
Abstract
Alkalophilic Cellulosimicrobium cellulans CKMX1 isolated from mushroom compost is first report on actinomycetes that has the ability to produce thermostable cellulase-free xylanase, which is an important industrial enzyme used in the pulp and paper industry. Strain CKMX1 was characterized by metabolic fingerprinting, whole-cell fatty acids methyl ester analysis and 16Sr DNA and found to be C. cellulans CKMX1.The enzyme was purified by gel permeation and anion exchange chromatography and had a molecular mass of 29 kDa. Xylanase activity was optimum at pH 8.0 and 55 °C. The enzyme was somewhat thermostable, retaining 50 % of the original activity after incubation at 50 °C for 30 min. The xylanase had K m and V max values of 2.64 mg/ml and 2,000 µmol/min/mg protein in oat spelt xylan, respectively. All metal ions except HgCl2, CoCl2 as well as CdCl2 were well tolerated and did not adversely affect xylanase activity. The deduced internal amino acid sequence of C. cellulans CKMX1 xylanase by matrix assisted laser desorption ionization-time of flight mass spectrometry resembled the sequence of β-1,4-endoxylanase, which is a member of glycoside hydrolase family 11. Some of the novel characteristics that make this enzyme potentially effective in xylan biodegradation could be useful for pulp and paper biobleaching are discussed in this manuscript.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cellulose and hemicelluloses are the two major components of lignocelluloses generated by photosynthesis. Xylan, composed of a linear backbone of 1,4-β-linked d-xylose units, is the major component of hemicellulose (Wong et al. 1988). Endo-xylanase (endo-1,4-β-xylanase, EC 3.2.1.8) and β -xylosidase (xylan-1,4-β-xylosidase, EC 3.2.1.37) are the two key enzymes (collectively xylanases) responsible for the hydrolysis of xylan. Endo-xylanase act on homopolymeric back bone of 1,4-linked β-d-xylopyranose producing xylooligomers (Collins et al. 2005), while β-xylanosidases act on these xylooligomers releasing xylose (Knob et al. 2010). However, a complete degradation requires the synergistic action of acetyl esterase to remove the acetyl substituents from the β-1,4-linked d-xylose backbone of xylan (Subramaniyan and Prema 2002).
Xylanase has great potential as a starting material for the production of useful end products such as xylooligosaccharides (XOs) (van Zyl et al. 2001). Xylanase and microorganisms that produce them have wide biotechnological and industrial applications. They are used in the management of waste, to degrade xylan to renewable fuels, production of bulk chemicals, enzymatic treatment of animal feeds to release pentose sugars, as food additives in baking industry, clarification of juices and ingredients in laundry detergents or fabric care compositions (Wong et al. 1988; Collins et al. 2002). One of the most important biotechnological applications of xylanase is its use in pulp bleaching primarily to reduce lignin and increase the brightness of the pulp (Viikari et al. 1994).
Also, not just only alkali and thermal-stable xylanases are “useful”; more and more studies focused on the cold-active enzymes. There is enduring need for a broad range of novel enzymes and biocatalysts which are required for various industrial and biotechnological applications. The cold environments are extremely diverse in nature with fluctuation in temperature, radiation and pressure. These environments present largely “untapped” bioresource and the microorganisms flourishing, there have great potential for isolation of new and novel enzymes (Chen et al. 2000, 2013).
So far, several xylanases have been isolated from different microorganisms, including bacteria (actinomycetes, eubacteria, and archaea), fungi, and yeasts (Shi et al. 2011; Walia et al. 2012a, b). Various xylan-degrading thermotolerant actinomycetes, such as Cellulosimicrobium (Kim et al. 2012), Actinomadura (Sriyapai et al. 2011), Nonomuraea (Leskinen et al. 2005), Microtetraspora (Berens et al. 1996), Streptomyces (Zhou et al. 2010) and Thermomonospora (George et al. 2001) have been purified and characterized. Some researchers seem, however, to have paid special attention to xylanases from actinomycetes origins particularly because of the promising potential they offer for a wide range of biotechnological applications particularly in the pulp and paper industry (Beg et al. 2000). In fact, in order for a xylanase to achieve actual industrial application, it should ideally fulfil a number of specific requirements that are highly desired in the marketplace. Some of these characteristics that pertain to operational standards, such as alkaline pH, high thermostability, high specific activity, and strong resistance to metal cations and chemicals. Other specifications relate to cellulase-free xylanase, cost-effectiveness, eco-friendliness and ease of use.
There have been reports since 1982 regarding the purification of xylanases from various microorganisms (Li et al. 2010; Mamo et al. 2006; Bataillon et al. 2000). However, the purification and characterization of xylanase from Cellulosimicrobium sp. requires special consideration due to its growing need in various industries. Despite the large flow of data on xylanases, however, only a few reports have so far dealt with the isolation of xylanases from Cellulosimicrobium sp. A few studies report on the purification and characterization of xylanases from Cellulosimicrobium cellulans (Kim et al. 2012). This is due to their limited potential for industrial application, since the xylanases previously purified from Cellulosimicrobium sp. had poor enzyme stability and catalytic activity. Low molecular mass and thermostable xylanases are of industrial importance because they can better diffuse into the biomass structure or fibrous pulp and can thus efficiently hydrolyzed xylan in biomass hydrolysis or pulp bleaching. The present study focuses on the purification to homogeneity and biochemical characterization of alkaline cellulase-free xylanase produced by the bacterium C. cellulans CKMX1 isolated from mushroom compost.
