
Abstract
The release of arsenic (As) from sediments poses a risk to human health especially at high pH levels. Despite this, the distribution and kinetic response of mobile As remains unclear under varying pH conditions. In this study, a microcosm incubation experiment was performed, using sediment cores in combination with dialysis (Peeper) and thin film diffusive gradients (DGT), to investigate the distributions of mobile As (soluble As in pore water and DGT-labile As) at high vertical resolutions (2–4 mm). Results show that the concentrations of soluble As present in the water column increased 1.5-fold with an increase in pH from 5.4 to 11.2. Both soluble As in pore water and DGT-labile As exhibited stable low-level distributions in the uppermost layer beneath the sediment-water interface, followed by increasing concentration distribution with decreasing layers to middle depths. The mean concentrations of mobile As species increased with increased water pH in both sediment profiles and with upward diffusion gradients, showing a 0.2-fold increase of soluble As in the top 20-mm layer and a 0.6-fold increase in deeper 20–52-mm layers, while DGT-labile As showed a 1.0- and 1.1-fold increase in these two layers, respectively. Modeling of DGT-induced flux in sediments (DIFS) showed that the desorption rate constant increased more rapidly than the absorption rate constant, resulting in the increased availability of solid As pools, therefore resupplying the soluble As in pore water from sediments.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
The element arsenic (As) is highly toxic to organisms and has been ranked by the International Agency for Research on Cancer (IARC) as a group 1 carcinogen (Wang and Mulligan 2006; Mittal et al. 2014; Roggenbeck et al. 2016). Additionally, arsenic compounds are considered a highly dangerous environmental contaminant (Mandal and Suzuki 2002; Kaur et al. 2011; Du et al. 2015), with the World Health Organization recommending a maximum permissible concentration of 10 μg L−1 in drinking water (Gorchev and Ozolins 2004). It has been established that sediments in high-As waters typically store high levels of As (Gao et al. 2013), acting as a major sink for As in surface aquifers and groundwater systems and therefore posing the risk of As release into water systems. Arsenic deposits in sediments have been found to be dominated by inorganic As(V) and As(III), which exist in neutral form (H3AsO3) or as oxyanions (H2AsO4 − and HAsO4 2−) under circumneutral pH conditions (Smedley and Kinniburgh 2002).
The mobility of As in sediments depends on multiple processes including oxidation/reduction, complexation/precipitation, adsorption/desorption, and dissolution of As-bearing phases (Matera et al. 2001; Lock et al. 2017). pH is a critical and fundamental variable in influencing the elemental biogeochemistry of sediments. Sediment surface charge is pH-dependent and controlled by the transfer of protons between pore water and mineral surfaces (Wang and Liang 2015). When the environmental pH is lower, equal to, or higher than the pH point of zero charge (pHzpc) on the mineral surface, the surface charge will be characterized as positive, zero, or negative, respectively. Accordingly, negatively charged As(V) will be adsorbed on to the mineral surfaces at a differing rate and strength, due to electrostatic attraction or repulsion. The main As-bearing minerals in aquifer sediments are Fe(III) (hydr)oxides which are predominantly positively charged at pH 3.7–7.0 and negatively charged at pH 8.0–10.0 (Dixit and Hering 2003; Guo et al. 2014; Zhang et al. 2004). Therefore, the adsorption of As(V) in sediments is generally stronger under acidic conditions and weaker under alkaline conditions, while the effect of pH on As(III) is weakened due to its incomplete ionized state (Mamindy-Pajany et al. 2011; Maji et al. 2007). Rubinos et al. (2011) observed an interactive effect between pH, As, and time, where the short-term remobilization of As in sediments occurred via desorption under natural pH conditions (especially in the presence of phosphate), whereas dissolution of the solid phase may be involved under both acidic and alkaline conditions. Ma et al. (2015) further found that the pH played a key role in weak As adsorption processes, especially with water-soluble As, due to reduced pH causing an adsorption intensity increase from water-soluble form to the amorphous (hydr)oxide-associated form. Despite much advancement in this field in recent years, there are still only few reports on the kinetics of As release in sediments under varying pH conditions.
