
Abstract
Infectious bursal disease (IBD) virus (IBDV) serotype 1 is the causative agent of IBD, a highly contagious immunosuppressive disease of young chickens. In this study, we examined IBDV infection in apparently healthy 21 guinea fowls and 20 pigeons obtained in Tanzania and Zambia by virus neutralization test (VNT) and reverse transcription polymerase chain reaction (RT-PCR) for the VP2 hypervariable region (VP2-HVR) of IBDV. Two guinea fowls (9.5%) in Tanzania were RT-PCR and VNT positive for IBDV, and 1 pigeon (5%) in Tanzania was RT-PCR positive and VNT negative. Phylogenetic analysis based on the nucleotide sequences of the PCR products indicated that segment A of IBDV detected from one guinea fowl and a pigeon belonged to the very virulent genotype of European/Asian type, while the other IBDV detected from a guinea fowl belonged to the classical genotype. To our knowledge, this is the first report of detection of the IBDV genome in free-living pigeons and guinea fowls. The detection of IBDV from apparently healthy guinea fowls and pigeons elucidates the role of wild birds in the epidemiology of IBDV.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Infectious bursal disease virus (IBDV) is the causative agent of infectious bursal disease (IBD) of young chickens [6]. IBDV is classified into the genus Avibirnavirus of the family Birnaviridae [4]. The virus is extremely lymphocidal and infects IgM-bearing B-lymphocytes in the bursa of Fabricius [19] leading to immunosuppression. Two distinct serotypes, 1 and 2, exist in IBDV. Serotype 1 viruses are pathogenic to chickens and are further classified into classical virulent, antigenic variant, and very virulent (VV) IBDVs based on their virulence or antigenicity. Serotype 2 viruses are mainly isolated from turkey and are non-pathogenic to chickens [8].
IBDV genome consists of two segments of double-stranded RNA (dsRNA), segments A (3.4 kb) and B (2.8 kb). The large segment A encodes two viral proteins, a 17–21-kDa non-structural viral protein 5 (VP5) and a 110-kDa precursor polyprotein (NH2–VP2–VP4–VP3–COOH), which is processed into mature VP2 (37 to 42 kDa), VP3 (32 to 35 kDa), and VP4 (24 to 28 kDa). The smaller segment B encodes VP1 (90 kDa), an RNA-dependent RNA polymerase. The hypervariable region (HVR), which spans amino acids from position 206 to 350 within VP2 (VP2-HVR), is known to be critical for determination of the conformational epitopes responsible for recognition of virus neutralizing antibodies in VP2 [2, 25]. The VP2-HVR has the highest amino acid sequence variation among serotype 1 strains [10, 12], and the nucleotide and deduced amino acid sequences within this region are widely used for molecular diagnosis and genotyping of IBDVs [9].
Since 1987, pathotypic IBDV variants with enhanced virulence, called very virulent IBDVs (VV-IBDVs), emerged in Europe, and have spread to many places of the world. These strains were called as “European VV-IBDV.” Recently, Kasanga et al. [12] described the existence of the VV-IBDV variants in Tanzania, which were genetically different from the European VV-IBDVs. These Tanzanian VV-IBDVs were closely related to some strains isolated in the western part of Africa and were thus called “African VV-IBDV” [11, 12]. However, the origins of the European and African VV-IBDVs are not clearly known.
There is some speculation that unknown IBDVs distributed in wild birds contributed to the evolvement and/or emergence of new IBDV strains [7, 28]. In addition, Nigerian researchers reported that VV-IBDVs originated from Africa [18]. However, there is no information about the distribution of IBDV in wild birds in Africa. The only information presently available is based on a serological survey [16]. Furthermore, chickens in many parts of Africa are likely to be in contact with captive guinea fowls and free-living pigeons.
In this study, we conducted a serological and molecular epidemiological survey of IBDV in captive and free-living guinea fowls and pigeons in selected parts of Tanzania and Zambia to investigate the involvement of wild birds in the evolution and epidemiology of IBDV. Our findings indicate that IBDVs detected in guinea fowls and pigeons are genetically closely related to some chicken isolates in the VP2-HVR, suggesting that wild birds could be involved in evolution and epidemiology of IBDV in Africa.