Materials and methods
Strain isolation
Alkalophilic C. cellulans CKMX1 was isolated from mushroom compost taken from Directorate of Mushroom Research, Solan. Xylan degrading bacteria were isolated by the enrichment technique. The most predominant bacterial colonies capable of good growth on basal salt medium (BSM) with the following composition (g/l): Na2HPO4, 6.0; KH2PO4, 3.0; NaCl, 0.5; NH4Cl, 1.0, 1 M MgSO4 (2 ml) and 1 M CaCl2 (0.1 ml) were picked and purified. An isolate yielding a clear hydrolysis zone on BSM containing 0.5 % xylan by Congo red plate assay. The bacterial culture was grown and maintained in BSM, pH 8.0, containing 0.5 % xylan. The bacterial culture was maintained in 30 % glycerol at −20 °C.
Morphological characterization
Morphological characteristics of isolates including colony morphology, Gram’s reaction, cell shape, presence of spores were investigated (Sneath 1994).
Metabolic fingerprinting
Metabolic fingerprinting was done by using Bergey’s mannual of systematic bacteriology (Sneath 1994) and commercial kits i.e. KB009 Hi carbohydrate™ kit (HiMedia,India).
Whole-cell fatty acids methyl ester (FAME) analysis
The isolates were identified based on whole-cell fatty acids, derivatized to methyl esters, and analyzed by gas chromatography using the Sherlock Microbial Identification System (MIS-MIDI, USA). The FAME profiles were compared with the TSBA50 aerobe library general system software v5.0. Qualitative and quantitative differences in the fatty acid profiles were used to compute the distance for each strain relative to the strains in the library (Sasser 1990; Sasser and Wichman 1991). FAME analysis was performed at Institute of Microbial Technology (IMTECH), Chandigarh. India.
Molecular taxonomic characterization
Bacterial isolate was grown in nutrient broth at 35 °C overnight. Bacterial cells were harvested by centrifugation at 5,000×g for 5 min and DNA was isolated from these bacterial cells by using Real genomic DNA extraction kit (Taiwan). The isolated DNA was finally suspended in 100 µl of elution buffer and quantified on 1 % agarose gel. The total genomic DNA was kept at −20 °C before use (Sambrook and Russel 2001).
Species level identification of strain was conducted by 16S rDNA sequence comparison. PCR reaction was carried out in 20 µl reaction containing ~50 ng of template DNA, 20 pmol of each primer fC1 (5′-GCAAGTCGAGCGGACAGATGGGAGC-3′) and reverse primer rC2 (5′-AACTCTCGTGGTGTGACGGGCGGTG-3′), 0.2 mM dNTPs and 1 U Taq polymerase (Genei Bangalore) in 1× PCR buffer. Reaction were cycled 35 times as 94 °C for 30 s, 58 °C for 30 s, 72 °C for 1 min 30 s followed by final extension at 72 °C for 10 min. The PCR products were analyzed on 1 % agarose gel in 1× TAE buffer, run at 100 V for 1 h. Gels were stained with ethidium bromide and photographed. Amplified PCR products were eluted from the gel using gel extraction kit (Real genomic (Hi Yield ™ Gel/PCR DNA Extraction Kit), eluted fragment was then sequenced (Xcleris, India) using PCR primers.
Sequence analysis
The sequence was aligned with corresponding sequences of 16S rDNA from the database using BLAST (Altschul et al. 1997). Multiple alignments were generated by the MULTALIN program (Corpet 1988). Phylogenetic tree was constructed with the help of ClustalW (Higgins et al. 1994) Tree was viewed with the help of TreeView (Page 1996).
Protein determination
The concentration of protein in culture supernatant was estimated using the Lowry’s method of protein estimation with bovine serum albumin as a standard.
Enzyme activity assay
Xylanase activity was assayed using 1 % oat spelt xylan (Sigma, St, Louis, MO) in 0.2 M Tris–HCl buffer (pH 8.0) The reaction mixture contained 0.5 ml 1 % oat spelt xylan in Tris–HCl buffer (0.2 M, pH 8.0) and 0.5 ml diluted enzyme. It was incubated at 50 °C for 5 min in a water bath with occasional shaking. The release of reducing sugars was determined using the 3,5-dinitrosalicylic acid (DNSA) method with a xylose standard curve according to the colorimetric method of Miller (1959). After incubation, 3 ml DNSA reagent was added into the test tubes, which stopped the enzymatic reaction. The tubes were immersed in boiling water bath and removed after 15 min when colour development was completed. Tubes were cooled to room temperature. The contents were transferred to a 25 ml volumetric flask and final volume made up to 25 ml with distilled water. Optical density was read at 540 nm in a Spectronic-20 by using appropriate blank. One unit (IU) of enzyme activity was defined as the amount of enzyme required to liberate 1 μmol reducing sugars per minute per ml under given assay conditions. Xylanase activity is expressed as U/g dry bacterial pomace (DBP).
Enzyme preparation
Solid state fermentation was carried out in Erlenmeyer flasks (250 ml) containing 10 g substrate (apple pomace) and 25 ml mineral salt solution (BSM) at pH 8.0 were autoclaved at 15 psi pressure for 20 min, cooled and inoculated with 2 ml bacterial suspension (OD 1.0 at 540 nm). After mixing, the flasks were incubated at 35 °C for 3 days. After 72 h, the flasks were taken out and the contents were extracted with 50 ml sterilized buffer (0.2 M, pH 8.0, Tris HCl). The flasks were kept in shaker for half an hour to ensure thorough mixing of apple pomace with the buffer. The culture supernatant was obtained following centrifugation at 10,000 rpm for 20 min at 4 °C. Ammonium sulphate was added to the supernatant to 80 % saturation. The mixture was left overnight and then the precipitate was recovered by centrifugation at 10,000 g for 20 min at 4 °C. The precipitate was dissolved in 50 mM phosphate buffer (pH 8.0) and dialysed against the same buffer for 48 h. Dialysis was performed using tubing cellulose membrane with a molecular weight cut off of 10–15 kDa (Sigma). The dialysed fraction was the crude enzyme extract used for further purification.
