
Abstract
This study assessed the suitability of passive sampler extracts for use with a GC-MS-database rapid screening technique for around 940 organic chemicals. Chemcatcher™ passive sampler systems containing either Empore™ SDB-XC or C18FF disks were deployed at 21 riverine sites in and near Melbourne, Victoria, Australia, for a period of 28 days during September–October 2008. Methanolic elution of the SDB-XC and C18FF disks produced an extract that, after evaporation and inversion into hexane, was compatible with the GC-MS-database method enabling over 30 chemicals to be observed. The sources of the non-agricultural chemicals are still unclear, but this study was conducted in a relatively dry season where total rainfall was approximately 40 % lower than the long-term mean for the catchment during the study period. Thus, the risks may be greater in wetter seasons, as greater quantities of chemicals are likely to reach waterways as the frequency, extent and intensity of surface run-off events increase. This study provides valuable information for policy and decision-makers, both in Australia and other regions of the world, in that passive sampling can be conveniently used prior to analysis by multi-residue techniques to produce data to assess the likely risks trace organic chemicals pose to aquatic ecosystems.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Managing the effects of trace organic chemical contaminants in waterways requires information, often far more than we (as scientists and/or managers) can afford to directly measure at all the places at all of the times, and for all the chemicals of interest. The measurement of nutrients and salts in surface and groundwater is now routine and offered cheaply by many commercial laboratories; however, this is not so for many other contaminants of concern, including many pesticides, pharmaceutical and personal care products (PPCPs) and industrial chemicals. Measurement of trace organic chemicals (TrOCs) in waters can be expensive in Australia, which inhibits monitoring by water authorities (as providers of water), catchment management authorities (as custodians of the natural environment) and consumers (e.g. irrigators, industry and household and domestic water users), potentially resulting in increased risk to the natural environment. Strategies and/or tools are therefore required to focus monitoring and risk assessment programs in a cost-effective manner; the use of new sampling methods and preliminary screening of samples using rapid assessment tools are thus an increasingly attractive prospect for waterways managers.
When assessing organic substances, many analytical methods may have to be used to cover a large number of known compounds (Gómez et al. 2011), with concomitant financial implications associated with operating multiple, definitive chemical analytical screens using gas and/or liquid chromatography-based methods for volatile, semi-volatile non-polar and polar organic industrial, agricultural and domestic chemicals. In 2005, Kadokami et al. reported a new screening method that combined a mass-structure database with gas chromatography-mass spectrometry (GC-MS) to create a system that can screen samples for more than 940 semi-volatile compounds, including numerous halogenated and non-halogenated hydrocarbons, polynuclear aromatic hydrocarbons (PAHs), polychlorinated biphenyl compounds (PCBs), pharmaceutical and personal care products (PPCPs) and pesticides. The Kadokami et al. (2005) automated identification and quantification system with database (AIQS-DB) method was originally developed for screening unknown pollutants in environmental samples after incidents, e.g. fish kills, food contamination scares or the unknown number of harmful chemicals discharged into the environment after disasters such as large-scale fires and earthquakes, and has since successfully been applied to both water and sediment samples from rivers and estuaries in Japan (Kadokami et al. 2009; Pan et al. 2014), in Vietnam (Kadokami et al. 2012; Duong et al. 2014) and Victorian wastewater treatment plant effluents (Allinson et al. 2012). Consequently, when applied to river water, as in this study, this GC-MS-database (DB) method can give a good indication of the presence of some TrOCs that would not otherwise be identified if samples were to be subjected to only a very limited number of analytical screens for a limited number of target chemicals.