The traditional batch sorption approach has commonly been adopted in investigating the effects of pH on As mobility, in which the sediments used experimentally were mixed to form a homogeneous sample prior to testing. However, sediments have been shown to possess high heterogeneity in the distribution of chemicals and substances involved in biogeochemical processes (Ding et al. 2015; Santner et al. 2015; Stockdale et al. 2009). It has been found that various biogeochemical processes affect the partition of As in sediments, including iron oxyhydroxide redox cycling, the availability of manganese oxides and organic matter, the thickness of the oxic layer and its co-precipitation with sulfides or iron sulfides, and macrozoobenthos bioturbation and hydrodynamics (Aziz et al. 2017; Bennett et al. 2012a; Yao et al. 2016). These processes may cause large variation in the distribution of labile As, in both vertical and horizontal directions of sediment profiles. A series of vertical profile investigations at the millimeter scale have been performed by Sun et al. (2016, 2017) and Wang et al. (2016) using a passive sampling technique of diffusive gradients in thin films (DGT). These studies observed that the maximal concentration of DGT-labile As was detected in deep sediment layers, with concentrations one to two orders of magnitude greater than those detected at the sediment-water interface (SWI). The competitive effects of phosphate on the promotion of As mobilization in sediments have also been confirmed at the millimeter scale and are visible up to a depth of 60 mm (Sun et al. 2017). Therefore, high-resolution investigations are required to understand the effects of pH on As release in sediments, taking spatial dynamics and the heterogeneity of effect into consideration. The high-resolution dialysis technique Peeper is valuable in assessments of this kind, where in combination with DGT; this technique provides the distribution of soluble As in pore water from sediments at a resolution of 2 to 4 mm (Ding et al. 2010a; Xu et al. 2012).
The purpose of this study was to reveal the effects of pH on the mobility of As in sediment profiles. An incubation experiment was performed using sediment cores, where the water column pH was conditioned from weakly acidic to strongly alkaline conditions, respectively. High-resolution Peeper and DGT techniques were used to obtain the high-resolution distributions of pore water-soluble As and labile As in sediments, with DGT-induced fluxes in sediments (DIFS) additionally used to simulate the kinetics of As release from labile solid As pools, under varying pH conditions.
2 Materials and Methods
2.1 Preparation of the DGT and Peeper Probes
Zr-oxide DGT was provided by EasySensor Ltd. (Nanjing, China; www.easysensor.net) and was used to measure DGT-labile As in sediments at a 2.0-mm vertical spatial resolution (Ding et al. 2010a; Sun et al. 2014). The probe was assembled according to the method by Ding et al. (2015). A Zr-oxide binding gel was sequentially covered by an agarose diffusive gel and a polyvinylidene fluoride (PVDF) membrane (Durapore®, Millipore; 0.10-mm thickness, 0.45-μm pore size), then sealed using a flat DGT holder (Ding et al. 2016). The assembled DGT probe was soaked in 0.01 M NaCl and deoxygenated with nitrogen for a minimum of 16 h prior to deployment in sediments.
The Peeper technique was used to measure soluble As in pore water at a 4.0-mm vertical spatial resolution (Ding et al. 2010b). The Peeper was constructed of perspex plates (20-cm length) with 30 equally spaced chambers, each containing 200-μL volume of deionized water and covered with a 0.45 μm PVDF membrane. Following this, the Peeper was soaked in 0.01 M NaCl and deoxygenated with nitrogen for at least 16 h prior to deployment in sediments.
2.2 Experimental Setup
2.2.1 Sediment Core Incubation
Sediments used in experiments were collected from Meiliang Bay of eutrophic Taihu Lake (31°30′31.1″N, 120°10′31.0″E), the third largest freshwater lake in China. A total of 21 sediment cores (11-cm diameter; 40-cm length) were collected using a gravity corer (11 cm × 50 cm, Rigo Co., Japan) on 26 April 2016, with water samples also collected in the same sites. Sediment cores were then sectioned at 2-cm intervals and the sediment layers from corresponding depths were pooled together and completely homogenized to eliminate lateral heterogeneity in natural sediments (Ding et al. 2015). Sediments were sieved with a 0.6-mm pore-size mesh to remove macrofauna and large particles. Sediments were then aliquoted into 21 perspex tubes in accordance with their original depths, then placed in seven tanks filled to a depth of 45 cm with filtered lake water. The seven microcosm tanks were incubated at 25 °C for 2 months, and water was aerated by pumped for 5 min every hour to maintain O2 saturation during the entire incubation period.
At the end of the pre-incubation phase, the pH values in the microcosm water columns were adjusted with 0.1 M NaOH or 0.1 M HCl, with pH adjustment performed in stages from its original level of pH 8.1 to pH 5.4, pH 6.5, pH 7.1, pH 8.8, pH 10.0, and pH 11.2, respectively. Each microcosm tank was maintained for 20 days, where during each treatment the pH was re-conditioned daily to ensure stability.
2.2.2 Sampling
On the 18th day of incubation, each pH treatment column had a Peeper probe inserted into the sediment core, then following 24 h the DGT probe was also inserted into the same core. Both probes were left in situ for a further 24 h, at which point the Peeper and DGT probes were retrieved. The surface of the Peeper filter membrane was cleaned sequentially using wet filter papers and deionized water. Plastic membranes were then placed on the surfaces of the sampler filter membranes to prevent oxidation of pore water samples. Pore water samples were immediately collected using a pipette, with a volume of about 200 μL extracted from each chamber. The DGT probes were stored in sterile plastic bags and kept moist prior to analysis.