Purification of xylanase
All purification steps were performed at 4 °C. The crude enzyme extract was purified using gel filtration chromatography and ion exchange chromatography. For gel filtration chromatography, the crude enzyme solution was applied to Sephadex G-100 column (2.0 × 30 cm) (Genei Bangalore). The enzyme was eluted with 20 mM Tris–HCl buffer (pH 8.0) at a flow rate of 0.5 ml/min and 2 ml fractions were collected. The active fractions from the column that exhibited xylanase activity were pooled and concentrated using a Millipore Ultrafiltration with molecular weight cut off at 10 kDa (Millipore, Billerica, USA). The concentrate was then dialysed against 50 mM Tris–HCl buffer (pH 8.0) with five changes and further purified by anion exchange chromatography. The enzyme solution was applied to DEAE-Cellulose column (1.0 × 15 cm) equilibrated with elution buffer (20 mM Tris HCl, pH 8.0; 0.05 M NaCl). Elution was performed with a linear gradient of 0.1–0.5 M NaCl in a same buffer at a flow rate of 0.2 ml/min. The purified samples were collected in a collection tube. Fractions exhibiting xylanase activity were pooled and concentrated using an Millipore Ultrafiltration with molecular weight cut off at 10 kDa were used as described later.
SDS-PAGE and zymogram analysis
The molecular mass of the purified xylanase was estimated by SDS-PAGE, which was performed using 12 % polyacrylamide gel by the method described by Matsuoka et al. (2007). Zymogram analysis was performed using 0.1 % oat-spelt xylan (w/v) incorporated into the polyacrylamide during gel preparation. Following sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE), gels were washed four times for 30 min at 4 °C in renaturation buffer (100 mM KH2PO4/NaOH, pH 7.0). The first two washes contained 25 % (v/v) isopropanol. Clearing zones that correspond to regions of enzyme activity were visualised using 0.5 % (w/v) Congo red. The xylanase band was revealed against the dark background by treatment with 0.5 % acetic acid solution.
Characterization of xylanase
Temperature optimum and thermal stability
The optimum temperature of the purified xylanase of C. cellulans CKMX1 was determined in 0.2 M Tris–HCl buffer, pH 8.0 by varying reaction temperature from 35 to 70 °C. The thermal stability was also assessed at 50–60 °C for 0–3 h in 0.1 M Tris- HCl buffer, pH 8.0 and activity was measured after an interval of 10 min.
pH optimum
The optimum pH of the purified xylanase of C. cellulans CKMX1 was determined by incubating the purified enzyme for 1 h at 35 °C in the following buffer (0.1 M): citrate phosphate buffer (pH 4.0–5.0), potassium phosphate buffer (pH 6.0–7.0) and Tris–HCl buffer (pH 8.0–10.0) and enzyme activity was assayed.
Effect of metallic ions and enzyme inhibitors on xylanase activity
The influence of metal ions and enzyme inhibitors on xylanase activity was investigated under the standard assay conditions. The purified xylanase was incubated in the presence of 1 mM solution of metallic ions (MnSO4, ZnSO4, NaCl, MnCl2, CoCl2, CdCl2, CaCl2, HgCl2 and inhibitors (EDTA, DTT, 2-mercaptoethanol) for 1 h at room temperature (25 ± 2 °C) and the activity of purified xylanase was investigated. The activity was expressed as a percentage of the activity level in the presence of metallic ions or inhibitors. Xylanase activity measured in the absence of any compound was taken as control (100 %). Residual activity was measured using the enzyme assay described above.
Substrate specificity and kinetic measurements
The kinetic parameters (K m and V max ) of the purified xylanase was deduced by determining initial reaction velocity of the enzyme at various concentration of substrate (1–20 mg/ml) and then plotting 1/[S] versus 1/[V] in Lineweaver–Burk plot.
Determination of internal amino acid sequence of xylanase by MALDI-TOF MS
The internal amino acid sequence of xylanase was determined by subjecting the digested mixture of peptides to an enzyme i.e. trypsin to matrix assisted laser desorption ionization-time of flight mass spectrometry (MALDI-TOF MS) analysis for peptide mass fingerprinting and protein sequencing of the digestion products by mass spectrometry using Bruker Ultraflex MALDI-TOF MS and a MALDI-spectrum was obtained (Zhang et al. 2001). The mass-to-charge ratio of the resulting peptides gave information to identify the proteins in the sample. This was done by using the matrix-assisted laser desorption ionization mass spectrometry (MALDI-MS) procedure. The experimentally obtained mass-to-charge values was matched against theoretical obtained mass to-charge values data from already identified protein sequences and a score depending on the correlation was given. The analysis was performed at Merck Millipore Bioscience at Bangalore. The data was analyzed using MASCOT search engine.
Results
Isolate identification
Strain CKMX1 was isolated from mushroom compost and showed the clear zones on oat-spelt xylan agar plates following staining with 1 % Congo red solution, indicating that it secretes considerable amounts of xylanase. In solid state fermentation (SSF), strain CKMX1 produced cellulase-free xylanases i.e. CMCase, FPase, avicelase and β-glucosidase were not detected in the enzyme supernatant (Data not shown) (Walia et al. 2012b).