Collection of grab (or spot) samples is most commonly used to characterise TrOC residues in surface waters, although integrative sampling with passive samplers (or passive sampling) is becoming a more commonly used alternative. A ‘passive sampler’ can be defined as a device that is able to acquire a sample from discrete location without the active media transport induced by pumping or purge techniques (ITRC 2006). Hence, most passive samplers consist of a receiving phase with high affinity for organic contaminants, separated from the aquatic environment by a diffusion limiting membrane. Some of the most commonly used devices that rely on diffusion and sorption to accumulate analytes in the sampler are semi-permeable membrane devices (SPMDs) and passive in situ samplers (such as the Chemcatcher™ system (CC)). Passive samplers had been little utilised on natural water samples in Australia at the inception of this study. In Victoria, one of the first detailed studies using passive samplers occurred between 2004 and 2006 when Rose and Kibria (2007) deployed SPMDs containing trimethyl pentane in irrigation canals in northern Victoria. The monitoring found three insecticides (endosulfan, chlorpyrifos and parathion methyl) in the passive sampler solvents. Elsewhere in Australia, a number of relatively recent studies have detected residues of atrazine, diuron, hexazinone and simazine in rivers and estuaries on the eastern seaboard of Australia using the CC passive sampling system (Escher et al. 2006; Muller et al. 2008; Shaw et al. 2009a, b, 2010; Stephens et al. 2009; O’Brien et al. 2011), and Mueller et al. (2011) tracked POPs in the Brisbane River, but prior to this study, there had been no significant studies investigating TrOCs in surface waters in Victoria using the CC system.
The sampling rates of chemicals into passive samplers are dependent on a range of factors, both intrinsic to the passive samplers themselves and extrinsic factors (Leonard et al. 2002). Extrinsic environmental factors that may affect uptake include flow rate, water temperature and changes in the sorptive properties of the water (due to dissolved organic matter and suspended particulates; Leonard et al. 2002). Intrinsic factors that may affect uptake include the polarity of the contaminant (as measured by its octanol-water partition coefficient, Kow), the diffusivity of the molecules that have to pass through the aqueous boundary layer, sampler design, exposure time and concentrations of chemicals in the surrounding water. Perhaps of most importance is the type of receiving phase used in the passive sampler relative to target analytes, and whether the receiving phases are used with rate limiting membranes or without them (‘naked’). The purpose of rate limiting membranes is literally that suggested by their name—to reduce the rate at which chemicals are adsorbed by the receiving phase. For instance, the sampling rates (Rs) reported by Shaw et al. (2009a, b) when using naked sulphonated polystyrene divinylbenzene-reverse phase sulphonated disks (Empore™ SDB-RPS disks; Rs: atrazine 0.44 L/day; simazine 0.36 L/day) were about three times higher than when the same experiment was conducted with SDB-RPS disks and a polyether sulphone (PES) membrane (Rs: atrazine and simazine, 0.140 L/day). By reducing the sampling rates, the length of time before all available sites in the receiving phase are saturated is increased. This in turn allows for lengthier exposure periods in the field. In that context, SDB-RPS disks were used naked for 10 days for monitoring a polar herbicide (amitrole) in New South Wales by Sánchez-Bayo et al. (2013), whereas Allinson et al. (2014) were able to use Empore™ SDB-XC disks covered by a PES membrane for 28 days to target similarly polar herbicides in Victorian surface waters. At the inception of this study, receiving phases appeared to be being chosen by expert opinion based on their use to extract target analytes from water samples in the laboratory, although since that time, there has been some systematic study comparing the performance of different receiving phases in the field (e.g. SDB-RPS, SDB-XC and C-18 disks for the monitoring of nonylphenolethoxylates and nonylphenol in aquatic ecosystems; Ahkola et al. 2014).
In recognition of the potential risks that TrOCs pose to aquatic ecosystems, and the lack of information on the levels of such chemicals in Victorian freshwaters, this study was initiated to compare the utility of two different, but common receiving phases for the CC passive sampler system under field conditions in conjunction with a ‘single-shot’ multi-residue testing method. To that end, CCs were deployed at 21 sites in urban, peri-urban and rural waterways in and around Melbourne in September–October 2008 and retrieved samples prepared for measurement of approximately 940 semi-volatile organic chemicals by GC-MS-DB.
2 Materials and Methods
2.1 Study Sites
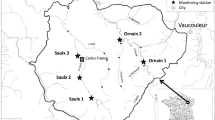
Twenty-one sites were monitored in and around Melbourne for this study (Fig. 1), which was conducted independent of, but in conjunction with a larger, complementary multi-disciplinary study of waterways in and around Melbourne. In that context, this manuscript is a companion piece to the papers by Wightwick et al. (2012, 2013) and Allinson et al. (2014) who reported the levels of fungicides, metals and herbicides, respectively, in the surface waters and sediments in riverine ecosystems in the Middle and Upper Yarra catchment area, some of which were used for this study. The sites were selected for a variety of reasons: either because they were already part of Melbourne Water’s Yarra River water quality monitoring program (e.g. site 23), were in a section of very high regional importance (as defined by PPWRRHS 2007; e.g. site 18), were downstream of a tributary with of very high regional importance (e.g. site 1), downstream of a tributary with known industrial pollution or waste water treatment plant (WWTP) impacts (e.g. site 3, 16, 17) or being sampled as part of other concurrent Melbourne Water funded research (e.g. sites 22, 23 and 24 used by Schäfer et al. 2011).

