2, rps14, and rps16), and others (accD, clpP, and matK). We detected no RNA editing sites in the ccsA, petD, petL, psaB, psbB, rpl2, rpl20, rpl23, rps8, and ycf3 genes. The highest numbers of potential editing sites were found in the NADH dehydrogenase genes, which was consistent with the previous findings in tobacco, maize, rice, and other plants (Corneille et al. 2000; Kim et al. 2019; Tsudzuki et al. 2001; Yang et al. 2020), i.e., ndhB gene was the highest in frequency at 11–12 sites, followed by the ndhD gene at eight sites. Most editing sites were distributed at the 1st and 2nd codon positions (Table S2) as observed in the chloroplast genome of the hornwort Anthoceros formosae (Kugita et al. 2003) and the mitochondrial genes of Arabidopsis (Giegé and Brennicke 1999), whereas the mitochondrial genes of Physarum polycephalum showed the different pattern of codon bias with more editing at the 3rd codon position than at the 1st and 2nd positons within coding regions (Mahendran et al. 1991). The highest conversions in the editing frequencies of codons associated with the corresponding amino acid changes were represented by the changes from serine (S) to leucine (L) (average confidence score of 22.935) followed by proline (P) to leucine (L) (average confidence score of 8.86) (Fig. S2).
The divergence level of nucleotide diversity among the eight plastomes of wild Malus species was visualized by plotting with mVISTA (Frazer et al. 2004), using the plastome of M. tschonoskii of section Docyniopsis as a reference. The results exhibited a high degree of synteny and gene order conservation in the mVISTA graph (Fig. 3). The LSC region was the most divergent, whereas the two IR regions were highly conserved. Most noncoding and intron regions were found to be more divergent and variable than the coding regions; however, several protein-coding regions of accD, rpoA, ycf1, and ndhF were relatively divergent. The overall nucleotide diversity (Pi) among eight plastomes showed an average Pi value of 0.00167 with 938 polymorphic sites, ranging from 0 to 0.01546, which was quite low, albeit similar to other genera of Rosaceae (i.e., Rosa at 0.00154) (Jeon and Kim 2019). The genetic polymorphisms in different regions of the chloroplast genome vary substantially, and wild Malus species harbored relatively higher nucleotide polymorphisms in both the LSC and SSC regions compared to those in the IR regions as observed similarly in Panax species (Jiang et al. 2018). They showed higher Pi values in the LSC (Pi = 0.002192) and SSC (Pi = 0.002394) regions, while obviously low values were found in the IR regions (Pi = 0.00045). Seven divergence hotspots among eight wild Malus plastomes are suggested as potential chloroplast markers: six intergenic regions (trnK-rps16, trnR-atpA, petN-psbM, trnT-psbD, psbZ-trnG, and ndhC-trnV) and one protein-coding region (ycf1) (Fig. 4).

Comparison of the chloroplast genomes of eight Malus species visualized by mVISTA. Gray arrows indicate genes with their orientation and position. Genome regions are color coded as pink blocks for the conserved coding genes (exon), blue blocks for introns, and peach blocks for noncoding sequences in intergenic regions (CNS). Thick lines below the alignment indicate the quadripartite regions of genomes; the LSC region is green, IR regions, aqua blue, and SSC region, orange

Seven most variable hotspot regions found in eight plastomes of wild Malus species by sliding window analysis. Six intergenic regions of trnK-rps16, trnR-atpA, petN-psbM, trnT-psbD, psbZ-trnG, ndhC-trnV, and one coding gene of ycf1
All of eight Malus cp genomes contained comparable numbers and distribution patterns of repeated sequences. Simple sequence repeats (SSRs) have high polymorphisms due to large variations in motifs and number of repetitions. Because of their high level of polymorphisms and genome-wide distribution, they have been used for powerful tools to measure genetic diversity and address the population genetic issues, such as gene flow, parentage, and population structure (Wang et al. 2009). In this study, we detected 103-114 SSRs by MISA based on search parameters set for 1-15 (mono-nucleotide motifs with 15 minimum numbers of repetition), 2-5, 3-3, 4-3, 5-3, and 6-3. The majority of the SSRs were tri-nucleotide motifs (61–68 SSRs, 60%) followed by di-nucleotide (17–20, 18%), and mono-nucleotide (14–18, 15%) (Fig. S3A). The most abundant repeat motif was “AAT/ATT” (22%) followed by “AAG/CTT” (21%) in all eight genomes (Fig. S3B, Table S3). SSRs were distributed most frequently in the intergenic regions (62%), followed by coding regions (31%), with much lower numbers found in the noncoding introns (7%) in each cp genome (Table S4). The coding regions with highest number of SSRs were ycf genes, eight in ycf1 (two pseudogenized, six coded in SSC and IR), and six SSRs (three duplicated in each IR) in ycf2. Considering the quadripartite regional occupancy of SSRs, the IR and SSC regions were lower in overall SSR frequency compared with the LSC region, 16% from the SSC region and 11% from each of both IR regions versus 62% from the LSC region (Table S4). Additionally, we found 49 pairs of large repeats in each cp genome (excluding duplicated IR region) using the parameters of maximum computed repeats = 50, minimum repeat size = 8 bp, and hamming distance = 1 by REPuter. They contained 23–31 forward, 2–12 reverse, and 13–20 palindromic matches of repeats (Fig. S4A). Lengths of 21–25 repeats were the most frequent (49%) followed by lengths of 26–30 repeats (21%), while longer repeats of 31–35 (8%), 36–40 (14%), and 41< (8%) were rarer than shorter ones (Fig. S4B).