Characterization of Cellulosimicrobium cellulans CKMX1
Phenotypic and metabolic characteristics of Cellulosimicrobium cellulans CKMX1
The strain CKMX1 was characterized initially according to morphological, physiological and biochemical characteristics (Table 1). The isolated colonies on basal salt medium after 48 h of incubation were cream colour, circular with a smooth surface, convex elevation, 0.1–0.5 mm in diameter and entire edged. The morphological characteristics of the strain were as follows: substrate hyphae were present, the cells were Gram positive, non-spore forming and non-motile. Young cultures were composed of straight or curved rods. The rods occurred singly or in chains.
The physiological and biochemical characteristics of the isolated strain CKMX1 are given in Table 1. The isolate is an aerobic, catalase producing strain. Nitrate was reduced to nitrite and starch was hydrolyzed. The isolate tested positive for gelatine liquefaction. The strain was positive for cellulolytic and esculin hydrolysis and could utilize a wide array of carbohydrates (assessed using a KB009 Hicarbohydrate™ Kit (HiMedia, India)), including xylose, maltose, fructose, dextrose, galactose, trehalose, sucrose, l-arabinose, mannose, inulin, sodium gluconate, glycerol, salicin, cellobiose, xylitol, d-arabinose, as sole carbon source.
FAME analysis
FAME analysis showed the composition of cell-wall fatty acids of isolate. The fatty acids Iso-C14:0, C14:0, Iso-C15:0, Anteiso-C15:0, C16:0, Iso-C17:0, Anteiso-C17:0 was present in the isolate (Table 2). The analysis showed the highest similarity of the isolate with C. cellulans as per MIDI system (Microbial Identification System, Inc.). Isolate exhibited a specific fatty acid composition, making it a “microbial fingerprint.”
Phylogenetic analysis of Cellulosimicrobium cellulans CKMX1 according to 16Sr DNA sequence
Universal primers were used successfully to amplify 16S rDNA from bacterial isolate CKMX1, yielding an amplicon of the expected size, i.e. ~1136 bp. The sequence of 16S rDNA from CKMX1 was then analyzed using BLASTn analysis (http://www.ncbi.nlm.nih.gov/blast) and was found to have 97 % homology with several C. cellulans strains reported from different parts of the world. The 16S rDNA sequence of CKMX1 was also compared with the corresponding sequences of different Cellulosimicrobium sp. reported from different parts of the world. Sequence analysis revealed that CKMX1 belongs to C. cellulans strain CKMX1 as it showed maximum homology (97 %) with C. cellulans strain AMP-11 (accession no. HM104377).
To trace out the evolutionary patterns of the test isolate and to determine the relationship with other selected sequences at NCBI, a phylogenetic tree was also constructed using the neighbor-joining (J) method of mathematical averages (UPGMA) among 16Sr DNA sequence of CKMX1 and the corresponding sequence of eight different Cellulosimicrobium sp. Strain CKMX1 was united with quite high statistical support by the bootstrap estimates for 1,000 replications. The resulting phylogenetic tree (Fig. 1) also verified CKMX1 as C. cellulans strain CKMX1 as the strain CKMX1 clustered closely with C. cellulans with high boot strap value (80 %). The 16S rDNA sequence of the strain has been deposited in the GenBank database under accession number JN135476. Based on above morphological, biochemical and molecular characterization, the strain CKMX1 was identified as C. cellulans CKMX1.

Neighbour-joining tree based on 16S rRNA gene sequences showing the phylogenetic relationship of strain CKMX1. The numbers at the nodes indicate the levels of bootstrap support based on data for 1,000 replicates; values inferred greater than 50 % are only presented. The scale bar indicates 10 substitutions per nucleotide position
Enzyme purification
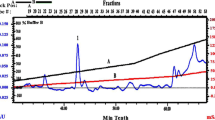
After 3 days of cultivation of C. cellulans CKMX1 in an alkaline medium in the presence of apple pomace as a substrate for xylanase production in SSF, the extracellular xylanase was detected at 940.30 U/g DBP in the culture supernatant (Table 3). The crude enzyme, obtained as a cell-free supernatant, was precipitated using ammonium sulphate to 80 % saturation. The crude extract from the culture medium was taken through the two-step purification of gel filtration and ion-exchange chromatography. The profiles of elution in gel filtration chromatography exhibited various protein peaks, and a major xylanase peak (fraction no. 42-54) (Fig. 2); the active fractions were pooled, concentrated and injected into the ion exchange column, the xylanase was eluted as a single peak, showing that purer xylanase was obtained (fraction no. 66-72) (Fig. 3). Table 3 represents the purification of xylanase. The purified xylanase exhibited a specific activity of 36.62 U/mg. An overall recovery of 20.28 % and 5.92-fold purification of CKMX1 xylanase were observed. The purified xylanase protein appeared as single band on SDS-PAGE with an estimated molecular mass of 29 kDa, respectively (Fig. 4a). A zymogram of the xylanase exhibited a band with significant activity corresponding to 29 kDa (Fig. 4b). The Congo red interacts strongly with polysaccharides containing contiguous β (1-4) linked reducing sugar units. The resulting dye-glucan complexes are intensely coloured making them very sensitive methods of detection of such polysaccharides. Hence (Fig. 4b) in the presence of active xylanase, the substrate xylan in the gel is hydrolysed and so there is a less intense colouration (Teather and Wood 1982).