2.3 Chemical Analysis
The retrieved Zr-oxide DGT was analyzed according to the procedure by Sun et al. (2014). Briefly, the retrieved Zr-oxide gel was sliced at 2.0-mm intervals and each gel slice was washed with 0.4 mL of 1.0 M NaOH eluent, with the extract then used for analysis of DGT-labile As concentrations.
The 200 mL pore water samples collected from retrieved Peeper chambers were immediately used for the analysis of soluble As. Concentrations of soluble As in all samples (pore water samples, DGT eluent samples, and water samples) were determined using hydride generation coupled with hydride generation atomic fluorescence spectrometry (HG-AFS) (AF-610D, Beijing Rayleigh Analytical Instrument Corporation, China) (Sun et al. 2014).
2.4 Data Analysis
DGT-labile As concentrations were calculated as a time-averaged concentration at the diffusion layer-sediment interface (C DGT) according to Eq. (1):
where Δg (cm) represents the thickness of the diffusion layer, D (cm2 s−1) represents the diffusion coefficient of the analyte in the diffusion layer, t (s) represents the deployment time, A (cm2) represents the exposure area of the gel, and M (μg) represents the corresponding accumulated mass over the deployment time.
The R ratio was calculated to be the mean DGT-labile As concentration, relative to the mean soluble As concentration (Eq. (2)). The distribution coefficient for As was established between solid and soluble phases (K d), calculated as the ratio of labile (easily exchangeable) solid phase concentration (C s), to soluble As concentration (C SOL) (Eq. (3)). The value of T c, representing the characteristic time for the perturbed system to reach a 63% equilibrium position, was derived based on the DGT-induced fluxes in sediments (DIFS) model for known R and K d (Harper et al. 1998, 2000). The desorption rate constant (k −1) was determined as outlined in Eq. (4), and the adsorption rate constant (k 1) was calculated as shown in Eq. (5).
3 Results
3.1 Water Column Soluble as Concentrations
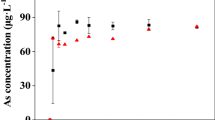
Figure 1 outlines the variation in soluble As concentrations in water columns with different pH treatments. The concentration of soluble As increased according to a rise in water column pH values from 5.4 to 11.2, with the increase in detectable soluble As concentration most pronounced with a pH increase from 8.1 to 11.2. The maximum As concentration at pH 11.2 was 0.254 μg L−1, 2.5-fold higher than the minimum As concentration (0.103 μg L−1) detected at pH 5.44.

Concentration of soluble As in the water columns maintained under varying pH conditions
3.2 Soluble and DGT-Labile As
Figure 2 shows the vertical distributions of soluble As in pore water, under varying pH conditions. The concentrations of soluble As remained largely constant, although a small decline in concentrations can be observed in the top 20-mm sediment layer, with a notable increase to peak As concentrations at the middle depths, followed by a decreasing trend in concentrations according to further sediment depth. The depths containing maximal As concentrations in treatments of pH 8.8, 10.0, and 11.2 occurred at a depth of about − 36 mm (below the sediment-water interface (SWI)), with maximal As concentrations observed at deeper depths (− 60 mm) in lower pH treatments. An increasing trend was observed in soluble As concentrations with increasing pH conditions, with this effect most notable from the SWI to a depth of − 52 mm. The gradient of upward distribution of soluble As also became more distinct from the depth of − 20 mm. Alkaline pH treatments presented more pronounced increases in soluble As concentrations, consistent with the changes observed in soluble As concentrations in the water column with increased pH, where the mean concentration of soluble As detected in the top 20-mm layer and deeper 20–52-mm layers increased from 4.34 to 5.38 μg L−1 and from 6.70 to 10.94 μg L−1, respectively (Table 1).

Vertical distributions of soluble As in pore water under varying pH conditions
Figure 3 outlines the vertical distribution of DGT-labile As concentrations in sediments under varying pH conditions. The concentration of DGT-labile As remained relatively constant with a slight increase observed in the top 20-mm sediment layer, followed by a significant increase to maximal concentrations at a depth of between − 30 to − 34 mm. Concentrations then remained relatively constant to the bottom of sediment columns for treatments under pH 5.44–8.14 conditions, or with higher pH treatments remained constant to a depth of − 40 to − 46 mm and then gradually decreased in concentration to the bottom of sediments columns. An increasing trend in concentration of labile As was apparent with increasing pH, with this effect being most notable between the SWI and depths of − 52 mm. The gradient of upward distribution of labile As concentrations became steeper from the depth of − 20 mm to its maximal point. The mean concentration of DGT-labile P detected in the top 20-mm layer and deeper 20–52-mm layers increased from 0.28 to 0.57 μg L−1 and 2.46 to 5.12 μg L−1, respectively (Table 1). The most pronounced increase was visible under alkaline pH treatment conditions, which is consistent with the change in soluble As concentrations observed in water columns.