Selective pressure in genes or genomic regions is inferred by the proportion of amino acid substitutions driven by natural selection during chloroplast genome evolution. Purifying selection removes deleterious variations, while positive selection fixes beneficial variation in the population and promotes the emergence of new phenotypes, offering fitness advantages in adaptation to the environment (Choudhuri 2014). Comparison of synonymous and nonsynonymous substitution rates can reveal the direction and strength of natural selection acting on the protein level. The rate of synonymous substitutions (dS), which is similar for many different genes, is significantly higher than that of nonsynonymous substitutions (dN), and the genes under positive selection are considered to have an evolutionary character in that dN is greater than dS (Endo et al. 1996). Therefore, the ratio of nonsynonymous substitution and synonymous substitution rates (denoted as ω=dN/dS) has been widely used as a genomic signature of selective pressure acting on a protein-coding gene, with ω=1 indicating neutral mutations; ω < 1, purifying selection; and ω > 1, diversifying positive selection (Yang et al. 2000). We identified that one of NADH dehydrogenase subunit genes of photosynthesis, ndhD gene, potentially evolved under positive selection in eight Malus plastomes by calculating the dN/dS ratio using various site-specific substitution models implemented in EasyCodeML (Gao et al. 2019; Yang 1997). Support for the gene under positive selection was identified, as codon substitution alternative model M8 (beta and ω > 1) provides a better fit than the null model M7 (beta in the interval 0 < ω < 1) from the pairwise comparison of likelihood ratio test (LRT) at significant level with p-value below 0.05 (Yang et al. 2000). Positively selected site in ndhD gene was suggested based on the posterior probability calculated by the Bayes empirical Bayes (BEB) method (Yang et al. 2005) with cutoff = 0.95 indicated with asterisk (*) in Table 4. The ndhD gene was included in the previously reported six genes (accD, rbcL, rps3, ndhB, ndhD, and ndhF) as undergoing positive selection in other Rosaceae plants (Yang et al. 2020). The critical importance of the genes that a plastome carries and its high conservativeness has contributed to the traditional view that purifying selection is the predominant force shaping chloroplast evolution due to functional limitations; however, the latest empirical evidence is no longer supportive of this hypothesis, and is now pointing to adaptive plastome variation (Bock et al. 2014). Recent advances in sequencing technology have contributed to an increase in the interest in taking advantage of the study of plastomes in phylogenetics, phylogeography, and population genetics. Variable genes potentially evolving under positive selection have occurred in the plastomes of a few other plant groups; three genes (rps2, rbcL, and ndhG) have been identified in Paulownia (Li et al. 2020a), five (rbcL, clpP, atpF, ycf1, and ycf2) in Panax (Jiang et al. 2018), three (clpP, ycf1, and ycf2) in the tribe Sileneae (Sloan et al. 2014), and in other angiosperms (Park et al. 2018; Piot et al. 2018; Wang et al. 2019). The genes identified as positively selected might undergo certain functional diversification in local adaptation during their evolutionary history that, in previous studies, has been discussed mainly as photosynthetic performance under variable environments of temperature and moisture (Bock et al. 2014).
Phylogenetic analysis
Maximum likelihood (ML) analysis computed by IQ-TREE (Nguyen et al. 2015) enabled us to build robust phylogenetic relationships among wild Malus crabapple species based on the best-fit model, “TVM+F+I” (Fig. 5). The ML tree was reconstructed using the aligned sequences of 79 protein-coding genes from 23 representative Malus plastomes with Pyrus pyrifolia as outgroup. We selected plastomes of wild crabapple collections (rather than germplasm resources) deposited in the GenBank database, in addition to the four plastomes assembled in this study, to elucidate the phylogenetic relationships among wild Malus species. Previous studies revealed many unresolved branches and low bootstrap supports within Malus due to the partial usage of conservative chloroplast genomes (Forte et al. 2002; Harris et al. 2002; Robinson et al. 2001; Volk et al. 2015). Our current plastome phylogenomic analysis provided greater resolution with high bootstrap values in the relationships among major sections of the genus Malus. In the ML tree, Malus was divided into two clades; the first clade included the species of sections Chloromeles, Eriolobus, and M. florentina of section Sorbomalus, while the other clade comprised primarily the species of sections Docyniopsis, Malus, and Sorbomalus. M. florentina is a single species in the series Florentinae of section Sorbomalus, and has been placed under Sorbomalus owing to its morphological similarity of lobed leaves. It has previously been suggested to raise the taxonomic rank of M. florentina to section Florentinae, because, based on phytochemical and molecular studies, it showed greater similarity to the sections of Docyniopsis and Eriolobus than to other Sorbomalus species (Qian et al. 2008).