Protein and xylanase activity profile of fractions of Sephadex G-100 column chromatography of the ammonium sulphate fractionation of C. cellulans CKMX1

Protein and xylanase activity profile of fractions of DEAE column chromatography of xylanase rich pooled fractions of gel filtration chromatography

a Sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS-PAGE) (12 %) of xylanase preparations at various stages of purification. M Medium range molecular weight marker; Lane A crude enzyme (10 µg); Lane B ammonium sulphate fractionation (10 µg); Lane C Sephadex G-100 column chromatography pooled fractions (10 µg); Lane D DEAE column chromatography pooled fractions (10 µg); b zymogram of purified xylanase
Effect of pH and temperature on activity and stability of xylanase
The temperature optimum of purified xylanase was 60 °C (Fig. 5a) and the enzyme was stable over the range of 50–60 °C (Fig. 5b). The enzyme stability declined rapidly as the temperature increased above 50 °C, and retained 50 % activity at 50 °C and pH 8.0 with in the first 30 min and after that a gradual decrease was found and it was rapidly inactivated at 60 °C after 30 min (Fig. 5b). The xylanase purified from the C. cellulans CKMX1 was more stable at 50 °C than 55 and 60 °C.

a Effect of reaction temperature on xylanase activity of C. cellulans CKMX1 and reactions were performed for 5 min at various temperature 35–70 °C. b Effect of time on thermal stability of C. cellulans CKMX1. The enzyme was preincubated at 50 °C (circles), 55 °C (squares) and 60 °C (triangles). After incubation, all samples were analyzed for xylanase activity, using oat spelt xylan as a substrate 0.5 % under standard conditions. c Effect of buffer system and pH on xylanase activity. Reaction was carried out at 55 °C using oat spelt xylan as substrate 0.5 % for 5 min in citrate buffer (pH 4.0–5.0), potassium phosphate buffer (pH 6.0–7.0) and Tris–HCl buffer (pH 8.0–10.0)
The pH optimum of the xylanase was measured at constant temperature (55 °C) and substrate concentration (0.5 %). The purified xylanase had a broad optimal pH range (5–9), with maximum activity at pH 8.0 (Fig. 5c). More than 85 % of the maximum observed activity of the enzyme was reached between pH 6 and 8. The activity of the enzyme beyond this range was low. At pH 7.0 or pH 8.0, the enzyme activity was retained more than 85 %; however, at pH 10.0, it reduced to only 50 % (Fig. 5c).
Effect of metallic ions and enzyme inhibitors on xylanase activity
Various cations and compounds were assayed at a concentration of 1 mM to investigate their effects on xylanase activity (Table 4). Among eleven metal ions and inhibitors tested, CaCl2 addition to the purified enzyme increased the activity i.e. 8.90 %; HgCl2 caused maximum inhibition i.e. 67.85 % while CoCl2 and CdCl2 caused 36 and 26.85 %, respectively. The other metal ions tested had no appreciable effect on the activity of this xylanase. The metalloenzymes (metalloproteins) can be readily distinguished from other proteins by treatment with metal chelating agents such as EDTA, which chelate metal ion cofactor and inactivate the enzyme. However, in the present study, EDTA could not significantly reduce the activity of purified xylanase of C. cellulans CKMX1. DTT and 2-mercaptoethanol are reducing agents and break the disulphide bond between two cysteine residues of protein and thus interfere with the folding of enzyme protein leading to its inactivation and these reagents did not reduce the activity of purified xylanase of C. cellulans CKMX1.
Enzyme kinetics
The kinetic parameters K m and V max of the enzyme were determined from Lineweaver–Burk double reciprocal plots of xylanase activity at 60 °C using various concentrations of oat-spelt xylan as substrate (Fig. 6). A rapid increase in the rate of reaction was observed with an increase in the concentration of xylan up to 8 %, beyond which the reaction rate stabilized. The K m value of xylanase was 2.64 mg/ml and V max was 2,000 µmol/min/mg protein, respectively (data not shown).

Lineweaver–Burk plot of purified xylanase of C. cellulans CKMX1
Internal amino acid sequence of xylanase determined by MALDI-TOF MS
An amino acid sequence of the purified enzyme from the digested protein analysed by MALDI-TOF was identified as PRVLARRRGERDEVGRDRTRPGLVLLGGRGPGRELRGRAG. The results of a MASCOT from Matrix-Science search indicated that this partial amino acid sequence showed 85 % homology with endo-beta-1,4-xylanase (xylK-1) gene (accession number ACY75514.1) of the glycoside hydrolase family 11 from Cellulosimicrobium sp. Glycoside hydrolases family 11 (EC 3.2.1-) constitute a large group of enzymes that hydrolyse the glycosidic bond between at least two carbohydrates or between a carbohydrate and a non-carbohydrate moiety, which is monospecific and that consists solely of xylanase (Collins et al. 2005; Henrissat 1991).
Discussion
This is the first report on the purification and characterization of Cellulosimicrobium celllulans CKMX1 isolated from mushroom compost. In this study, we characterized a strain of C. cellulans CKMX1 capable of producing cellulase-free xylanase. According to the morphological, biochemical, FAME analysis and 16S rDNA sequencing isolate was identified as a strain of C. cellulans CKMX1. C. cellulans CKMX1 is one of the unusual bacterial strains that produce low molecular weight cellulase-free, alkali stable xylanase (1,4-d-xylan xylohydrolase, E.C. 3.2.1.8). However, the production of cellulases is invariably associated with the xylanases produced by Cellulosimicrobium sp. reported earlier. In solid state fermentation (SSF), strain CKMX1 produced cellulase-free xylanases i.e. CMCase, FPase, avicelase and β-glucosidase were not detected in the enzyme supernatant (Walia et al. 2012b). So, the important characteristic of this strain to produce low molecular weight cellulase-free alkali stable xylanase makes this enzyme suitable for pulp and paper biobleaching industry. In this work, we have produced cellulase free xylanase from C. cellulans CKMX1 at an activity level of 940.30 U/g DBP in the culture supernatant which has a pH optimum of 8.0 at 55 °C and enzyme was stable over the range of 50–60 °C.