Vertical distributions of DGT-labile As in sediments under varying pH conditions
3.3 Resupply and Kinetics of As Release
The vertical distributions of R ratio (C DGT/C SOL) are presented in Fig. 4. R values remained at a relatively constant low level (around 0.1) between the SWI and a depth of − 16 mm, followed by a notable increase in R values in the middle depths of between − 24 and − 36 mm. R values then showed a decreasing trend with continued progression to the base of the sediment vertical profile. Increased water column pH caused a significant increase in R value, with this effect being most distinct from the SWI to a depth of − 32 mm. The maximum R value observed increased from 0.46 under pH 5.4 conditions to 0.76 under pH 11.2 conditions.

Vertical distribution of R values in sediments under varying pH conditions
The kinetic parameters utilized to interpret the release of As from solid labile sediment As pools under varying pH treatment conditions are listed in Table 2. According to increased water column pH, R values increased from 0.35 to 0.49, K d decreased from 1.22 to 0.82 (× 102 cm3 g−1), T c decreased from 18.49 to 12.52 s, k 1 increased from 5.41 to 7.99 (× 10−2 s−1), and k −1 increased from 2.54 to 5.59 (× 10−4 s−1).
4 Discussion
4.1 Distribution of As in Sediments and its Relationship with Water Column As
The distribution of soluble As in pore water, as well as DGT-labile As, has previously been characterized in both freshwater and marine sediments to a high vertical resolution (2–4 mm) (Bennett et al. 2012b; Sun et al. 2016, 2017; Yao et al. 2016). Distribution pattern generally shows an initial phase of increasing As concentrations in shallower depth layers, generally followed by a phase of decreasing concentrations at progressively deeper layers. This pattern of distribution was also observed in the present study (Figs. 2 and 3), where initially stable and low-level concentration distributions were apparent for pore water As and DGT-labile As in the upper 20-mm layer. This effect can be attributed to the presence of adsorbed, co-precipitated, and mineral As(V) species, via association with various sediment oxides and clay minerals under oxic conditions (Hafeznezami et al. 2016; Jia et al. 2007). The phase of increasing As concentration observed in upper sediment layers can be attributed to reductive dissolution of As-bearing Fe(III) (hydr)oxides during the transition of sediment redox potential status from oxic to anoxic (Bose and Sharma 2002; Pedersen et al. 2006; Aguilar et al. 2007). This effect was confirmed by the observation of a simultaneous increase in both dissolved Fe(II) concentrations in pore water and DGT-labile As concentrations (Bennett et al. 2012a). The phase of decreasing As concentrations observed in deeper anoxic layers may be attributed to the immobilization of inorganic As(III) by Fe(II)-, or Fe(II)/Fe(III)-bearing minerals (e.g., magnetite, siderite, and green rust), which form concomitantly during microbial reduction of Fe(III) (Handley et al. 2013; Root et al. 2007; Sun et al. 2016).
The release of As from sediments is regarded as a significant risk to water quality in surface aquifers and groundwater systems. Unfortunately, it remains difficult to directly investigate this process under field conditions, as the concentration of soluble As in the vicinity of the SWI cannot be accurately detected using traditional ex situ methods, which typically include a collection of sediment samples followed by chemical analysis. In the present study, the concentrations of both soluble As in pore water and DGT-labile As in sediments were measured using passive techniques with high vertical resolutions, which allows identification of the drivers of change in water column As concentrations, in relation to sediment concentrations. A significant variation in the concentration of soluble As was observed in the water column (Fig. 1). As the microcosm tanks used in experiments were closed systems except that the water column contacted with air, the variation in As concentrations in the water column can be assumed to be due to differences in responses of sediments under varying pH environments. In order to verify this interaction, the relationships between As concentrations in the water column and in sediment were investigated. Analysis showed significant positive correlations between the concentration of soluble As in the water column and the concentration of both soluble As pore water and DGT-labile As, with correlations observed in both the upper 20-mm layer and deeper 20–52-mm layers (Fig. 5). These findings confirm a direct As exchange pathway exists between the water column and sediment, with the mobility of As in sediments defining the As concentration of the water column.

Relationship between As concentrations in the water column and concentrations in the upper 20-mm sediment layer or deeper 20–52-mm sediment layers, respectively
It has been established that soluble As in pore water is the most mobile form of sedimentary As, releasing As into the water column through upward diffusion if concentrations in the upper sediment layer are greater than the concentration of soluble As in the water column (O’Day et al. 2004). The depletion of soluble As from pore water induces the release of weakly solid-bound As, resupplying the concentration of soluble As in pore water. As supported by DGT measurement data establishing concentrations of both soluble As in pore water and weakly bound solid As, DGT-labile As can be regarded as a highly mobile form of As (Bennett et al. 2012a; Sun et al. 2016; Wang et al. 2016). Therefore, the variation in concentrations of soluble As and DGT-labile As reflects the mobility of As in sediment and the significant potential for release into the water column.