Maximum likelihood tree inferred from 79 protein-coding genes of 23 Malus (Rosaceae) taxa using Pyrus pyrifolia as outgroup. Bootstrap values over 50%, based on 1000 replicates, are shown on each node. The species indicated in red are the four Malus plastomes newly sequenced in this study. The subclade of red square contains the species belonging to the series Baccatae (section Malus) and series Sieboldianae (section Sorbomalus) except four species specified in blue
The traditional classification of several sections and series of the genus Malus was not supported by chloroplast genome-wide phylogeny in this study. First, section Chloromeles was not monophyletic, as M. coronaria and M. ioensis were nested within clade I, whereas M. angustifolia was included in the subclade of Baccatae/Sieboldianae within clade II. This unexpected placement of M. angustifolia was also reported in previous study (Liu et al. 2019) owing to its close relationship to M. baccata. Section Docyniopsis was not monophyletic either, although the species of section Docyniopsis were nested in the same clade II. The occurrence of non-monophyly in the core Malus group (i.e., section Malus and Sorbomalus) was more complex, as both sections of Malus and Sorbomalus were polyphyletic, and their species were intermixed. Specifically, the species of series Baccatae (section Malus) and series Sieboldianae (section Sorbomalus) were closely related to each other within the strongly supported subclade Baccatae/Sieboldianae (100% bootstrap value), even though they are traditionally placed under different sections. Within the subclade Baccatae/Sieboldianae, the 1st group included the species of Baccatae of section Malus (M. halliana and M. hupehensis) and the species of section Sorbomalus (M. toringo of series Sieboldianae from China and M. toringoides of series Kansuenses). M. toringoides has been suggested to have a hybrid origin with paternal contribution most likely from M. transitoria (Feng et al. 2007; Tang et al. 2014), for which this study is quite supportive, based on the fact that M. toringoides was not genetically close to M. transitoria in maternally inherited chloroplast phylogeny, but was closely related to M. toringo. This study could not decisively determine its maternal parent, as the plastomes of the species from section Malus (M. sikkimensis of series Baccatae or M spectabilis of series Malus) that had been previously suggested as its maternal parent (Tang et al. 2014) were not available for comparison with M. toringo in our analyses. The 2nd group in subclade Baccatae/Sieboldianae primarily included M. baccata and M. toringo (collected from Korea and Japan) together with the species of series Malus of section Malus (M. micromalus and M. prunifolia), and M. angustifolia of section Chloromeles. There was a weakly supported interrelationship among the members of the 2nd group in subclade Baccatae/Sieboldianae.
The genetic similarity of Baccatae and Sieboldianae in addition to their morphological similarity has raised questions concerning their systematic positioning (Forte et al. 2002; Harris et al. 2002; Robinson et al. 2001; Savelyeva and Kudryavtsev 2015; Savelyeva et al. 2017). Even from the highly resolved chloroplast genome-based phylogeny in this study, they were not separated from each other, although they taxonomically belong to two different sections of Malus and Sorbomalus. However, the taxonomic requirements to merge the series of Baccatae and Sieboldianae remain a subject of future studies considering both the maternally inherited chloroplast and the biparently inherited nuclear DNA-based phylogenies. Despite its high resolution, plastome phylogeny represents only the maternal line in Rosaceae as reported previously (Brettin et al. 2000; Kaneko et al. 1986; Matsumoto et al. 1997; Raspé 2001); therefore, nuclear DNA-based tree topology should be compared with chloroplast phylogeny to yield the most plausible conclusion with an unequivocal explanation of the new classification based on the congruency between them. One novel finding of the current whole plastome phylogenomic study is that M. toringo exhibited the geographic pattern of its plastome diversity, exposing two distinct chlorotypes. The accession of M. toringo sampled from China (MK571562) was clustered with the Chinese accessions of M. sieboldii (synonym of M. toringo) and M. toringoides within the 100% supported clade, while the other chlorotype was displayed by the accessions of M. toringo collected from Japan and Korea. They were nested into another clade (90% bootstrap value) separate from the Chinese chlorotype and were the most closely related to M. baccata sampled from the sympatric region in Korea. The geographic location of M. sieboldii (NC050059), which was included in the same clade, is not known, as it was sampled from the Royal Botanic Gardens, Kew, in the UK without specified source information (Li et al. 2020b). Although we identified two types of chloroplast genomes in M. toringo, questions remain as to its monophyly and species delimitation, requiring further in-depth investigation. Given the maternal inheritance of plastid genomes and the high frequency of hybridization and gene flow in Malus, it is plausible that our current results are due to past gene flow among congeneric species in sympatric regions. To disentangle the complex evolutionary history of the crabapple genus and the unexpected findings in M. toringo, it would be necessary to carry out detailed population genetic or phylogeographic studies, or both.