In this investigation, using ion exchange chromatography, gel permeation chromatography and Ultrafiltration, purified enzyme was separated from other proteins. The purified xylanase exhibited a specific activity of 36.62 U/mg. An overall recovery of 20.28 % and 5.92-fold purification of strain CKMX1 xylanase were observed. Cellulase-free xylanase obtained from a moderate thermophile Bacillus licheniformis A99 had a specific activity of 28.7 U/mg (Archana and Satyanarayana 2003). The specific activity of xylanase produced by Bacillus pumilus and Aspergillus ficuum was 298 and 288 U/mg, respectively (Panbangred et al. 1993; Lu et al. 2008). The specific activity of purified xylanase from various microorganisms varies from 28.7 to 1,697.7 U/mg of protein (Inagaki et al. 1998; Khandeparkar and Bhosle 2006b). A zymogram of the xylanase exhibited a band with significant activity corresponding to 29 kDa (Fig. 4a, b). This finding is consistent with that of monomeric xylanase (15–67 kDa) reported in numerous investigations (Lu et al. 2008; Teather and Wood 1982; Nakamura et al. 1993; Waino and Ingvorsen 2003; Wejse et al. 2003).
The temperature optimum of purified xylanase was 60 °C and the enzyme was stable over the range of 50–60 °C (Fig. 5a, b). A similar range of optimal temperatures has been identified for a low molecular weight xylanase from B. pumilus SSP-34 (Subramaniyan 2012). A stability test revealed that the xylanase was stable for 4 h at 50 °C. Alkalophiles are the most likely source of enzymes tolerant to high salt and high temperatures (Sanchez-Porro et al. 2003). On the basis of its properties, the strain CKMX1 xylanase may be suited to these particular conditions. The pH optimum of the xylanase was measured at constant temperature (55 °C) and substrate concentration (0.5 %). The purified xylanase had a broad optimal pH range (6–9), with maximum activity at pH 8.0 (Fig. 5c). The pH optimum for xylanase isolated from many bacteria is mainly in the neutral pH range. Xylanase isolated from Halorhabdus utahensis and Bacillus sp. SPS-0 have an optimum pH of 6.0–8.0 (Waino and Ingvorsen 2003; Bataillon et al. 1998).
Various cations and compounds were assayed at a concentration of 1 mM to investigate their effects on xylanase activity (Table 4). However, in the present study, EDTA could not significantly reduce the activity of purified xylanase of C. cellulans CKMX1. DTT and 2-mercaptoethanol are reducing agents and break the disulphide bond between two cysteine residues of protein and thus interfere with the folding of enzyme protein leading to its inactivation and these reagents did not reduce the activity of purified xylanase of C. cellulans CKMX1. Similar results were found in case of cellulase-free xylanase of a moderate thermophile B. licheniformis A99, where HgCl2 and AgNO3 completely inhibited the xylanase activity whereas it was unaffected by EDTA treatment too (Archana and Satyanarayana 2003).
The kinetic parameters K m and V max of the enzyme were determined from Lineweaver–Burk double reciprocal plots of xylanase activity at 60 °C using various concentrations of oat-spelt xylan as substrate (Fig. 6). The K m value of xylanase was 2.64 mg/ml and V max was 2,000 µmol/min/mg protein, respectively (data not shown). This K m value was lower than those obtained for Bacillus halodurans (Mamo et al. 2006) and Enterobacter sp. MTCC 5112 (Khandeparkar and Bhosle 2006a), indicating that the xylanase from C. cellulans had higher affinity for oat-spelt xylan. There are only few exceptions from Bacillus sp., for example, B. stearothermophilus had K m value 1.63 mg/ml (Khasin et al. 1993). Aeromonas caviae 9.4 mg/ml (Kubata et al. 1992) and Streptomyces T-7 10 mg/ml (Keskar 1992) were the few ones with higher K m values. Similar results were also found with extremely thermophilic xylanases from Thermotoga thermarum with molecular weight of 120 and 40 kDa, have low K m value (Tsenga et al. 2002).
An amino acid sequence from the digested protein was analysed by MALDI-TOF to identify the purified enzyme. The peptide sequence alignment showed areas of sequence homology with endo-β-1,4-xylanase belonging to the glycoside hydrolase family 11 (Henrissat 1991).
In this study, we isolated a novel strain of C. cellulans CKMX1 from mushroom compost that has the ability to produce thermostable xylanase without cellulase activity. The time course for xylanase accumulation by the C. cellulans CKMX1 in solid state fermentation showed that the highest xylanase activity reached 940.30 U/g in a basal salt medium with oat-spelt xylan used as a substrate after 72 h of cultivation. The enzyme showed a broad pH activity profile and optimal pH and temperature for the purified 29 kDa xylanase were pH 8 and 60 °C, respectively. Overall, the findings indicate that the xylanase presented in the current work is endowed with a number of promising properties that are highly valued in the pulp and paper industry.