Materials and methods
Birds, sera, and viruses
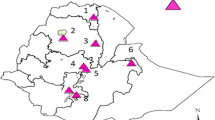
We examined 21 apparently healthy guinea fowls (Numida meleagris) and 20 apparently healthy pigeons (Columba livia) for IBDV infection in 2005. Ten of the guinea fowls were obtained in Zambia and all the other birds were obtained in Tanzania. All birds were obtained from different geographical regions in Tanzania and Zambia. The birds obtained were free-living and captive in areas where no IBD outbreaks were reported to occur (Table 1).
Serum was prepared from blood collected in birds prior to the postmortem examination. Each serum sample was absorbed to a Nobuto blood filter strip (Advantec, Tokyo, Japan), air-dried according to the manufacturer’s instructions, and transported to Japan for serological studies.
Bursa tissues from clinically healthy birds were smeared on tissue sampling filter papers, inactivated with 99.5% ethanol, air-dried, and transported to Japan. The inactivation of the virus was done as described previously [15].
Complementary DNA (cDNA) synthesis
Total RNA was isolated from bursa-smeared filter paper using TRIZOL (Invitrogen, Carlsbad, CA, USA) as reported previously [15]. The first strand cDNA was synthesized using Rever Tra Ace reverse transcriptase (RT) (Toyobo, Osaka, Japan) and random primer Pd(N)6 (Toyobo). Briefly, 3 μl of resuspended dsRNA in RNase-free water was mixed with 1.5 μl of dimethyl sulfoxide (DMSO), incubated for 5 min at 97°C, then immediately chilled on ice. To this was added 2.5 μl of 5 × RT reaction buffer, 2.5 μl of 2.5 mM dNTPs, 0.5 μl of 25 mM Pd(N)6 random primer, 0.5 μl of RNase inhibitor, 0.25 μl of Rever Tra Ace reverse transcriptase enzyme (100 U/μl), and 0.25 μl of RNase-free water to a final volume of 11 μl. Reactions were incubated for 45 min at 42°C, then for 5 min at 94°C, and withheld at 4°C. The synthesized cDNAs were used as templates for reverse transcription polymerase chain reaction (RT-PCR).
Reverse transcription polymerase chain reaction (RT-PCR) for VP2-HVR
For detection of the VP2-HVR by RT-PCR, forward primer V1 (position 737–754: 5′-CCA GAG TCT ACA CCA TAA-3′) and reverse primer V2 (position 1189–1208: 5′-CCT GTT GCC ACT CTT TCG TA-3′) were used (numbering based on segment A nucleotide sequence of 52–70 classical strain (GenBank accession: D00869)). The V1V2 primer set was tested and found to be highly sensitive and specific for IBDV VP2-HVR since it could detect the Acc I-Spe I region of VP2-HVR in bursa tissue, embryonated chicken eggs, dried filter papers, and transformed E. coli without amplification of any non-specific gene(s) or DNAs. The cDNA templates were amplified using Thermus aquaticus Ex-Taq DNA polymerase (Takara, Shiga, Japan) and V1V2 primers in a TAKARA PCR Thermo Cycler GP (Takara) as previously described [12]. Amplified products were run on 1.2% agarose gel and visualized with ethidium bromide staining.
RT-PCR for part of VP1
The forward and reverse primers used to amplify segment B encoding 332 amino acids of the VP1 N-terminus were BF1 (position 1–22: 5′-CCT CTT CTT GAT GAT TCT ACC A-3′) and BR1 (position 1021–1040: 5′-GAC CAT ATG TTA CGG GTC TT-3′), where the position numbers are based on the Lukert classical strain (GenBank accession: AY918947). The BF1 and BR1 primers were designed based on the consensus sequences of genome segment B of serotypes 1 and 2 strains, which are available in GenBank. The cDNA templates were amplified using Thermus aquaticus Ex-Taq DNA polymerase (Takara, Japan) and BF1BR1 primers in a TAKARA PCR Thermo Cycler GP (Takara, Japan). Briefly, 5 μl of each cDNA reaction mix was mixed with 5 μl of 10× Ex-Taq DNA polymerase reaction buffer, 4 μl of 2.5 mM dNTPs, 2 μl of 25 mM BF1 and BR1 both primers, 0.25 μl of Ex-Taq DNA polymerase enzyme (5 U/μl), and 33.75 μl of diethyl pyrocarbonate-treated water to a final volume of 50 μl. Amplification consisted of 35 cycles for denaturing (30 s at 94°C), primer annealing (30 s at 55.6°C), and primer extension (30 s at 72°C), with a final single extension step of 10 min at 72°C. The amplified products were run on 1.0% agarose gel and visualized with ethidium bromide staining.