4.2 Effects of pH on the Release of As in Sediment
A notable increase in As concentration in both water column and sediments was observed, in accordance with increased pH of the water column from weakly acid to strongly alkali (Fig. 1 and Table 1). A similar phenomenon has previously been reported in analysis of As sorption onto sediments, metal oxide, and natural minerals (Ma et al. 2015; Mamindy-Pajany et al. 2011; Guerra et al. 2016). In the present study, analysis showed that in both the top 20-mm layer and deeper 20–52-mm layers, increased pH had a significant positive correlation with concentrations of soluble As in pore water and DGT-labile As in sediments (Fig. 6), with significance established to be lower than p < 0.01. These findings suggest that increase in water pH conditions results in an increase in available concentrations of both pore water As and DGT-labile sediment As. These findings are supported by the increase in upward distribution gradients for both soluble As and DGT-labile As, in relation to increased pH of water (Figs. 2 and 3).

Impact of pH on the relationship between available As concentrations in water column and both top 20-mm sediment layers and deeper 20–52-mm sediment layers, respectively
As aforementioned, the mobility of As has been affected by surface charge of As-bearing minerals which are controlled by pH through the electrostatic attraction or repulsion of As(III)/As(V) (Guo et al. 2014; Mamindy-Pajany et al. 2011; Zhang et al. 2004). Iron (hydr)oxides are regarded a major binding phase for As in sediments, due to their great abundance, large surface area, and strong binding affinity (Wang et al. 2010). Ma et al. (2015) found that readily reducible Fe minerals (such as ferrihydrite, lepidocrocite, goethite, hematite, and akageneite) are a major cause for variations observed in levels of As adsorption in sediments. Additionally, Mamindy-Pajany et al. (2011) found that the amount of As(V) adsorbed onto goethite and magnetite remained constant in the pH range of 2–8, decreasing by 20% when the pH was higher than 8. Therefore, As(V) can be readily desorbed from Fe (hydr)oxides under alkaline condition via electrostatic repulsion, as As(V) exists as a negatively charged oxyanionic species (e.g., AsO4 − or HAsO4 2−). This desorption process should mainly occur in the upper oxic sediment layer where As(V) is a dominant species (Guo et al. 2014), which may explain the increased soluble As concentrations in pore water and DGT-labile As concentrations in the top 20-mm layer of sediments, with increased water pH.
Pronounced increases were observed in soluble As concentrations in pore water and DGT-labile As concentrations in the upper 20-mm layer sediments. This suggests that the increase in water pH significantly promotes the release of As(III) in deeper anoxic sediment layers. Variation in pH showed a similar but weak effect on the adsorption of As(III) onto sediments and Fe (hydr)oxides in comparison with As(V), reflected by decreased adsorption affinity and capacity, in accordance with increased pH conditions (Maji et al. 2007; Qiao et al. 2012). It is of note that previous studies have revealed that variations in pH influence redox state, with an increase in pH conditions reducing the redox state by electron consumption (Hinsinger et al. 2003), would promote the reduction of As(V) to As(III), in accordance with the pH-induced reduction of redox state. It has been established that As(III) is desorbed more rapidly and extensively from metal oxides and minerals than As(V) species (Tufano et al. 2008).
4.3 Effects of pH on Kinetics and Release of As in Sediments
The increase in water column pH values resulted in a consistent increase in R and decrease in K d (Table 2), with DIFS modeling of As release kinetics (based on R and K d) showing a consistent decrease in T c and increased k 1 and k −1 (Harper et al. 2000). Further analysis established a significant positive correlation between pH and R, k 1, and k −1, while a negative correlation was established between pH and both K d and T c (Table 3). These findings demonstrate that change in pH conditions significantly affects the kinetics of As release in sediments.
The R ratio reflects the ability of available solid As pools, to resupply soluble As concentrations in pore water, where R values vary between 0 and 1, with a greater value suggesting a stronger ability to resupply available As. Therefore, an increase in R value in accordance with increased pH conditions implies a pH-dependent increase in the availability of solid As pools, resupplying soluble As concentrations in pore water. This is further supported by the decrease in K d, which reflects a reduction in the ability of solid sediments to retain As under equilibrium conditions (Zhou et al. 2005), which may be attributed to the increased negative charge on the solid surface and therefore repulsion of negatively charged As(V). T c defines the time required for partitioning components of K d to reach 63% of their equilibrium value (Harper et al. 2000), with decrease in T c value reflecting an increase in the resupply rate of available As from solid sediments, in accordance with increased water column pH conditions. Variations in T c depend on changes in the adsorption rate constant (k 1) and desorption rate constant (k −1) (Ernstberger et al. 2005), with increased k 1 and k −1 inducing a decrease in T c. However, the maximum k −1 observed at pH 11.2 was 2.1-fold higher than the minimum value observed at pH 5.44, which is greater than 1.5 times for k 1. Therefore, the As desorption rate from available solid sediment pools increased relatively to adsorption rate, which facilitated the release of As into pore water with increased pH of the water column as observed.
