
Abstract
We have determined the complete nucleotide and amino acid sequences of the Polish Pepino mosaic virus (PepMV) isolate marked as PepMV-PK. The PepMV-PK genome consists of a single positive-sense RNA strand of 6412-nucleotide-long that contains five open reading frames (ORFs). ORF1 encodes the putative viral polymerase (RdRp), ORFs 2–4 the triple gene block (TGB 1–3), and ORF5-coat protein CP. Two short untranslated regions flank the coding ones and there is a poly (A) tail at the 3′ end of the genomic RNA. Thus, the genome organization of PepMV-PK is that of a typical member of the genus Potexvirus. Phylogenetic analysis based on full-length genomes of PepMV sequences showed that PepMV-PK was most closely related to the Ch2 isolate from Chile. Comparison of PepMV-PK and Ch2 showed the following nucleotide identities: 98% for the RdRp, 99% for the CP genes, and 98, 99, and 98% for the TGB1, TGB2, and TBG3, respectively. This high level of nucleotide sequence identity between the Chilean and Polish PepMV-PK isolates suggest their common origin.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pepino mosaic virus (PepMV) was detected for the first time in 1974 in Peru on pepino (melon-pear, Solanum muricatum), and described in 1980 by Jones et al. [1]. It latently infected tomato and did not pose any apparent threat to this plant. The isolates of PepMV belong to different strains: Peruvian, European tomato, US1 and US2 [2]. The presence of PepMV in Europe was confirmed in 1999, but in contrast to the Peruvian strain, the virus exhibited a virulent pathogenicity toward tomato [3, 4]. The virus was also detected in tomato crops in North and South America. Very recently both the Peruvian strain as well as the US2 strains have been detected in Spain [5]. The recombinant isolates between the European tomato and US2 strains also occurred in the Spanish population of PepMV isolates. Confirmation of the existence of these recombinant strains in Europe is important, as is the fact whether they spread from a common source or were derived by independent recombination. Whole genome sequencing can be used to positively identify the strain variants and begin to address the issue of their origins.
In 2005 we isolated PepMV from Polish tomato fruits with the symptoms of distinctive lighter areas giving a marbled appearance. The isolate signed PepMV-PK seems to be different than the previously obtained PepMV isolate due to different biological and serological reactions [6]. This was confirmed by preliminary characterization of sequences of the triple gene block (TGB), coat protein (CP), and a part of polymerase gene (RdRp). Pairwise comparisons of PepMV-PK with other isolates from the GenBank database showed that the Polish isolate was completely different from the European strain, and shared the highest nucleotide sequence identity with the isolates Ch2 from Chile and US2 from USA.
In 2006 and 2007 we found more isolates of PepMV in different regions of Poland. Based on sequence analyses of a part of RdRp and the whole TGB and CP genes of these isolates we confirmed that they are very similar to PepMV-PK and belong to US2 strain. The presence of different PepMV strains, the possibility to create recombinant isolates and significant potential to cause damage to the protected tomato production prompted us to establish the whole sequence of the Polish isolate. Here we present the molecular characterization of the PepMV-PK genome sequence (GenBank accession number EF408821) and the affinities with other isolates of PepMV.
Materials and methods
Virus source and purification
The virus sources were plants infected with PepMV-PK after mechanical inoculation. The virus was isolated from Lycopersicon esculentum fruits with symptoms of mild yellow mosaics, transferred to a set of tested plants, and maintained in greenhouse conditions. To prepare inoculum for mechanical transmission, Nicotiana benthamiana plants were infected with infection symptoms and then grinded in 0.05 M phosphate buffer, pH 7.5; the sap was subsequently used for inoculation. The plants were tested for PepMV presence by the immunoenzymatic (ELISA) test.
The virus was purified from systemically infected N. benthamiana plants by extraction with 0.1 M citric buffer pH 7.5 and chloroform clarification, followed by low-speed centrifugation. The virions present in the supernatant were precipitated by the addition of Triton X-100 to 1% (w/v), NaCl to 1% (w/v), and polyethylene glycol 6,000 to 9% (w/v). The precipitate was redissolved in the citric buffer, sedimented by high–speed centrifugation and clarified by low-speed centrifugation. The partially purified virus was further centrifuged over a 10–40% sucrose density gradient. The virus sedimented in the sucrose gradient was collected and concentrated by a final high-speed centrifugation step. The concentration of purified virus was measured by spectrophotometer.
Preparation of total or viral RNA
Total RNA was extracted from infected L. esculentum plants by the method described by Chomczyński [7]. Viral RNA was extracted from the purified virus preparations (1 mg/ml). The virus was incubated for 30 min at 37°C in a buffer (50 mM Tris–HCl, 50 mM NaCl, 5 mM EDTA, 1% SDS, 1 mg/ml proteinase K), and then extracted three times with a mixture of phenol/chloroform/isoamyl alcohol (24:24:1). Next, the collected upper layer was extracted twice with a mixture of chloroform/isoamyl alcohol (24:1). The RNA was precipitated by three volumes of 96% ethanol and 0.1 volume of 3 M sodium acetate addition, followed by incubation for 30 min at −80°C and centrifugation. The obtained pellets were washed with 70% ethanol and dried in speed-vacuum. Finally, the RNA was dissolved in 20–30 μl of sterile water, and 2 μl of each sample were mixed with FORMAzol® and separated in 1% agarose gel.