Cloning and sequencing
The PCR products were gel purified using a Nucleospin DNA purification kit (Macherey-Nagel Inc., Easton PA, USA) and cloned into the plasmid pGEM-T-Easy vector (Promega, Madison WI, USA). Recombinant clones containing VP2-HVR were sequenced at Doragon Genomics center (TAKARA Bio, Mie, Japan). Both strands of at least three independent clones were sequenced per sample. The VP2-HVR nucleotide sequences of 1 pigeon and 2 guinea fowl isolates were assembled and edited using GENETYX-ATSQ software, version 5.0.0 (GENETYX Co., Tokyo, Japan), and deposited in the DDBJ/GenBank/EMBL database under accession numbers AB306714 (KTG-6), AB306715 (KTG-8), and AB306716 (KTP-13).
Sequence and phylogenetic analyses
The nucleotide and deduced amino acid sequences were analyzed with the aid of GENETYX-MAC software, version 14.0.1 (GENETYX Co., Tokyo, Japan). The nucleotide and amino acid sequences of detected viruses were subjected to BLAST searches [http://www.ddbj.nig.ac.jp/search/blast-e.html] to determine their identity with other strains. Phylogenetic analysis was performed using nucleotide alignment created in Clustal W version 1.8.3. The phylogenetic tree based on nucleotide sequences was constructed by the Neighbor-Joining (NJ) method implementing the Kimura two-parameter option [21] and the DNA parsimony program with PHYLIP package, version 3.67 [http://evolution.genetics.washington.edu/phylip.html] using 32 representative strains, including the sequences detected from pigeon and guinea fowls. The topological accuracy of the tree was estimated by the bootstrap method [13], with 1,000 replicates.
Virus neutralization test (VNT)
Virus neutralization (VN) antibody against IBDV serotype 1 was examined by the microneutralization method [20] using sera eluted from filter papers. Serum was eluted with 400 μl of sterile PBS leading to a 1:10 diluted stock sera, according to the manufacturer’s instructions (Advantec, Tokyo, Japan). The serum-adsorbed filter papers were sterilized with UV-light illumination for 15 min on each side prior to serum elution using a method standardized in our laboratory (Kasanga et al., a manuscript in preparation). Prior to the experiment, the VN titers of the native whole sera with known antibody titers against serotype 1 IBDV antigen were compared with their filter paper-eluted counterpart sera and found to correlate each other (data not shown). Then, the eluted sera of the test and control samples were serially diluted in a 96-well microtiter plate (Nunc. Roskilde, Denmark). One hundred TCID 50 [26] of the serotype 1 GBF-1E virus [27] was added to each well. The diluted serum containing the virus was incubated at 37°C for 1 h in the presence of 5% CO2. After the incubation, 50 μl of chicken embryo fibroblast (CEF) cell suspension adjusted to 5 × 105 cells/ml was added and cultured for 5 days. The VN titers were read as the reciprocal of the highest dilution at which no cytopathic effects were observed. VN titers of 80 or higher were considered positive as described previously [17].