5 Conclusions
The variation in pH conditions in the water column had a significant impact on the distribution of mobile As forms and their kinetic responses. Soluble As in the water column showed a 1.5-fold increase from pH 5.44 to pH 11.2, which was found to be directly related to As mobility in sediments. The increase in water column pH conditions induced an increase in concentration of pore water-soluble As and DGT-labile As in sediment profiles, with the mean values of soluble As showing a 0.2- and 0.6-fold increase in the upper 20-mm layer and deeper 20–52-mm layers, respectively, while the mean values of DGT-labile As showed a 1.0- and 1.1-fold increase in the two layers, respectively. Moreover, upward diffusion gradients for both mobile As species became gradually steeper, implying that the As release potential continually increased. Further DIFS modeling showed that the available solid As pools exhibit increased R values (0.49 to 0.35) and a decrease in K d (1.22 to 0.82), while the increase in As resupply ability was attributed to the increase in k −1 value [2.54 to 5.59 (× 10−4 s−1)].
References
Aguilar, J., Dorronsoro, C., Fernández, E., Fernández, J., García, I., Martín, F., Sierra, M., & Simón, M. (2007). Arsenic contamination in soils affected by a pyrite-mine spill (Aznalcóllar, SW Spain). Water, Air, and Soil Pollution, 180(1–4), 271–281.
Aziz, Z., Bostick, B. C., Zheng, Y., Huq, M. R., Rahman, M. M., Ahmed, K. M., & van Geen, A. (2017). Evidence of decoupling between arsenic and phosphate in shallow groundwater of Bangladesh and potential implications. Applied Geochemistry, 77, 167–177.
Bennett, W. W., Teasdale, P. R., Panther, J. G., Welsh, D. T., Zhao, H. J., & Jolley, D. F. (2012a). Investigating arsenic speciation and mobilization in sediments with DGT and DET: a mesocosm evaluation of oxic-anoxic transitions. Environmental Science & Technology, 46(7), 3981–3989.
Bennett, W. W., Teasdale, P. R., Welsh, D. T., Panther, J. G., Stewart, R. R., Price, H. L., & Jolley, D. F. (2012b). Inorganic arsenic and iron(II) distributions in sediment porewaters investigated by a combined DGT-colourimetric DET technique. Environmental Chemistry, 9(1), 31–40.
Bose, P., & Sharma, A. (2002). Role of iron in controlling speciation and mobilization of arsenic in subsurface environment. Water Research, 36(19), 4916–4926.
Ding, S. M., Qin, S., & Di, X. (2010a). Development of the DET technique for high-resolution determination of soluble reactive phosphate profiles in sediment pore waters. International Journal of Environmental and Analytical Chemistry, 90, 1130–1138.
Ding, S., Xu, D., Sun, Q., Yin, H., & Zhang, C. (2010b). Measurement of dissolved reactive phosphorus using the diffusive gradients in thin films technique with a high-capacity binding phase. Environmental Science & Technology, 44(21), 8169–8174.
Ding, S., Han, C., Wang, Y., Yao, L., Wang, Y., Xu, D., Sun, Q., Williams, P. N., & Zhang, C. (2015). In situ, high-resolution imaging of labile phosphorus in sediments of a large eutrophic lake. Water Research, 74(0), 100–109.
Ding, S., Xu, D., Wang, Y., Wang, Y., Li, Y., Gong, M., & Zhang, C. (2016). Simultaneous measurements of eight oxyanions using high-capacity diffusive gradients in thin films (Zr-oxide DGT) with a high-efficiency elution procedure. Environmental Science & Technology, 50(14), 7572.
Dixit, S., & Hering, J. G. (2003). Comparison of arsenic(V) and arsenic(III) sorption onto iron oxide minerals: implications for arsenic mobility. Environmental Science & Technology, 37(18), 4182–4189.
Du, M., Wei, D., Tan, Z., Lin, A., & Du, Y. (2015). The potential risk assessment for different arsenic species in the aquatic environment. Journal Environmental Sciences, 27(1), 1–8.
Ernstberger, H., Zhang, H., Tye, A., Young, S., & Davison, W. (2005). Desorption kinetics of Cd, Zn, and Ni measured in soils by DGT. Environmental Science & Technology, 39(6), 1591–1597.
Gao, X., Su, C., Wang, Y., & Hu, Q. (2013). Mobility of arsenic in aquifer sediments at Datong Basin, northern China: effect of bicarbonate and phosphate. Journal of Geochemical Exploration, 135, 93–103.