Approximate location of sampling sites in and around the city of Melbourne (M) in the Yarra River catchment (oval outline), Barwon River catchment (sites 21 and 24) and on the Mornington Peninsula (sites 22 and 23), in Victoria, Australia
Most of the study sites (75 %) were within the Yarra River catchment, with two study sites on the Mornington Peninsula and two sites in an agricultural location in the Barwon River catchment (sites 22, 23 and 21, 24, respectively). The Yarra River watershed is approximately 4000 km2 and home to approximately 2 million people in the city of Melbourne (the capital and most populous city in the state of Victoria). The city is located at the northern-most point of a large natural bay (Port Phillip Bay), with the city centre itself positioned on the estuary of the Yarra River (Fig. 1). The metropolitan area extends along the eastern and western shorelines of Port Phillip Bay and more than 25 km inland. The Yarra River flows 240 km from its headwaters to the sea in Port Philip Bay, and, in general, water quality is good in the upper catchment, but deteriorates downstream because of diffuse pollution from changed land use, particularly from agriculture and urban development (PPWRRHS 2007). Specifically, the upper sections of the Yarra River and its main tributaries (above site 1; Fig. 1) flow through forested, mountainous areas that have been reserved for water supply purposes for more than 100 years. Most of the land in the upper-middle section (above site 1) has been cleared for agriculture, although there is significant peri-urban development. The land downstream of site 1 is primarily urban and industrial. The lowest 10-km section of the Yarra River is estuarine. In the urbanised area of the Yarra River catchment, stormwater has a major impact on the river’s water quality. Several small (10,000–50,000 population equivalents) wastewater treatment plants (WWTPs) discharge treated effluent into the Yarra River (either directly or via a tributary), although most of Melbourne’s domestic and industrial sewage is transferred to two large WWTPs in the south and west of the city and after treatment discharged to the ocean.
2.2 Passive Sampling
A single type of passive sampler was used in this study, namely the Chemcatcher™ passive sampler system (CC). The CC system consists of machined polytetrafluoroethylene (PTFE) body that protects the chromatographic receiving phase (see Kingston et al. 2000). In this study, the CC system was fitted with either an Empore™ SDB-XC disk or an Empore™ C18FF disk (47 mm; 3 M, MN, USA) as the receiving phase and a polyethersulfone (PES) membrane (Sterlitech Corp, WA, USA) as the diffusion-limiting membrane. The SDB-XC and C18 disks were conditioned by soaking them in methanol (1 h) after which they were rinsed with deionised water on a disk extraction manifold; the PES membranes were conditioned in 50:50 methanol/water for 1 h then deionised water for 1 h. Samplers containing the two types of receiving phases were deployed in October 2008 for time-integrated monitoring to allow first a qualitative assessment (i.e. presence/absence) and then, where possible, a semi-quantitative assessment (i.e. based on estimated time-weighted average water concentrations) of semi-volatile chemicals in the catchment. For full details of CC preparation and field deployment, readers are directed to the Supplementary Information in Allinson et al. (2014).
Passive samplers were disassembled at DEPI Queenscliff Centre, and the receiving disk and PES membrane dried at 35 °C on a hotplate for approximately 1.5 h. Each disk was wrapped separately in aluminium foil, labelled, placed inside another labelled plastic bag and stored at 4 °C until analyte elution. One disk from each of the site replicates was haphazardly chosen and was eluted with methanol (5 mL) into a glass tube and the resulting solution evaporated to dryness with N2. The samples were reconstituted in hexane (1 mL), and in this dissolved form, the sample was transported to the University of Kitakyushu for multi-residue screening.