Results
Detection of IBDV by RT-PCR in wild birds
Two out of 21 guinea fowls (3 free-living and 18 captives) and 1 out of 20 pigeons (8 free-living and 12 captives) were RT-PCR positive for IBDV VP2-HVR (Fig. 1 and Table 1). Of the three detected IBDV genomes, two were from captive guinea fowls and one was from a free-living pigeon. The three virus genomes were detected in birds originated from three locations namely Kivuma, Kapere, and Magadu in Tanzania, and were designated as KTG-6 (guinea fowl, 6G), KTG-8 (guinea fowl, 8G), and KTP-13 (pigeon, 13P), respectively. These places were located at least 10 km apart in different geographical areas with no known IBD outbreaks in the surrounding area. In Tanzania, the frequencies of detection of IBDV by RT-PCR were 18.2% (2/11) in guinea fowls and 5% (1/20) in pigeons. No samples were positive in Zambia (0/10) (Table 1).

RT-PCR amplification of the IBDV VP2-HVR of some guinea fowl and pigeon bursal homogenate samples. The positive samples are 472 bp in size. Lane M, 100-bp molecular ladder; Lanes 1–10, identification numbers of tested samples namely 5G, 6G, 8G, 13P, 14P, 21P, 23P, 25P, 29P, 31P; Lane 11, negative control (distilled water); Lane 12, positive control (GEB-10 vaccine strain); G, guinea fowl; P, pigeon. Samples 6G, 8G, and 13P represent IBDV strains KTG-6, KTG-8, and KTP-13, respectively
RT-PCR for part of VP1
All guinea fowl and pigeon samples, including the VP2-HVR positives (Table 1), were negative for segment B amplification of the fragment, which corresponds to the one-third of the VP1 N-terminus. On the other hand, the corresponding segment B region of the chicken IBDV isolates detected in Tanzania and Zambia, which served as positive controls for this experiment, were successfully amplified with the same set of oligonucleotides (BF1BR1 primers) and RT-PCR conditions (data not shown).
Nucleotide sequences of KTG-6, KTG-8 and KTP-13
The VP2-HVR sequences of KTG-6, KTG-8, and KTP-13 were all 434 bp long (without primer sequences), and contained neither nucleotide deletion(s) nor insertion(s) in any of the sequences compared with that of the standard classical strain 52–70. The nucleotide sequence diversity among the three detected sequences ranged from 0.70% (between KTG-6 and KTP-13) to 7.14% (between KTG-8 and KTP-13). A BLAST search performed with nucleotide sequences revealed the maximum identity of 99.7% between KTG-6 and a very virulent strain Ehime91 isolated in Japan. The alignment of VP2-HVR nucleotide sequences of the guinea fowl (KTG-6 and KTG-8) and pigeon (KTP-13) isolates with that of selected reference strains is shown in Fig. 2a.

Nucleotide (a) and amino acid (b) sequence comparison of the VP2-HVR domain of selected serotype 1 and 2 IBDVs. Nucleotide and deduced amino acid alignment were performed by parallel editor option of GENETYX-MAC software version 14.0.1. Dots indicate positions where nucleotides or amino acids are identical to that of 52–70-reference strain. Nucleotide or amino acid differences with respect to 52–70 are indicated. Hydrophilic regions important for antigenicity of the virus are boxed. Dashes show where corresponding amino acids are missing. Numbers refer to nucleotide or amino acid position for each sequence. Nucleotide or amino acid residues conserved in all sequences are indicated as asterisks. Sequences detected in this study from guinea fowls and pigeon are marked with #. Sites where amino acids in KTP-13, KTG-6, or KTG-8 are specific or different from that of serotype 1 strains are marked with arrowheads. A: major hydrophilic region A; I: minor hydrophilic region 1; II: minor hydrophilic region 2; III: minor hydrophilic region 3; and B: major hydrophilic region B [3]. Strains used for comparison include serotype 1 strains (52–70, D00869: classical virulent; GEB-10, AB200987: classical attenuated vaccine strain; GBF-1, D16828: chicken embryo adapted; 002–73, AF148073: Australian classic; Variant E, D10065: antigenic variant; 88180, AJ001941: atypical very virulent; UK661, Z25480 and Giza2000, AY318758: European/Asian very virulent; and KMRG-48, AB200983: African very virulent) and serotype 2 strain OH, U30818
Deduced amino acid sequences of VP2-HVR
A BLAST search performed with deduced amino acid sequences revealed the maximum identity of 100% between KTG-6 and a very virulent strain UK661 isolated in UK. Comparison of the amino acid sequence of VP2-HVR of KTP-13, KTG-6, and KTG-8 with those of selected serotype 1 IBDV strains revealed unique amino acid residues at positions 278 (T) and 331 (E) in guinea fowl isolate KTG-8, and at positions 287 (G) and 321 (T) in pigeon isolate KTP-13 (Fig. 2b). The unique amino acid residues at positions 278 (T) in KTG-8 and 321 (T) in KTP-13 were also found in serotype 2 strain OH and serotype 1 atypical very virulent strain Giza2000, respectively (Fig. 2b). There were no unique amino acid residues in KTG-6. In addition, the KTG-6 and KTP-13 IBDVs conserved putative virulence marker amino acids at positions 222(A), 242(I), 256(I), 294(I), and 299(S), which are found in most reported VV-IBDVs [27]. The KTG-8 virus, on the other hand, conserved amino acid residues at positions 222(P), 242(V), 256(V), 294(L), and 299(N) similar to that of classical strains but different from that observed in VV-IBDVs (Fig. 2b).