Conclusion
In this study, we determined the complete plastome sequences of two wild crabapple species in the genus Malus (Rosaceae). As expected, we found highly conserved plastomes within the genus, including gene order and content, and a slow rate of evolution. The frequency of codon usage was biased toward high RSCU values of U and A at the third codon position, and we found that the highest numbers of potential editing sites were found in the ndhB gene followed by the ndhD gene with most editing sites at the 1st and 2nd codon positions. Comparative analysis among the eight wild Malus plastomes revealed seven divergence hotspots of six intergenic regions (trnK-rps16, trnR-atpA, petN-psbM, trnT-psbD, psbZ-trnG, and ndhC-trnV), and one protein-coding gene (ycf1) as potential chloroplast markers for phylogenetic studies of Malus species. We also identified that ndhD gene in eight Malus plastomes potentially evolved under positive selection. The results of phylogenetic analyses based on the aligned sequences of 79 protein-coding genes of 23 representative Malus plastomes provided high resolutions with strong bootstrap support in the relationships among major sections of the genus Malus. The genetic similarity between the series Baccatae and Sieboldianae from two different sections (Malus and Sorbomalus, respectively) was confirmed in this study. Interestingly, M. toringo exhibited the geographic pattern of its plastome diversity, revealing two distinct chlorotypes distributed in China and Japan/Korea.
Data Availability
The whole chloroplast genome data are deposited in the GenBank databank under the accession numbers MK571561 for M. baccata, and MK571562, MK571563, and MK571564 for M. toringo from China, Japan, and Korea, respectively.
References
Akiyama S, Thijsse G, Esser HJ, Ohba H (2014) Siebold and Zuccarini’s type specimens and original materials from Japan, Part 5. Angiosperms. Dicotyledoneae 4. J Jpn Bot 89:279–330
Bock DG, Andrew RL, Rieseberg LH (2014) On the adaptive value of cytoplasmic genomes in plants. Mol Ecol 23:4899–4911. https://doi.org/10.1111/mec.12920
Brettin TS, Karle R, Crowe EL, Iezzoni AF (2000) Chloroplast inheritance and DNA variation in sweet, sour, and ground cherry. J Hered 91:75–79
Brown SK (2012) Apple. In: Badenes ML, Byrne DH (eds) Fruit breeding. Springer, New York, pp 329–367
Brudno M, Do CB, Cooper GM, Kim MF, Davydov E, NISC Comparative Sequencing Program, Green ED, Sidow A, Batzoglou S (2003) LAGAN and Multi-LAGAN: efficient tools for large-scale multiple alignment of genomic DNA. Genome Res 13:721–731
Chateigner-Boutin AL, Small I (2011) Organellar RNA editing. Wiley Interdiscip Rev RNA 2:493–506
Chen X, Li S, Zhang D, Han M, Jin X, Zhao C, Wang S, Xing L, Ma J, Ji J, An N (2019) Sequencing of a wild apple (Malus baccata) genome unravels the differences between cultivated and wild apple species regarding disease resistance and cold tolerance. G3-Genes Genomes Genet 9:2051–2060
Cheng H, Li J, Zhang H, Cai B, Gao Z, Qiao Y, Mi L (2017) The complete chloroplast genome sequence of strawberry (Fragaria× ananassa Duch.) and comparison with related species of Rosaceae. PeerJ 5:e3919
Choudhuri S (2014) Fundamentals of Molecular Evolution. In: Bioinformatics for beginners: genes, genomes, molecular evolution, databases and analytical tools. Elsevier Inc., Oxford, pp 27–53
Coart E, Van Glabeke S, De Loose M, Larsen AS, Roldán-Ruiz I (2006) Chloroplast diversity in the genus Malus: new insights into the relationship between the European wild apple (Malus sylvestris (L.)) Mill. and the domesticated apple (Malus domestica Borkh.). Mol Ecol 15:2171–2182
Corneille S, Lutz K, Maliga P (2000) Conservation of RNA editing between rice and maize plastids: are most editing events dispensable? Mol Gen Genet 264:419–424
Cornille A, Gladieux P, Smulders MJM, Roldán-Ruiz I, Laurens F, Le Cam B, Anush Nersesyan A, Clavel J, Olonova M, Feugey L, Gabrielyan I, Zhang X-G, Tenaillon MI, Giraud T (2012) New insight into the history of domesticated apple: secondary contribution of the European wild apple to the genome of cultivated varieties. PLoS Genet 8:e1002703. https://doi.org/10.1371/journal.pgen.1002703
Cornille A, Giraud T, Smulders MJM, Roldán-Ruiz I, Gladieux P (2014) The domestication and evolutionary ecology of apples. Trends Genet 30:57–65
Daniell H, Lin CS, Yu M, Chang WJ (2016) Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol 17:134