The AIQS-DB method identifies and quantifies chemical substances by using a combination of retention times, mass spectra and internal standard calibration curves registered in the database. In order to obtain accurate results, a GC-MS has to be adjusted to designated conditions that are almost the same as the instrumental conditions when the database was constructed. The results obtained from performance check standards (Naginata criteria sample mix 3: Hayashi Pure Chemical, Osaka, Japan) are evaluated against three criteria (Kadokami et al. 2004, 2005): spectrum validity, inertness of column and inlet liner, and stability of response. When the results for performance check standards satisfy the criteria, the difference between the predicted and actual retention times is less than 3 s, and chemical concentrations obtained (excluding some highly polar compounds which are difficult to measure by GC) are comparable to those obtained by conventional internal standard methods (Kadokami et al. 2005, 2009). After redissolving the sample in hexane (1 mL) and adding of internal standards (Naginata IS mix 3: Hayashi Pure Chemical, Osaka, Japan), the extract was analyzed by GC-MS (QP-2010Plus; Shimadzu, Kyoto, Japan). The GC-MS conditions are described elsewhere (Kadokami et al. 2004). Identification and quantification of the 940 or so semi-volatile compounds were finally performed by AIQS-DB. For full details of the extraction method and chemical determination, see Kadokami et al. (2005, 2009) and Jinya et al. (2013).
The instrument detection limit (IDL) for the chemicals in the database is typically ≤0.01 ng. Given a final determination adjusted by internal standard, analyte masses on disks correspond to residue concentrations reported as μg/disk.
3 Results and Discussion
The main objective of this pilot study was to explore the utility of the GC-MS-DB method in combination with passive sampling. In that context, the disk extracts were compatible with the GC-MS-database rapid screening technology, enabling 30 chemicals to be observed (Table 1), including pesticides, anti-oxidants, PAHs, solvents and plant steroids. For convenience, the 942 chemicals in the GC-MS multi-residue method are divided into groups based on their probable source. Grouping is, at times, somewhat arbitrary given that many of the wide range of chemicals covered in the GC-MS screen have multiple uses and therefore sources. For instance, octanol might be considered a solvent or a fragrance, and phenol either an industrial chemical or a disinfectant or a natural product. Some chemicals in the screen, such as the phthalates and petroleum hydrocarbons, are ubiquitous in the built environment, including in laboratories, and their presence in reagent blank samples meant their presence on the CC disks could not reliably be assigned to real environmental contamination, and so such chemicals are not reported. Consequently, only a limited number of contaminants were reliably observed on the disks, perhaps reflecting the relatively uncontaminated waters into which most of the CCs were deployed. However, overall passive sampling using the CC system was eminently suited to providing field-concentrated samples for determination by the GC-MS-DB system.
There was little difference in the number of chemicals found in the disk extracts (SBD-XC, 18 chemicals; C18FF, 22 chemicals; Table 1) nor on the number of occasion residues were observed (SDB-XC, 41 detects; C18FF, 47 detects). For instance, there was little difference between the disks in the number of times one or zero chemicals were detected on the disks (SDB-XC, 67 % of analyses; C18FF, 62 % of analyses), and both disks were equally good for identification of (non)contaminated waters, with the data from both SDB-XC and C18FF disks highlighting the relative contamination of the city sites 16 and 17 compared to the rural sites (Table 2 and 3). Approximately half of all detected residues were observed on both disks, although the two disks did appear to have a tendency to sample chemicals with different polarity. For instance, the 22 chemicals sampled by the C18FF disk appeared more lipophilic than those sampled by the SXB-XC disk, although the variation in the data is such that there is no statistical difference between the two disks (C18FF average logKow of retained chemicals 4.0, range 1.4–9.7; SDB-XC average logKow 2.7, range −0.07–6.1). The detection of a wide range of chemicals with radically different polarities using the two different receiving phases is consistent with Jinya et al (2013), who reported that the number and concentrations of chemicals observed after extraction of water samples using an Empore™ SDB-XD disk were almost identical to those obtained from the same samples using a cartridge-type SPE method and a liquid–liquid extraction method; in that case, the log Kow values of the detected pollutants ranged from −0.07 (caffeine) to 14.6 (squalene).