Phylogeny of IBDVs detected in wild birds
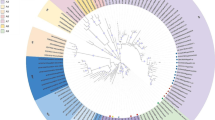
The phylogenetic trees constructed by NJ and DNA parsimony program with PHYLIP package had similar topologies (data not shown). In a phylogenetic tree based on the nucleotide sequences of VP2 HVR, two sequences KTG-6 and KTP-13 fell within the very virulent (VV) genotype and the other virus sequence, KTG-8, clustered within the classical genotype (Fig. 3). In the tree, the VV genotype is subdivided into two major clusters namely VV1 and VV2. The KTG-6 and KTP-13 belonged to the VV1 cluster comprising the African and European/Asian VV-IBDVs. In the VV1 cluster, the two viral sequences for KTG-6 and KTP-13 are closely related to PO7 and Tanzanian strains of the European/Asian VV type (Fig. 3). In the classical genotype cluster, the KTG-8 is closely related to the classical strain 2512 isolated in the USA with 96.8% nucleotide sequence similarity.

Phylogenetic tree constructed by DNA parsimony program and Neighbor-Joining methods based on 434-bp sequences in VP2 hypervariable domain of IBDV serotype 1 strains using nucleotide alignment created in Clustal W version 1.8.3. Sequences were obtained from 2 guinea fowls (in boldface), 1 pigeon (in boldface with # mark), and 29 chicken-origin IBDV sequences including Tanzanian strains [12]. The numbers at forks indicate the bootstrap values (1,000 replicates) greater than 750 in important junctions of the tree. VV: Very virulent genotype subdivided into clusters VV1 and VV2; C: Classic genotype subdivided into CV and AUS; CV: Classic and antigenic variant genotype; AUS: Australian classic genotype; VVA: Very virulent atypical; Italicized and underlined names: Represent Tanzanian VV-IBDVs in VV1 cluster; Accession numbers: VV1 (NIE/95/007/c, AJ86917; NIE/98/027/c, AJ586948; NIE/97/222/c, AJ586940; NIE/98/059/c, AJ586950; NIE/96/090/c, AJ586926; UK661, Z25480; Giza2000, AY318758; PO7, AY665672; MYGA, AJ238647; KTBR-18, AB200977; KMZA-28, AB200979; KDSM-35, AB200980; KMRG-26, AB200978; KDSM-02, AB200976; KMRG-00, AB200975); VV2 (KMZA-78, AB200979; KMRG-79, AB200986; KMRG-40, AB200982; KMRG-38, AB200981; KMRG-46, AB201125; KDSM-32, AB201124; KMRG-48, AB200983; KARS-53, AB200984); VVA (88180, AJ001941); CV (Cu-1, AF159219; 2512, DQ355819; Variant E, D10065; IBA, AJ586965); AUS (002–73, AF148073)
Serological response to IBDV antigen
Of the 41 serum samples examined, only two guinea fowl sera contained IBDV antibodies and were positive for VN test against serotype 1 IBDV antigen (titers > 1:80). The two VN-positive guinea fowls were also positive for IBDV VP2-HVR by RT-PCR with respective VN titers of 1:800 and 1:3,200 (Table 1). IBDV infection was serologically detected in 9.5% (2/21) of the guinea fowls and none of the pigeons. The positive and negative control sera used in this experiment were positive and negative for serotype 1 IBDV antigen, respectively (data not shown).