References
Altschul SF, Thomas LM, Alejandro AS, Jinghui Z, Zheng Z, Webb M, David JL (1997) Gapped BLAST and PSIBLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Archana A, Satyanarayana T (2003) Purification and characterization of a cellulase-free xylanase of a moderate thermophile Bacillus licheniformis A99. World J Microbiol Biotechnol 19:53–57
Bataillon M, Cardinali APN, Duchiron F (1998) Production of xylanase from a newly isolated alkalophilic thermophilic Bacillus sp. Biotechnol Lett 20:1067–1071
Bataillon M, Cardinali APN, Castillon N, Duchiron F (2000) Purification and characterization of a moderately thermostable xylanase from Bacillus sp. strain SPS-0. Enzyme Microb Technol 26:187–192
Beg QK, Bhushan B, Kapoor M, Hoondal GS (2000) Enhanced production of a thermostable xylanase from xylanase from Streptomyces sp. QG-11-3 and its application in biobleaching of eucalyptus kraft pulp. Enzyme Microb Technol 27:459–466
Berens S, Kaspari H, Klemme JH (1996) Purification and characterization of two different xylanases from the thermophilic actinomycete Microtetraspora flexuosa SIIX. Antonie Van Leeuwenhoek 69:235–241
Chen S, Qu Y, Liu X (2000) Purification and properties of alkaline xylanase from Bacillus pumilus A-30. Chin J Biochem Mol Biol 17(3):224–229
Chen S, Kaufman MG, Miazgowiczand KL, Bagdasarian M, Walker ED (2013) Molecular characterization of a cold-active recombinant xylanase from Flavobacterium johnsoniae and its applicability in xylan hydrolysis. Bioresour Technol 128:145–155
Collins T, Meuwis MA, Stals I, Claeyssens M, Feller G, Gerday C (2002) A novel family 8 xylanase, functional and physicochemical characterization. J Biol Chem 277:35133–35139
Collins T, Gerday C, Feller G (2005) Xylanases, xylanase families and extremophilic xylanases. FEMS Microbiol Rev 29:3–23
Corpet F (1988) Multiple sequence alignment with hierarchial clustering. Nucleic Acids Res 16:10881–10890
George SP, Ahmad A, Rao MB (2001) Studies on carboxymethyl cellulase produced by an alkalothermophilic actinomycete. Bioresour Technol 78:221–224
Henrissat B (1991) A classification of glycosyl hydrolases based on amino-acid sequence similarities. Biochem J 280:309–316
Higgins D, Thompson J, Gibson T, Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Inagaki K, Nakahira K, Mukai K, Tamura T, Tanaka H (1998) Gene cloning and characterization of an acidic xylanase from Acidobacterium capsulatum. Biosci Biotechnol Biochem 62:1061–1067
Keskar SS (1992) High activity xylanase from thermotolerant Streptomyces T7: cultural conditions and enzyme properties. Biotechnol Lett 14:481–486
Khandeparkar R, Bhosle N (2006a) Isolation, purification and characterization of the xylanase produced by Arthrobacter sp. MTCC 5214 when grown in solid state fermentation. Enzyme Microb Technol 39:732–742
Khandeparkar R, Bhosle N (2006b) Purification and characterization of thermoalkalophilic xylanase isolated from the Enterobacter sp. MTCC 5112. Res Microbiol 157:315–325
Khasin A, Alchanati I, Shoham Y (1993) Purification and characterization of a thermostable xylanase from Bacillus stearothermophilus T-6. Appl Environ Microbiol 59:1725–1730
Kim DY, Ham S-J, Kim HJ, Kim J, Lee M-H, Cho H-Y, Shin D-H, Rhee YH, Son K-H, Park H-Y (2012) Novel modular endo-β-1,4-xylanase with transglycosylation activity from Cellulosimicrobium sp. strain HY-13 that is homologous to inverting GH family 6 enzymes. Bioresour Technol 107:25–32
Knob A, Terrasan C, Carmona E (2010) β-Xylosidases from filamentous fungi: an overview. World J Microbiol Biotechnol 26:389–407
Kubata KB, Horitsu H, Kawai K, Takamizawa K, Suzuki T (1992) Xylanase of Aeromonas caviae ME-I isolated from the intestine of a herbivorous insect (Samia cynthia pryeri). Biosci Biotechnol Biochem 56:1463–1464
Leskinen S, Mantyla A, Fagerstrom R, Vehmaanpera J, Lantto R, Paloheimo M (2005) Thermostable xylanases, Xyn10A and Xyn11A, from the actinomycete Nonomuraea flexuosa: isolation of the genes and characterization of recombinant Xyn11A polypeptides produced in Trichoderma reesei. Appl Microbiol Biotechnol 67:495–505
Li X, She Y, Sun B, Song H, Zhu Y, Lv Y, Song H (2010) Purification and characterization of a cellulase-free, thermostable xylanase from Streptomyces rameus L2001 and its biobleaching effect on wheat straw pulp. Biochem Eng J 52:71–78
Lu F, Lu M, Lu Z, Bei X, Zhao H, Wang Y (2008) Purification and characterization of xylanase from Asprgillus ficuum AF-98. Bioresour Technol 99:5938–5941
Mamo G, Hatti-Kaul R, Mattiasson B (2006) A thermostable alkaline active endo-β-1-4-xylanase from Bacillus halodurans S7: purification and characterization. Enzyme Microb Technol 39:1492–1498
Matsuoka S, Yukawa H, Inmi M, Doi RH (2007) Synergistic interaction of Clostridium cellulovorans cellulosomal cellulase and HbpA. J Bacteriol 189:7190–7194
Miller GL (1959) Use of dinitrosalicylic acid reagent for determination of reducing sugars. Anal Biochem 31:426–428