Given the limited number of chemicals sampled by the CCs, it is natural to wonder whether this is due to an intrinsic property of the passive sampling system, or simply due to a lack of chemicals in the waters studied? Most of the waters studied are in peri-urban and rural areas, and there is little information on contamination by industrial and domestic organic chemicals in those waterways. In other words, in this case, we have no a priori information with which to judge the performance of the CC systems for more than half the chemicals in the GC-MS-AIQS-DB system. However, there is some information on pesticide contamination in the studied waterways. For instance, 98 pesticides were monitored at our study’s sites in 2008–2009 using four gas chromatography and two high-performance liquid chromatography-based analytical screens (Wightwick et al. 2012; Allinson et al. 2014; Schäfer et al. 2011). In that context, 32 pesticides were reported at one or more sites at low ng/L concentrations in September and October 2008, i.e. during the time our CCs were deployed. Eighty-seven percent of the pesticides in the six group-specific analytical screens used by Wightwick et al. (2012) and Allinson et al. (2014) are in the GC-MS-AIQS-DB screen used in this study. Almost all of the pesticides observed on the disks in this study were also observed in water samples by Wightwick et al. (2012) and/or Allinson et al. (2014). Moreover, all of the herbicides and fungicides (except procymidone) observed in this study were also observed in either SPMD solutions by Wightwick et al. (2012), or CC eluates by Allinson et al. (2014). The only pesticides observed on the SDB-XC disks used by Allinson et al. (2014), but not on the disks used in this study, were atrazine and hexazinone. Consequently, the CC system appears to have been sampling pesticides adequately, and the lack of industrial and domestic chemicals on the CC disks was most likely due to the relatively uncontaminated nature of most of the waters being surveyed.
Passive sampling has some advantages over grab water sampling in that the samplers need little attention apart from deployment and collection, and they also integrate and average exposures over time, thus enabling identification of events that may be missed by grab sampling (ITRC 2006; Gagnon et al. 2007). From a practical perspective, the in situ sampling and preconcentration obtained using passive sampling reduces the manual handling and breakage risks associated with transport of large numbers of large volume water samples in glass bottles from field to laboratory, and the number of laboratory operations required to obtain instrument-ready solutions for analysis. However, in natural waterways samplers may be subject to periodic high-energy flows, which can wash away the samplers, and be the subject of human inquisitiveness, which may see the samplers removed from the waterway; both these externalities can reduce the integrity of a sampling program, and both were experienced in this study. In addition, some chemicals may be missed, as they may not readily partition into the sampler, and once sampled, some chemicals may be desorbed into the water column. The major disadvantages, however, of passive sampling methods are that the data from the sampler may not be directly comparable to toxicity data based on water concentrations, and water concentrations may have to be extrapolated through the use of field or laboratory-derived sampling rates (Rs), few of which are available (Allinson et al. 2014). For instance, sampling rates are currently not available to derive time-weighted average water concentrations (TWAWCs) for most of the chemicals detected in the disk eluates. However, it was possible to estimate a field sampling rate for simazine (61 mL/day) using SDB-XC disk data for the sites studied in Allinson et al. (2014). The TWAWC generated for simazine (0.07 μg/L) was approximately one fifth of the actual mean simazine concentration (0.37 μg/L) for the same sites reported by Allinson et al. (2014).
4 Conclusions
The main objective of this pilot study was to explore the utility of the GC-MS-DB method in combination with passive sampling. In that context, the disk extracts were eminently compatible with the GC-MS-database rapid screening technology, enabling 30 chemicals to be observed in the surface waters in and around Melbourne. From a practical perspective, the in situ sampling and pre-concentration reduces the manual handling risks associated with transport of large numbers of large volume water samples in glass bottles from field to laboratory, and the number of laboratory operations required to obtain instrument-ready solutions for analysis. The sources of the non-agricultural chemicals on the samplers is still unclear, but this study was conducted in a relatively dry season where total rainfall was approximately 40 % lower than the long-term mean in the catchment during the study period. Thus, the risks may be greater in wetter seasons, as greater quantities of chemicals are likely to reach waterways as the frequency, extent and intensity of surface run-off events increase. This study provides valuable information for policy and decision-makers, both in Australia and other regions of the world, in that passive sampling can be conveniently used prior to analysis by multi-residue techniques to produce data to assess the likely risks trace organic chemicals pose to aquatic ecosystems.
References
Ahkola, H., Herve, S., & Knuutinen, J. (2014). Study of different Chemcatcher configurations in the monitoring of nonylphenolethoxylates and nonylphenol in aquatic environment. Environmental Science and Pollution Research, 21, 9182–9192.