Discussion
Although there has been some speculation on the contribution of wild birds to the evolution and/or emergence of new IBDVs strains [7, 28], as well as on the possible African origin of VV-IBDVs [18], there is presently no direct evidence of IBDV in wild birds in Africa. Our findings indicate that the IBDVs detected in guinea fowls and pigeon were genetically closely related to some chicken isolates, suggesting that wild birds could be involved in the evolution and epidemiology of IBDV in Africa.
In the current study, we detected serotype 1-specific IBDV antibodies and nucleotide sequences corresponding to the VP2-HVR in apparently healthy guinea fowls and pigeon in Tanzania (Table 1). Studies conducted by other researchers in Nigeria [16], Australia [24], and Japan [17] revealed the presence of serotype 1 IBDV antibodies in wild birds including carrion crows (Corvus corone) and rock pigeons (Columba livia), which were found around barns and domestic chicken flocks. Another study conducted in Nigeria showed that guinea fowl keets are susceptible to experimental infection with IBDV and can transmit IBD to in-contact sentinel chickens [1]. Phylogenetic analysis based on VP2-HVR indicated that KTG-6 and KTP-13 were closely related to the chicken-origin IBDV strain MYGA isolated in Cuba, PO7 isolated in Tunisia, and the Tanzanian VV-IBDVs of European/Asian type clustered in VV1 (Fig. 3). The close phylogenetic relationships of KTG-6 and KTP-13 with the Tanzanian chicken-origin IBDV isolates in VV1 raise the possibility that cross infection with IBDV between commercial chickens and wild birds had occurred. Together with the previous data, our findings imply that the virus is directly or indirectly transmitted between wild birds and domestic chickens.
One of the pigeon samples was RT-PCR positive for IBDV but VN negative, whereas two guinea fowl samples were both RT-PCR and VNT positive for serotype 1 IBDV (Table 1). Although previous research showed seroconversion for serotype 1 IBDV in pigeon [17], the RT-PCR-positive pigeon may have been in the early stage of IBDV infection such that the antibody titer against IBDV was still low and could not neutralize the virus antigen in VNT.
The VP2-HVR nucleotide sequences of the PCR products of detected viruses, KTG-6, KTG-8, and KTP-13, were different from that of the Tanzanian and Zambian chicken-origin IBDV strains, which were handled at the same time during RNA extraction, RT-PCR, cloning, and sequencing procedures (data not shown). This observation implies that the detected viral genomic RNAs were not a result of contamination with other IBDV samples, which were handled together either at the working bench or in various machines used throughout experiments in the laboratory. On the other hand, the nucleotide sequence of KTG-6 showed high identity (99.7%) with that of Ehime91, a Japanese VV-IBDV strain. Although there was high identity between Ehime91 and KTG-6, the Ehime91 virus was not used in our laboratory during conduction of this study. Therefore, this observation shows that the KTG-6 nucleotide sequence was not a result of contamination with Ehime91 virus.
The detection of serotype 1 IBDV antibodies and IBDV genomic RNA from apparently healthy pigeons (Columba livia) and guinea fowls (Numida meleagris) (Table 1) indicates that at least some species of pigeons and guinea fowls can be subclinically infected with IBDV. Homing pigeons can fly long distances of up to 1,800 km without loosing the direction of their nests [22]. Previously, we detected VV-IBDVs in Tanzania in eastern Africa, which were genetically closely related to some strains isolated in Nigeria in the western Africa [12]. Although the invasive pathways of VV-IBDVs in Tanzania and other countries have not been clearly defined, the detections of VV-IBDV genotype in guinea fowls and pigeons raise the possibility that wild birds carry the virus to distant places during flying activities and contribute to the spread of IBDV to many parts of the world. Therefore, wild birds may act as reservoirs of viruses and play a role in the epidemiology of IBDV.