Nakamura S, Wakabayashi K, Nakai R, Aono R, Horikoshi K (1993) Purification and some properties of an alkaline xylanase from alkaliphilic Bacillus sp. strain 41M-1. Appl Environ Microbiol 59:2311–2316
Page RDM (1996) TREEVIEW: an application to display phylogenetic trees on personal computers. Comput Appl Biosci 12:357–358
Panbangred W, Shinmyp A, Kinoshita S, Okada H (1993) Purification and properties of endoxylanase produced by Bacillus pumilus. Agric Biol Chem 47:957–963
Sambrook J, Russel DW (2001) Molecular cloning: a laboratory manual. Cold spring Harbor Laboratory, New York
Sanchez-Porro C, Martin S, Mellado E, Ventosa A (2003) Diversity of moderately halophilic bacteria producing extracellular hydrolytic enzymes. J Appl Microbiol 94:295–300
Sasser M (1990) Technical note 102. Tracking a strain using the Microbial Identification System. MIS, Newark
Sasser M, Wichman MD (1991) Identification of microorganisms through use of gas chromatography and high-performance liquid chromatography. In: Balows A, Hausler WJ Jr, Herrman KL, Isenberg HD, Shadomy HJ (eds) Manual of clinical microbiology, 5th edn. American Society for Microbiology, Washington
Shi QQ, Sun J, Yu HL, Li CX, Bao J, Xu JH (2011) Catalytic performance of corn stover hydrolysis by a new isolate Penicillium sp. ECU0913 producing both cellulase and xylanase. Appl Biochem Biotechnol 164:816–830
Sneath PHA (1994) Endospore forming gram positive rods and cocci. In: Hensyl WM (ed) Bergey’s manual of systematic bacteriology, 9th edn. Williams and Wilkins, Philadelphia
Sriyapai T, Somyoonsap P, Matsui K, Kawai F, Chansiri K (2011) Cloning of a thermostable xylanase from Actinomadura sp. S14 and its expression in Escherichia coli and Pichia pastoris. J Biosci Bioeng 111:528–536
Subramaniyan S (2012) Isolation and characterisation of low molecular weight xylanase from Bacillus pumilus SSP-34. Appl Biochem Biotechnol 166:1831–1842
Subramaniyan S, Prema P (2002) Biotechnology of microbial xylanases: enzymology, molecular biology, and application. Degradation of xylan to d-xylose by recombinant Saccharomyces cerevisiae coexpressing the Aspergillus niger beta-xylosidase (xlnD) and the Trichoderma reesei xylanase II (xyn2) genes. Crit Rev Biotechnol 22:33–64
Teather RM, Wood PJ (1982) Use of Congo red-polysaccharide interactions in enumeration and characterization of cellulolytic bacteria from the bovine rumen. Appl Environ Microbiol 43:777–780
Tsenga MJ, Yap MN, Ratanakhanokchai K, Kyu KL, Chen ST (2002) Purification and characterization of two cellulase-free xylanases from an alkaliphilic Bacillus firmus. Enzyme Microb Technol 30:590–595
van Zyl WH, La Grange DC, Pretorius IS, Claeyssens M (2001) Degradation of xylan to d-xylose by recombinant Saccharomyces cerevisiae coexpressing the Aspergillus niger-Xylosidase (xlnD) and the Trichoderma reesei xylanase II (xyn2) genes. Appl Environ Microbiol 67:5512–5519
Viikari L, Kantelinen A, Sundquist J, Linko M (1994) Xylanases in bleaching: from an idea to the industry. FEMS Microbiol Rev 13:335–350
Waino M, Ingvorsen K (2003) Production of β-xylanase and β-xylosidase by the extremely halophilic archaeon Halorhabdus utahensis. Extremophiles 7:87–93
Walia A, Mehta P, Chauhan A, Shirkot CK (2012a) Production of alkalophilic xylanases by Paenibacillus polymyxa CKWX1 isolated from decomposing wood. Proc Natl Acad Sci India Sect B Biol Sci 83(2):215–223
Walia A, Mehta P, Chauhan A, Shirkot CK (2012b) Optimization of cellulase-free xylanase production by alkalophilic Cellulosimicrobium sp. CKMX1 in solid-state fermentation of apple pomace using central composite design and response surface methodology. Ann Microbiol 63:187–198
Wejse PL, Ingvorsen K, Mortensen KK (2003) Purification and characterization of two extremely halotolerant xylanases from a novel halophilic bacterium. Extremophiles 7:423–431
Wong KKY, Tan LUL, Saddler JN (1988) Multiplicity of β-1,4 xylanase in microorganisms: functions and applications. Microbiol Rev 52:305–317
Zhang Q, Zou H, Guo Z, Zhang Q, Chen X, Ni J (2001) Matrix-assisted laser desorption/ionization mass spectrometry using porous silicon and silica gel as matrix. Rapid Commun Mass Spectrom 15:217–223
Zhou J, Shi P, Zhang R, Huang H, Meng K, Yang P (2010) Symbiotic Streptomyces sp. TN119 GH 11 xylanase: a new pH-stable, protease- and SDS-resistant xylanase. J Ind Microbiol Biotechnol 38:523–530
Acknowledgments
We thank Department of Science and Technology, Ministry of Science and Technology, Govt. of India, for providing contingency grant through Inspire Fellowship.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Walia, A., Mehta, P., Chauhan, A. et al. Purification and characterization of cellulase-free low molecular weight endo β-1,4 xylanase from an alkalophilic Cellulosimicrobium cellulans CKMX1 isolated from mushroom compost. World J Microbiol Biotechnol 30, 2597–2608 (2014). https://doi.org/10.1007/s11274-014-1683-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11274-014-1683-3