Allinson, M., Nakajima, D., Kamata, R., Shiraishi, F., Goto, S., Salzman, S. A., & Allinson, G. (2012). A pilot survey of 39 Victorian WWTP effluents using a high speed luminescent umu test in conjunction with a novel GC-MS-database technique for automatic identification of micropollutants. Water Science and Technology, 66(4), 768–774.
Allinson, G., Bui, A., Zhang, P., Rose, G., Wightwick, A. M., Allinson, M., & Pettigrove, V. (2014). Investigation of 10 herbicides in surface waters of a horticultural production catchment in south-eastern Australia. Archives of Environmental Contamination and Toxicology, 67(3), 358–373.
Duong, H. T., Kadokami, K., Pan, S., Matsuura, N., & Nguyen, T. Q. (2014). Screening and analysis of 940 organic micro-pollutants in river sediments in Vietnam using an auto- mated identification and quantification database system for GC–MS. Chemosphere, 107, 462–472.
Escher, B. I., Quayle, P., Muller, R., Schreiber, U., & Mueller, J. F. (2006). Passive sampling of herbicides combined with effect analysis in algae using a novel high-throughput phytotoxicity assay (Maxi-Imaging-PAM). Journal of Environmental Monitoring, 8, 456–464.
Gagnon, B., Marcoux, G., Leduc, R., Pouet, M.-F., & Thomas, O. (2007). Emerging tools and sustainability of water - quality monitoring. TRAC Trends in Analytical Chemistry, 26, 308–314.
Gómez, M. J., Herrera, S., Solé, D., García-Calvo, E., & Fernández-Alba, A. R. (2011). Contaminants in wastewater and river water by stir bar sorptive extraction followed by comprehensive two-dimensional gas chromatography-time-of-flight mass spectrometry. Analytical Chemistry, 83, 2638–2647.
ITRC. (2006). Technology overview of passive sampler technologies. DSP-4. Washington, D.C: Interstate Technology and Regulatory Council, Authoring Team. Available on-line: www.itrcweb.orgLast accessed 26 November 2014.
Jinya, D., Iwamura, T., & Kadokami, K. (2013). Comprehensive analytical method for semi-volatile organic compounds in water samples by combination of disk-type solid-phase extraction and gas chromatography–mass spectrometry database system. Analytical Sciences, 29, 483–486.
Kadokami, K., Tanada, K., Taneda, K., & Nakagawa, K. (2004). Development of a novel database for simultaneous determination of hazardous chemicals. Bunseki Kagaku, 53, 581–588 (in Japanese).
Kadokami, K., Tanada, K., Taneda, K., & Nakagawa, K. (2005). Novel gas chromatography–mass spectrometry database for automatic identification and quantification of micropollutants. Journal of Chromatography A, 1089(1-2), 219–226.
Kadokami, K., Jinya, D., & Iwamura, T. (2009). Surveyon 882 organic micro-pollutants in rivers throughout Japan by automated identification and quantification systemwith a gas chromatography – mass spectrometry database. Kankyo Kagaku, 19(3), 351–360 (in Japanese).
Kadokami, K., Pan, S., Hanh, D. T., Li, X., & Miyazaki, T. (2012). Development of a comprehensive analytical method for semi-volatile organic compounds in sediments by using an Automated Identification and Quantification System with a GC-MS Database. Analytical Sciences, 28, 1183–1189.
Kingston, J. K., Greenwood, R., Mills, G. A., Morrison, G. M., & BjörklundPersson, L. (2000). Development of a novel passive sampling system for the time averaged measurement of a range of organic pollutants in aquatic environments. Journal of Environmental Monitoring, 2, 487–495.
Leonard, A. W., Hyne, R. V., & Pablo, F. (2002). Trimethylpentane - containing passive samplers for predicting time-integrated concentrations of pesticides in water: Laboratory and field studies. Environmental Toxicology and Chemistry, 21(12), 2591–2599.
Mueller, J. F., Mortimer, M. R., O’Brien, J., Komarova, T., & Carter, S. (2011). A cleaner river: Long term use of semipermeable membrane devices demonstrate that concentrations of selected organochlorines and PAHs in the Brisbane River estuary, Queensland have reduced substantially over the past decade. Marine Pollution Bulletin, 63, 73–76.