In the VP2-HVR, the deduced amino acid residues at positions 278 (T) and 331 (E) in KTG-8, and at position 287 (G) in KTP-13 are unique in these viruses when compared with serotype 1 strains (Fig. 2b). These unique residues suggest that the viruses underwent some mutational changes in guinea fowl and pigeon hosts, respectively, leading to the observed unique amino acid differences. In addition, the amino acid residues at positions 287 (G) and 321 (T) in KTP-13 are within the minor hydrophilic region 2 and major hydrophilic region B of VP2-HVR, respectively (Fig. 2b). The antigenicity of the virus is known to be determined by amino acid substitutions within the hydrophilic regions of the capsid protein [3], and amino acid changes at position 321 (A to T or V) was recently shown to have a major impact on virus antigenicity [5, 14]. In these studies [5, 14], amino acid substitution at position 321 (A to T) resulted in modification of the virus ability to escape virus neutralizing antibodies, which were directed to the major hydrophilic region B of the Egyptian isolate 99323 and Italian isolate 213622. Together with the previous data, our finding on the presence of unique amino acid residue at position 321 (T) of KTP-13 suggests that this virus is antigenically different from most serotype 1 IBDVs derived from chickens. Cross-neutralization studies may be needed to confirm this speculation.
We previously showed that African and European/Asian VV-IBDVs in chickens were present in Tanzania [12]. In the current molecular epidemiological investigation based on the VP2-HVR, we detected a classical genotype strain KTG-8 and the very virulent segment A genotype strains KTG-6 and KTP13 from captive guinea fowls kept under a free-range system and from a free-living pigeon. Although there is no evidence of the classical IBDV genotype among chickens in Tanzania, the detection of classical strain, KTG-8, from guinea fowl, which was kept in close contact with chickens, suggests that the classical serotype 1 IBDVs are present in chickens in Tanzania.
We were unable to amplify a part of genome segment B of KTG-6, KTG-8, and KTP-13 although the corresponding segment B regions of the chicken isolates were successfully amplified (data not shown). The primer set used, BF1 and BR1, was designed based on the genomic sequences of IBDVs that were originally derived from chickens. Because there has been genetic recombination and genomic reassortment of IBDV in nature between genome segments A and B [23], our inability to amplify a part of genome segment B of KTG-6, KTG-8, and KTP-13 may be because segment B has undergone genetic recombination. Therefore, the genetic nature and origin of genome segment B of KTG-6, KTG-8, and KTP-13 remain to be investigated.
In conclusion, we have detected a part of classical and very virulent IBD virus genome from apparently healthy pigeons and guinea fowls, which are genetically closely related to the IBDVs isolated from chickens. This close relationship of wild bird and chicken-origin IBDV based on the VP2-HVR indicates that wild birds can play a role in the epidemiology and spread of IBDV in nature. Further studies are needed to determine the full-length genome segments A and B of the guinea fowl and pigeon-origin IBDVs, and to isolate the viruses from wild birds to elucidate the evolutionary pathway, biological characteristics, and pathogenicity of IBDV strains in infected wild birds.