Gorchev, H. G., & Ozolins, G. (2004). WHO guidelines for drinking-water quality. World Health Organization.
Guerra, P., Gonzalez, C., Escauriaza, C., Pizarro, G., & Pasten, P. (2016). Incomplete mixing in the fate and transport of arsenic at a river affected by acid drainage. Water, Air, and Soil Pollution, 227(3), 1–20.
Guo, H., Wen, D., Liu, Z., Jia, Y., & Guo, Q. (2014). A review of high arsenic groundwater in Mainland and Taiwan, China: distribution, characteristics and geochemical processes. Applied Geochemistry, 41(1), 196–217.
Hafeznezami, S., Zimmer-Faust, A. G., Dunne, A., Tran, T., Yang, C., & Lam, J. (2016). Adsorption and desorption of arsenate on sandy sediments from contaminated and uncontaminated saturated zones: kinetic and equilibrium modeling. Environmental Pollution, 215, 290–301.
Handley, K. M., McBeth, J. M., Charnock, J. M., Vaughan, D. J., Wincott, P. L., Polya, D. A., & Lloyd, J. R. (2013). Effect of iron redox transformations on arsenic solid-phase associations in an arsenic-rich, ferruginous hydrothermal sediment. Geochimica et Cosmochimica Acta, 102(2), 124–142.
Harper, M. P., Davison, W., Zhang, H., & Tych, W. (1998). Kinetics of metal exchange between solids and solutions in sediments and soils interpreted from DGT measured fluxes. Geochimica et Cosmochimica Acta, 62(16), 2757–2770.
Harper, M. P., Davison, W., & Tych, W. (2000). DIFS—a modelling and simulation tool for DGT induced trace metal remobilisation in sediments and soils. Environmental Modelling & Software, 15(1), 55–66.
Hinsinger, P., Plassard, C., Tang, C., & Jaillard, B. (2003). Origins of root-mediated pH changes in the rhizosphere and their responses to environmental constraints: a review. Plant and Soil, 248(1), 43–59.
Jia, Y., Xu, L., Wang, X., & Demopoulos, G. P. (2007). Infrared spectroscopic and X-ray diffraction characterization of the nature of adsorbed arsenate on ferrihydrite. Geochimica et Cosmochimica Acta, 71(7), 1643–1654.
Kaur, S., Kamli, M. R., & Ali, A. (2011). Role of arsenic and its resistance in nature. Canadian Journal of Microbiology, 57, 769–774.
Lock, A., Wallschläger, D., Belzile, N., Spiers, G., & Gueguen, C. (2017). Rates and processes affecting As speciation and mobility in lake sediments during aging. Journal of Environmental Sciences. https://doi.org/10.1016/j.jes.2017.05.003.
Ma, J., Guo, H., Lei, M., Zhou, X., Li, F., & Yu. (2015). Arsenic adsorption and its fractions on aquifer sediment: effect of pH, arsenic species, and iron/manganese minerals. Water, Air, and Soil Pollution, 226(260), 1–15.
Maji, S. K., Pal, A., Pal, T., & Adak, A. (2007). Adsorption thermodynamics of arsenic on laterite soil. Journal of Surface Science & Technology, 22(3), 161–176.
Mamindy-Pajany, Y., Hurel, C., Marmier, N., & Roméo, M. (2011). Arsenic (V) adsorption from aqueous solution onto goethite, hematite, magnetite and zero-valent iron: effects of pH, concentration and reversibility. Desalination, 281(20), 93–99.
Mandal, B. K., & Suzuki, K. T. (2002). Arsenic round the world: a review. Talanta, 58(1), 201–235.
Matera, V., Hécho, I. L., Selim, H. M., & Sparks, D. L. (2001). Heavy metals release in soils (pp. 207–235). USA: Vadose Zone Journal.
Mittal, P., Chhillar, D., Dhattarwal, S. K., Singla, K., Merry, V., & Singh, S. (2014). Criminal arsenic poisoning: a case report. Anil Aggrawals Internet Journal of Forensic Medicine & Toxicology, 15 (2), 11 p.
O'Day, P. A., Vlassopoulos, D., Root, R., & Rivera, N. (2004). The influence of sulfur and iron on dissolved arsenic concentrations in the shallow subsurface under changing redox conditions. Proceedings of the National Academy of Sciences of the United States of America, 101(38), 13703–13708.
Pedersen, H. D., Postma, D., & Jakobsen, R. (2006). Release of arsenic associated with the reduction and transformation of iron oxides. Geochimica et Cosmochimica Acta, 70(16), 4116–4129.
Qiao, J., Jiang, Z., Sun, B., Sun, Y., & Wang, Q. (2012). Arsenate and arsenite removal by FeCl 3 : effects of pH, As/Fe ratio, initial As concentration and co-existing solutes. Separation and Purification Technology, 92(1), 106–114.