Muller, R., Schreiber, U., Escher, B. I., Quayle, P., Bengtson Nash, S. M., & Mueller, J. F. (2008). Rapid exposure assessment of PSII herbicides in surface water using a novel chlorophyll a fluorescence imaging assay. Science of the Total Environment, 401, 51–59.
O’Brien, D., Bartkow, M., & Mueller, J. F. (2011). Determination of deployment specific chemical uptake rates for SDB-RPDEmpore disk using a passive flow monitor (PFM). Chemosphere, 83, 1290–1295.
Pan, S., Kadokami, K., Li, X., Duong, H. T., & Horiguchi, T. (2014). Target and screening analysis of 940 micro-pollutants in sediments in Tokyo Bay, Japan. Chemosphere, 99, 109–116.
PPWRRHS. (2007). Port Philip and Westernport Regional River Health Strategy. Melbourne: Melbourne Water and Port Philip and Westernport Catchment Management Authority. Available online: www.melbournewater.com.au Last Accessed 15 June 2011.
Rose, G., & Kibria, G. (2007). Pesticide and heavy metal residues in Goulburn Murray irrigation water 2004-2006. Australasian Journal of Ecotoxicology, 13, 65–79.
Sánchez-Bayo, F., Hyne, R. V., Kibria, G., & Doble, P. (2013). Calibration and field application of Chemcatcher® passive samplers for detecting amitrole residues in agricultural drain waters. Bulletin of Environmental Contamination and Toxicology, 90, 635–639.
Schäfer, R. B., Pettigrove, V., Rose, G., Allinson, G., Wightwick, A., von der Ohe, P. C., Shimeta, J., & Kefford, B. J. (2011). Effects of pesticides monitored with three sampling methods in 24 sites on macroinvertebrates and microorganisms. Environmental Science and Technology, 45(4), 1665–1672.
Shaw, M., Eaglesham, G., & Mueller, J. F. (2009a). Uptake and release of polar compounds in SDB-RPSEmpore™ disks; implications for their use as passive samplers. Chemosphere, 75, 1–7.
Shaw, M., Negri, A., Fabricius, K., & Mueller, J. F. (2009b). Predicting water toxicity: Pairing passive sampling with bioassays on the Great Barrier Reef. Aquatic Toxicology, 95, 108–116.
Shaw, M., Furnas, M. J., Fabricius, K., Haynes, D., Carter, S., Eaglesham, G., & Mueller, J. F. (2010). Monitoring pesticides in the Great Barrier Reef. Marine Pollution Bulletin, 60, 113–122.
Stephens, B. S., Kapernick, A. P., Eaglesham, G., & Mueller, J. F. (2009). Event monitoring of herbicides with naked and membrane-covered Empore disk integrative passive sampling devices. Marine Pollution Bulletin, 58, 1116–1122.
Wightwick, A., Bui, A., Zhang, P., Rose, G., Allinson, M., Myers, J., Reichman, S. M., Menzies, N. W., Pettigrove, V., & Allinson, G. (2012). Environmental fate of fungicides in surface waters of a horticultural production catchment in southeastern Australia. Archives of Environmental Contamination and Toxicology, 62, 380–390.
Wightwick, A., Croatto, G., Reichman, S. M., Menzies, N. W., Pettigrove, V., & Allinson, G. (2013). Horticultural use of copper-based fungicides has not increased copper concentrations in sediments in the mid- and upper Yarra Valley. Water, Air and Soil Pollution, 224(12), 1701. doi:10.1007/s11270-013-1701-3.
Acknowledgments
This study was funded by the Department of Primary Industries (Projects #08160 and 06889), with additional resources from the University of Kitakyushu. The authors would like to thank members of GA’s Agrochemicals Research team for 'putting in the hard yards' to make this project a success under short notice and at times trying circumstances, especially Adam Wightwick. We also thank members of the Centre for Aquatic Pollution Identification and Management (CAPIM) for their assistance with sampling.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Allinson, G., Allinson, M. & Kadokami, K. Combining Passive Sampling with a GC-MS-Database Screening Tool to Assess Trace Organic Contamination of Rivers: a Pilot Study in Melbourne, Australia. Water Air Soil Pollut 226, 230 (2015). https://doi.org/10.1007/s11270-015-2423-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11270-015-2423-5