Dickson EE (2015) Malus toringo. Flora of North America @ efloras.org. FNA Vol. 9. http://www.efloras.org/florataxon.aspx?flora_id=1&taxon_id=242331492. Accessed 18 Jan. 2021
Endo T, Ikeo K, Gojobori T (1996) Large-scale search for genes on which positive selection may operate. Mol Biol Evol 13:685–690
Feng TT, Zhou ZQ, Tang JM, Cheng MH, Zhou SL (2007) ITS sequence variation supports the hybrid origin of Malus toringoides Hughes. Botany 85:659–666
Forte AV, Ignatov AN, Ponomarenko VV, Dorokhov DB, Savel’ev NI (2002) Phylogeny of the Malus (apple tree) species, inferred from its morphological traits and molecular DNA analysis. Genetika 38:1357–1369
Frazer KA, Pachter L, Poliakov A, Rubin EM, Dubchak I (2004) VISTA: computational tools for comparative genomics. Nucleic Acids Res 32:W273–W279
Freyer R, Kiefer-Meyer MC, Kössel H (1997) Occurrence of plastid RNA editing in all major lineages of land plants. Proc Natl Acad Sci U S A 94:6285–6290
Gao F, Chen C, Arab DA, Du Z, He Y, Ho SYW (2019) EasyCodeML: a visual tool for analysis of selection using CodeML. Ecol Evol 9:3891–3898
Giegé P, Brennicke A (1999) RNA editing in Arabidopsis mitochondria effects 441 C to U changes in ORFs. Proc Natl Acad Sci U S A 96:15324–15329
Guo W, Grewe F, Mower JP (2015) Variable frequency of plastid RNA editing among ferns and repeated loss of uridine-to-cytidine editing from vascular plants. PLoS One 10:e0117075
Gu C, Spongberg SA (2003) Malus. In: Wu ZY, Raven PH, Hong DY (eds) Flora of China, vol 9. Science Press and Missouri Botanical Garden Press, Beijing and St. Louis, pp 179–189
Harris SA, Robinson JP, Juniper BE (2002) Genetic clues to the origin of the apple. Trends Genet 18:426–430. https://doi.org/10.1016/S0168-9525(02)02689-6
Hokanson SC, Lamboy WF, Szewc-McFadden AK, McFerson JR (2001) Microsatellite (SSR) variation in a collection of Malus (apple) species and hybrids. Euphytica 118:281–294. https://doi.org/10.1023/A:1017591202215
Huckins CA (1972) A revision of the sections of the genus Malus Miller. Ph.D. dissertation, Cornell University, Ithaca
Iketani H, Ohashi H (2001) Malus. In: Iwatsuki K, Boufford E, Ohba H (eds) Flora of Japan, Vol. IIb, Angiospermae, Dicotyledoneae, Archichlamydeae (b). Kodansha, Tokyo, pp 120–123
Jansen RK, Saski C, Lee SB, Hansen AK, Daniell H (2011) Complete plastid genome sequences of three Rosids (Castanea, Prunus, Theobroma): evidence for at least two independent transfers of rpl22 to the nucleus. Mol Biol Evol 28:835–847. https://doi.org/10.1093/molbev/msq261
Jeon JH, Kim S-C (2019) Comparative analysis of the complete chloroplast genome sequences of three closely related East-Asian wild Roses (Rosa sect. Synstylae; Rosaceae). Genes 10:23. https://doi.org/10.3390/genes10010023
Jiang NG, Wang LC, Li XL (1996) A new sect. of Malus Mill.- Sect. Baccatus and its classification. J SW Agrie Univ (Chongqing, China) 18: 144-147 (In Chinese)
Jiang P, Shi FX, Li MR, Liu B, Wen J, Xiao H, Li LF (2018) Positive selection driving cytoplasmic genome evolution of the medicinally important ginseng plant genus Panax. Front Plant Sci 9:359
Kalyaanamoorthy S, Minh BQ, Wong TK, von Haeseler A, Jermiin LS (2017) ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods 14:587–589
Kaneko T, Terachi T, Tsunewaki K (1986) Studies on the origin of crop species by restriction endonuclease analysis of organellar DNA. II. Restriction analysis of cpDNA of 11 Prunus species. Jpn J Genet 61:157–168
Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol Biol Evol 30:772–780
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Meintjes P, Drummond A (2012) Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649
Kim S-H, Yang JY, Park JS, Yamada T, Maki M, Kim S-C (2019) Comparison of whole plastome sequences between thermogenic skunk cabbage Symplocarpus renifolius and nonthermogenic S. nipponicus (Orontioideae; Araceae) in East Asia. Int J Mol Sci 20:4678. https://doi.org/10.3390/ijms20194678
Korban SS, Skirvin RM (1984) Nomenclature of the cultivated apple. HortScience 19:177–180
Kugita M, Yamamoto Y, Fujikawa T, Matsumoto T, Yoshinaga K (2003) RNA editing in hornwort chloroplasts makes more than half the genes functional. Nucleic Acids Res 31:2417–2423
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis v7.0 for bigger datasets. Mol Biol Evol 33:1870–1874
Kurtz S, Choudhuri JV, Ohlebusch E, Schleiermacher C, Stoye J, Giegerich R (2001) REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res 29:4633–4642