References
O.A. Adewuyi, O.A. Durojaiye, D.F. Adene, Zentralbl. Veterinarmed. 36, 43–48 (1989)
C.D. Bayliss, U. Spies, K. Shaw, R.W. Peters, A. Papageorgiou, H. Muller, M.E. Boursnell, J. Gen. Virol. 71, 1303–1312 (1990)
X. Cui, H.S. Nagesha, I.H. Holmes, J. Virol. Methods 114, 109–112 (2003)
P. Dobos, B.J. Hill, R. Hallett, D.T. Kells, H. Becht, D. Teninges, J. Virol. 32, 593–605 (1979)
N. Eterradossi, C. Gauthier, I. Reda, S. Comte, G. Rivallan, D. Toquin, C. de Boisseson J. Lamande, V. Jestin, Y. Morin, C. Cazaban, P.M. Borne, Avian Pathol. 33, 423–431 (2004)
K. Hirai, S. Shimakura, J. Virol. 14, 957–964 (1974)
C.C. Hon, T.Y. Lam, A. Drummond, A. Rambaut, Y.F. Lee, C.W. Yip, F. Zeng, P.Y. Lam, P.T. Ng, F.C. Leung, J. Virol. 80, 8503–8509 (2006)
N.M. Ismail, Y.M. Saif, P.D. Moorhead, Avian Dis. 32, 757–759 (1988)
D.J. Jackwood, S.E. Sommer, Avian Dis. 43, 310–314 (1999)
D.J. Jackwood, S.E. Sommer, Avian Dis. 49, 246–251 (2005)
C.J. Kasanga, in MVM Thesis in Veterinary Microbiology (Virology). (Sokoine University of Agriculture, Morogoro, Tanzania, 2002)
C.J. Kasanga, T. Yamaguchi, P.N. Wambura, A.D. Maeda-Machang’u, K. Ohya, H. Fukushi, Arch. Virol. 152, 783–790 (2007)
M. Kimura, J. Mol. Evol. 16, 111–120 (1980)
A.M. Martin, F. Fallacara, I. Barbieri, G. Tosi, G. Rivallan, N. Eterradossi, R. Ceruti, P. Cordioli, Avian Dis. 51, 863–872 (2007)
M.T. Maw, T. Yamaguchi, C.J. Kasanga, K. Terasaki, H. Fukushi, Avian Dis. 50, 556–560 (2006)
D.R. Nawathe, O. Onunkwo, I.M. Smith, Vet. Rec. 102, 444 (1978)
M. Ogawa, T. Wakuda, T. Yamaguchi, K. Murata, A. Setiyono, H. Fukushi, K. Hirai, J. Vet. Med. Sci. 60, 1277–1279 (1998)
A.A. Owoade, M.N. Mulders, J. Kohnen, W. Ammerlaan, C.P. Muller, Arch. Virol. 149, 653–672 (2004)
J.M. Sharma, I.J. Kim, S. Rautenschlein, H.Y. Yeh, Dev. Comp. Immunol. 24, 223–235 (2000)
J.K. Skeeles, P.D. Lukert, E.V. De Buysscher, O.J. Fletcher, J. Brown, Avian Dis. 23, 95–106 (1979)
J. Thompson, D. Higgins, T. Gibson, Nucleic Acids Res. 22, 4673–4680 (1994)
C. Walcott, J. Exp. Biol. 199, 21–27 (1996)
Y. Wei, J. Li, J. Zheng, H. Xu, L. Li, L. Yu, Biochem. Biophys. Res. Commun. 350, 277–287 (2006)
G.E. Wilcox, R.L. Flower, W. Baxendale, V.W. Smith, Aust. Vet. J. 60, 86–87 (1983)
T. Yamaguchi, K. Iwata, M. Kobayashi, M. Ogawa, H. Fukushi, K. Hirai, Arch. Virol. 141, 1493–1507 (1996)
T. Yamaguchi, T. Kondo, Y. Inoshima, M. Ogawa, M. Miyoshi, T. Yanai, T. Masegi, H. Fukushi, K. Hirai, Avian Dis. 40, 501–509 (1996)
T. Yamaguchi, M. Ogawa, Y. Inoshima, M. Miyoshi, H. Fukushi, K. Hirai, Virology. 223, 219–223 (1996)
T. Yamaguchi, M. Ogawa, M. Miyoshi, Y. Inoshima, H. Fukushi, K. Hirai, Arch. Virol. 142, 1441–1458 (1997)
Acknowledgments
We thank the participating farmers in Tanzania and Zambia. The assistance of Mr Edson Rugaimukamu, Mr Geofrey Mulungu, and Mr Jonas Fitwangile of Sokoine University of Agriculture, and Ms. Christine and Ms. Caroline of the University of Zambia, in sample collection is highly appreciated. This work was partly funded by Grants-in-Aid for Basic Scientific Research (A) 17255010 and (C) 18580308 from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kasanga, C.J., Yamaguchi, T., Wambura, P.N. et al. Detection of infectious bursal disease virus (IBDV) genome in free-living pigeon and guinea fowl in Africa suggests involvement of wild birds in the epidemiology of IBDV. Virus Genes 36, 521–529 (2008). https://doi.org/10.1007/s11262-008-0219-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-008-0219-z