Roggenbeck, B. A., Banerjee, M., & Leslie, E. M. (2016). Cellular arsenic transport pathways in mammals. Journal of Environmental Sciences, 49, 38–58.
Root, R. A., Dixit, S., Campbell, K. M., Jew, A. D., Hering, J. G., & O'Day, P. A. (2007). Arsenic sequestration by sorption processes in high-iron sediments. Geochimica et Cosmochimica Acta, 71(23), 5782–5803.
Rubinos, D. A., Iglesias, L., Díaz-Fierros, F., & Barral, M. T. (2011). Interacting effect of pH, phosphate and time on the release of arsenic from polluted river sediments (Anllóns River, Spain). Aquatic Geochemistry, 17(3), 281–306.
Santner, J., Larsen, M., Kreuzeder, A., & Glud, R. N. (2015). Two decades of chemical imaging of solutes in sediments and soils—a review. Analytica Chimica Acta, 878, 9–42.
Smedley, P. L., & Kinniburgh, D. G. (2002). A review of the source, behaviour and distribution of arsenic in natural waters. Applied Geochemistry, 17(5), 517–568.
Stockdale, A., Davison, W., & Zhang, H. (2009). Micro-scale biogeochemical heterogeneity in sediments: a review of available technology and observed evidence. Earth-Science Reviews, 92(1–2), 81–97.
Sun, Q., Chen, J., Zhang, H., Ding, S., Li, Z., & Williams. (2014). Improved diffusive gradients in thin films (DGT) measurement of total dissolved inorganic arsenic in waters and soils using a hydrous zirconium oxide binding layer. Analytical Chemistry, 86(6), 3060–3067.
Sun, Q., Ding, S., Wang, Y., Xu, L., Wang, D., Chen, J., & Zhang, C. (2016). In-situ characterization and assessment of arsenic mobility in lake sediments. Environmental Pollution, 214, 314–323.
Sun, Q., Ding, S., Zhang, L., Cheng, M., & Zhang, C. (2017). A millimeter-scale observation of the competitive effect of phosphate on promotion of arsenic mobilization in sediments. Chemosphere, 180, 285–294.
Tufano, K. J., Reyes, C., Saltikov, C. W., & Fendorf, S. (2008). Reductive processes controlling arsenic retention: revealing the relative importance of iron and arsenic reduction. Environmental Science & Technology, 42(22), 8283–8289.
Wang, Z. H., & Liang, Y. (2015). Growth and alkaline phosphatase activity of Chattonella marina and Heterosigma akashiwo in response to phosphorus limitation. Journal of Environmental Sciences, 28(2), 1–7.
Wang, S., & Mulligan, C. N. (2006). Effect of natural organic matter on arsenic release from soils and sediments into groundwater. Environmental Geochemistry and Health, 28(3), 197–214.
Wang, S., Cao, X., Lin, C., & Chen, X. (2010). Arsenic content and fractionation in the surface sediments of the Guangzhou section of the Pearl River in Southern China. Journal of Hazardous Materials, 183(1), 264–270.
Wang, C., Yao, Y., Wang, P., Hou, J., Qian, J., Yuan, Y., & Fan, X. (2016). In situ high-resolution evaluation of labile arsenic and mercury in sediment of a large shallow lake. Science of the Total Environment, 541, 83–91.
Xu, D., Wu, W., Ding, S., Sun, Q., & Zhang, C. (2012). A high-resolution dialysis technique for rapid determination of dissolved reactive phosphate and ferrous iron in pore water of sediments. Science of the Total Environment, 421, 245–252.
Yao, Y., Wang, C., Wang, P., Hou, J., Wang, T., Liu, C., & Yuan, Y. (2016). In situ, high resolution ZrO-Chelex DGT for the investigation of iron-coupled inactivation of arsenic in sediments by macrozoobenthos bioturbation and hydrodynamic interactions. Science of the Total Environment, 562, 451–462.
Zhang, W., Singh, P., Paling, E. I., & Delides, S. (2004). Arsenic removal from contaminated water by natural iron ores. Minerals Engineering, 17(4), 517–524.
Zhou, A., Tang, H., & Wang, D. (2005). Phosphorus adsorption on natural sediments: modeling and effects of pH and sediment composition. Water Research, 39(7), 1245–1254.
Acknowledgements
This study was jointly sponsored by the National Natural Science Foundation of China (41621002, 41571465, 41322011) and National High-level Personnel of Special Support Program.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Chen, X., Sun, Q., Ding, S. et al. Mobile Arsenic Distribution and Release Kinetics in Sediment Profiles under Varying pH Conditions. Water Air Soil Pollut 228, 413 (2017). https://doi.org/10.1007/s11270-017-3601-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11270-017-3601-4