Lagesen K, Hallin P, Rødland EA, Stærfeldt HH, Rognes T, Ussery DW (2007) RNammer: consistent annotation of rRNA genes in genomic sequences. Nucleic Acids Res 35:3100–3108
Langenfelds V (1991) Apple tree systematics. In: Rija, Zinatne, pp 119–195 (in Russian)
Laslett D, Canback B (2004) ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res 32:11–16
Lee ST (2007) Maloideae. In: Park C-W (ed) The genera of vascular plants of Korea. Academy Publishing Co., Seoul, pp 573–584
Li P, Lou G, Cai X, Zhang B, Cheng Y, Wang H (2020a) Comparison of the complete plastomes and the phylogenetic analysis of Paulownia species. Sci Rep 10:2225. https://doi.org/10.1038/s41598-020-59204-y
Li Y, Liu Y, Xu C, Li F, Wang L, Zhou S (2020b) The complete chloroplast genome sequence of Malus toringo (Rosaceae). Mitochondrial DNA Part B-Resour 5:2832–2833
Librado P, Rozas J (2009) DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452
Liu BB, Hong DY, Zhou SL, Xu C, Dong WP, Johnson G, Wen J (2019) Phylogenomic analyses of the Photinia complex support the recognition of a new genus Phippsiomeles and the resurrection of a redefined Stranvaesia in Maleae (Rosaceae). J Syst Evol 57:678–694
Lohse M, Drechsel O, Bock R (2009) Organellar genome DRAW (OGDRAW): a tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr Genet 25:1451–1452
Mahendran R, Spottswood MR, Miller DL (1991) RNA editing by cytidine insertion in mitochondria of Physarum polycephalum. Nature 349:434–438
Matsumoto S, Wakita H, Soejima J (1997) Chloroplast DNA probes as an aid in the molecular classification of Malus species. Sci Hortic 70:81–86
Menezes APA, Resende-Moreira LC, Buzatti RSO, Nazareno AG, Carlsen M, Lobo FP, Kalapothakis E, Lovato MB (2018) Chloroplast genomes of Byrsonima species (Malpighiaceae): comparative analysis and screening of high divergence sequences. Sci Rep 8:1–12
Morgan J, Richards A (2003) The new book of apples. Ebury Press, London
Mower JP (2009) The PREP suite: predictive RNA editors for plant mitochondrial genes, chloroplast genes and user-defined alignments. Nucleic Acids Res 37:W253–W259
Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ (2015) IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol 32:268–274
Nikiforova SV, Cavalieri D, Velasco R, Goremykin V (2013) Phylogenetic analysis of 47 chloroplast genomes clarifies the contribution of wild species to the domesticated apple maternal line. Mol Biol Evol 30:1751–1760
Njuguna W, Liston A, Cronn R, Ashman TL, Bassil N (2013) Insights into phylogeny, sex function and age of Fragaria based on whole chloroplast genome sequencing. Mol Phylogenet Evol 66:17–29
Orešič M, Shalloway D (1998) Specific correlations between relative synonymous codon usage and protein secondary structure. J Mol Biol 281:31–48
Park I, Yang S, Kim WJ, Noh P, Lee HO, Moon BC (2018) The complete chloroplast genomes of six Ipomoea species and indel marker development for the discrimination of authentic Pharbitidis semen (Seeds of I. nil or I. purpurea). Front Plant Sci 9:965. https://doi.org/10.3389/fpls.2018.00965
Parks M, Cronn R, Liston A (2009) Increasing phylogenetic resolution at low taxonomic levels using massively parallel sequencing of chloroplast genomes. BMC Biol 7:84
Phipps JB, Robertson KR, Smith PG, Rohrer JR (1990) A checklist of the subfamily Maloideae (Rosaceae). Can J Bot 68:2209–2269
Piot A, Hackel J, Christin PA, Besnard G (2018) One-third of the plastid genes evolved under positive selection in PACMAD grasses. Planta 247:255–266. https://doi.org/10.1007/s00425-017-2781-x
USDA Natural Resources Conservation Service Website (n.d.). https://plants.usda.gov/core/profile?symbol=MABA. Accessed on 31 Aug., 2020
Qian GZ, Li LF, Tang GG (2006) A new section in Malus (Rosaceae) from China. Ann Bot Fenn 43:68–73
Qian GZ, Liu LF, Hong DY, Tang GG (2008) Taxonomic study of Malus section Florentinae (Rosaceae). Bot J Linn Soc 158:223–227. https://doi.org/10.1111/j.1095-8339.2008.00841.x
Raspé O (2001) Inheritance of the chloroplast genome in Sorbus aucuparia L. (Rosaceae). J Hered 92:507–509
Rehder A (1974) Manual of cultivated trees and shrubs exclusive of the subtropical and warm temperate regions, 2nd edn. Macmillan, New York
Robinson JP, Harris SA, Juniper BE (2001) Taxonomy of the genus Malus Mill. (Rosaceae) with emphasis on the cultivated apple, Malus domestica Borkh. Plant Syst Evol 226:35–58
Savelyeva EN, Kudryavtsev AM (2015) AFLP analysis of genetic diversity in the genus Malus Mill. (Apple). Russ J Genet 51:966–973
Savelyeva E, Kalegina A, Boris K, Kochieva E, Kudryavtsev A (2017) Retrotransposon-based sequence-specific amplified polymorphism markers for the analysis of genetic diversity and phylogeny in Malus Mill. (Rosaceae). Genet Resour Crop Evol 64:1499–1511
Scheffler K, Seoighe C (2005) A Bayesian model comparison approach to inferring positive selection. Mol Biol Evol 22:2531–2540
Sloan DB, Triant DA, Forrester NJ, Bergner LM, Wu M, Taylor DR (2014) A recurring syndrome of accelerated plastid genome evolution in the angiosperm tribe Sileneae (Caryophyllaceae). Mol Phylogenet Evol 72:82–89. https://doi.org/10.1016/j.ympev.2013.12.004
Tang L, Li J, Tan S, Li MX, Ma X, Zhou ZQ (2014) New insights into the hybrid origin of Malus toringoides and its close relatives based on a single-copy nuclear gene SbeI and three chloroplast fragments. J Syst Evol 52:477–486
Terakami S, Matsumura Y, Kurita K, Kanamori H, Katayose Y, Yamamoto T, Katayama H (2012) Complete sequence of the chloroplast genome from pear (Pyrus pyrifolia): genome structure and comparative analysis. Tree Genet Genomes 8:841–854. https://doi.org/10.1007/s11295-012-0469-8
Tsudzuki T, Wakasugi T, Sugiura M (2001) Comparative analysis of RNA editing sites in higher plant chloroplasts. J Mol Evol 53:327–332
Velasco R, Zharkikh A, Affourtit J, Dhingra A, Cestaro A, Kalyanaraman A, Fontana P, Bhatnagar SK, Troggio M, Pruss D, Salvi S, Pindo M, Baldi P, Castelletti S, Cavaiuolo M, Coppola G, Costa F, Cova V, Dal Ri A, Goremykin V, Komjanc M, Longhi S, Magnago P, Malacarne G, Malnoy M, Micheletti D, Moretto M, Perazzolli M, Si-Ammour A, Vezzulli S, Zini E, Eldredge G, Fitzgerald LM, Gutin N, Lanchbury J, Macalma T, Mitchell JT, Reid J, Wardell B, Kodira C, Chen Z, Desany B, Niazi F, Palmer M, Koepke T, Jiwan D, Schaeffer S, Krishnan V, Wu C, Chu VT, King ST, Vick J, Tao Q, Mraz A, Stormo A, Stormo K, Bogden R, Ederle D, Stella A, Vecchietti A, Kater MM, Masiero S, Lasserre P, Lespinasse Y, Allan AC, Bus V, Chagné D, Crowhurst RN, Gleave AP, Lavezzo E, Fawcett JA, Proost S, Rouzé P, Sterck L, Toppo S, Lazzari B, Hellens RP, Durel CE, Gutin A, Bumgarner RE, Gardiner SE, Skolnick M, Egholm M, van de Peer Y, Salamini F, Viola R (2010) The genome of the domesticated apple (Malus domestica Borkh.). Nat Genet 42:833–839. https://doi.org/10.1038/ng.654
Volk GM, Henk AD, Baldo A, Fazio G, Chao CT, Richards CM (2015) Chloroplast heterogeneity and historical admixture within the genus Malus. Am J Bot 102:1198–1208
Wang L, Zhang H, Jiang M, Chen H, Huang L, Liu C (2019) Complete plastome sequence of Iodes cirrhosa Turcz., the first in the Icacinaceae, comparative genomic analyses and possible split of Idoes species in response to climate changes. PeerJ 7:e6663. https://doi.org/10.7717/peerj.6663
Wang ML, Barkley NA, Jenkins TM (2009) Microsatellite markers in plants and insects. Part I: Applications of biotechnology. Genes Genomes Genomics 3:54–67
Williams AH (1982) Chemical evidence from the flavonoids relevant to the classification of Malus species. Bot J Linn Soc 84:1–39
Wyman SK, Jansen RK, Boore JL (2004) Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20:3252–3255
Yang JY, Pak J-H, Kim S-C (2018) The complete plastome sequence of Rubus takesimensis endemic to Ulleung Island, Korea: insights into molecular evolution of anagenetically derived species in Rubus (Rosaceae). Gene 668:221–228. https://doi.org/10.1016/j.gene.2018.05.071
Yang JY, Kang GH, Pak JH, Kim SC (2020) Characterization and comparison of two complete plastomes of Rosaceae species (Potentilla dickinsii var. glabrata and Spiraea insularis) endemic to Ulleung Island, Korea. Int J Mol Sci 21:4933
Yang Z (1997) PAML: a program package for phylogenetic analysis by maximum likelihood. Bioinformatics 13:555–556
Yang Z, Nielsen R, Goldman N, Pedersen AMK (2000) Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics 155:431–449
Yang Z, Wong WS, Nielsen R (2005) Bayes empirical Bayes inference of amino acid sites under positive selection. Mol Biol Evol 22:1107–1118
Zerbino DR, Birney E (2008) Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18:821–829
Acknowledgements
We thank Pan Li for his assistance with fieldwork in China, and Seon-Hee Kim, Seon A Yun, and Ji-Hyeon Jeon for field assistance in China, Japan, and Korea. We are also indebted to the anonymous reviewers for their helpful comments and suggestions on the earlier version of the manuscript.
Funding
This work was supported by “A Study on Utilization of Digitalized DNA Sequences in Plant Resources Based on Next Generation Sequence Analysis” (project number NIBR201803201) and “A study on plant resources based on DNA sequences utilizing Next Generation Sequencing technique (2nd year)” (project number NIBR201905201), which were funded by the National Institute of Biological Resources, Korea.
Author information
Authors and Affiliations
Contributions
Conceptualization, S-C.K. and M-S.C.; resources, M-S.C., M.M. and T.Y.; formal analysis, M-S.C. and J.K.; writing – original draft preparation, M-S.C.; writing – review and editing, S-C.K and M.M. All authors have read and agree with the published version of the manuscript. The authors declare that they have no conflict of interest.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Communicated by G.G. Vendramin
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Figure S1.
Relative synonymous codon usage (RSCU) of the protein-coding genes in eight chloroplast genomes of wild Malus species. Amino acids are shown on the x-axis, and the stacked RSCU values in each bar column are shown on y-axis. Each amino acid contains eight clustered bar columns representing eight plastomes; columns 1 through 8 within each amino acid column group represent M. toringo (Korea), M. toringo (Japan), M. toringo (China), M. Baccata, M. angustifolia, M. sieversii, M. tschonoskii, and M. trilobata, respectively (PNG 111 kb)
Figure S2.
Amino acid changes in predicted RNA editing sites in eight Malus plastomes. The scores (the proportion of sites that have the same amino acid at that position) of each editing site are shown on the y-axis. Color bricks indicate the RNA editing effect: Alanine to Valine, A→V; Histidine to Tyrosine, H→Y; Leucine to Phenylalanine, L→F; Proline to Leucine, P→L; Proline to Serine, P→S; Arginine to Cysteine, R→C; Arginine to Tryptophan, R→W; Serine to Phenylalanine, S→F; Serine to Leucine, S→L; Threonine to Isoleucine, T→I; and Threonine to Methionine, T→M (PNG 83 kb)
Figure S3.
Simple sequence repeat (SSR) numbers per motif length and motif type of the eight Malus plastomes. (A) Number of SSR motif. (B) Number of SSR motif type (PNG 248 kb)
Figure S4.
Repeat numbers per repeat type and repeat length of eight Malus plastomes. (A) Number of repeats in the distribution of forward, reverse, complement, and palindromic repeats. (B) Number of different repeat lengths (PNG 186 kb)
Table S1.
Codon-anticodon recognition pattern and codon usage for eight Malus chloroplast genomes of M. toringo, M. baccata, M. angustifolia, M. sieversii, M. tschonoskii, and M. trilobata. (XLSX 24 kb)
Table S2.
Predicted RNA editing sites in eight chloroplast genomes of wild Malus crabapples of M. toringo, M. baccata, M. angustifolia, M. sieversii, M. tschonoskii, and M. trilobata. (XLSX 32 kb)
Table S3.
The frequency of SSR candidates of eight chloroplast genomes of wild Malus crabapples of M. toringo, M. baccata, M. angustifolia, M. sieversii, M. tschonoskii, and M. trilobata. (XLSX 13 kb)
Table S4.
The SSR candidates of eight chloroplast genomes of wild Malus crabapples of M. toringo, M. baccata, M. angustifolia, M. sieversii, M. tschonoskii, and M. trilobata. (XLSX 23 kb)
Rights and permissions
About this article

Cite this article
Cho, MS., Kim, J.H., Yamada, T. et al. Plastome characterization and comparative analyses of wild crabapples (Malus baccata and M. toringo): insights into infraspecific plastome variation and phylogenetic relationships. Tree Genetics & Genomes 17, 41 (2021). https://doi.org/10.1007/s11295-021-01520-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11295-021-01520-z