Reverse transcription polymerase chain reaction (RT-PCR)
The amplification of PepMV-PK genome was performed in few steps with the literature primers as well as with those designed on the Ch2 sequence and the previously obtained PepMV-PK sequence [5] (Table 1). The reverse transcription was carried out using the extracted viral RNA (1μg/ μl), Superscript II Reverse Transcriptase (Invitrogen) and oligodT22 as a primer, according to the manufacturer’s instructions. Three overlapping cDNA fragments covering the entire CP gene, TGB and the part of polymerase gene were further amplified by polymerase chain reaction (PCR), using Taq DNA polymerase (Fermentas) and appropriate primers [5] (Fig. 1). In the first step a 600 nt region of the RdRp gene was amplified using Pep3 and Pep4 primers, a 1317 nt region encompassing the complete TGB was amplified using PepUSTGB-D and PepUSTGB-R primers; an 845 nt region, including the complete CP gene, was amplified using PepCP-D and PepCP-R primers. Briefly, 50 μl of reaction was carried out in 0.2 ml tubes containing: PCR buffer 10X, 125 μM each dNTPs, 2.5 μM each primers, and 5 U Taq polymerase. About 1 μl of RT mix was used as a template per 50 μl reaction. PCR conditions were the following: 35 cycles of 94°C, 47–55°C depending on specific primers (30 sek) and 72°C (1 min/kb of expected products), final step of 72°C 7 min. The reactions were carried out in a Biometra cycler. In the second step the reverse transcription polymerase chain reaction (RT-PCR) to amplify about 4350 nt of RdRp gene was performed with primers RdRpF and RdRpR, designed using the Oligo Analyzer software (http://www.uku.fi/∼kuulasma/OligoSoftware), based on Ch2 RdRp gene sequence and the previously obtained part of RdRp PepMV-PK sequence (Table 1). To amplify the 4350 nt of the RdRp, the Expand Long System (Roche) was used following vendor’s instructions. PCR conditions were as follows: 30 cycles at 94°C for 1 min., 55°C for 1 min., 68°C for 4 min., and 1 cycle at 68°C for 10 min.

The genome structure and cDNA clones obtained for sequencing of PepMV-PK
The 5′ and 3′ ends of PepMV-PK were obtained using the RACE kit (Invitrogen), following the manufacturer’s recommendation. All primer sequences used to obtain each of the overlapping cDNA clones of PepMV-PK are collected in Table 1.
Molecular cloning, nucleotide sequencing, and phylogenetic analysis
The amplified RT-PCR products for a part of RdRp (about 600 nt), TGB block, CP and 5′ and 3′ ends were purified using a Gel Extraction Kit (Qiagen), and then cloned using the pGEM-T-Easy Kit (Promega). Escherichia coli DH5α (Invitrogen) were transformed with the ligated vectors, and plasmid DNA were isolated using the Qiagen Miniprep Kit (Qiagen). Three independently purified recombinant plasmids were sequenced on an ABI automatic sequencer using standard T7 forward and Sp6 reverse primers. In the second part of RdRp, RT-PCR products were cloned into TOPO XL-PCR vector and electrocompetent One SHOT Top10 cells (Invitrogen), according to manufacturer’s instructions. Plasmids were purified with the Qiagen Miniprep Kit (Qiagen), and the DNA concentration was established by spectrophotometry.
The selected clones were sequenced with M13 forward and reverse primers and specific primers followed by internal sequence of RdRp clones. The sequences were determined at least three times using a Big Dye terminator Cycle Sequencing kit (Applied Biosystems). The complete genome sequence was deposited in the EMBL/GenBank database under accession number EF408821. Phylogenetic analyses were conducted with PepMV-PK and other available PepMV isolates using the amino acids sequences deduced from the genomes.
Complete virus sequences were selected from the NCBI Entrez system for phylogenetic analysis: France (Fr AJ438767), United Kingdom (UK AF340024), Spain (Sp13 AF484251; LE2000 AJ6066359; LE2002 AJ606360), United States (US1 AY509926; US2 AY509927), Chile (Ch1 DQ000984; Ch2 DQ000985), Peruvian isolates from Lycopersicum peruvianum (LP2001 AJ606361), and Solanum muricatum (SM74 AM109896). The phylogenetic analyses was performed using the BioEdit [8] and Mega 3.1 [9] software. The multiple sequence alignments were performed using ClustalW [10]. The obtained amino acid (aa) sequences and translations of open reading frames (ORFs) were generated with the ExPASy tools (http://www.expasy.org/tools/dna.html).
Phylogenetic analysis was carried out by the neighbor-joining method as implemented in the Mega package. Bootstrap values for phylogenetic comparisons are based on 1,000 pseudoreplicates. Single nucleotide polymorphism (SNP) was analyzed using the DNAsp program [11].
Results
Full-length sequencing of PepMV-PK using a RT-PCR-based stepwise strategy was employed to characterize the virus genome sequence. In the first step we amplified a part of RdRp, full-length CP gene and the TGB block; for all RT-PCR reactions we obtained the proper sized products. This step generated about ten clones for all parts of the genome, of which two were selected randomly and sequenced in both orientations. In the second step we subsequently obtained the remainder of the RdRp gene using the primers specific for the 5′ end of the Ch2 isolate of RdRp and the reverse primer specific for the previously obtained part of RdRp of the Polish isolate. This step generated long RT-PCR products of about 4,350 nt length. We obtained about 20 clones for RdRp and three clones were selected randomly for sequencing. The 5′ and 3′ genome RNA were sequenced from the products derived from the RACE system kit.
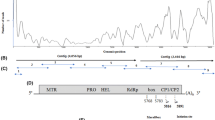
Excluding the polyA tail, the genomic RNA of PepMV-PK was 6,412 nt long. The genome organization, motif and domains typical for Potexviruses and described for other PepMV isolates were all found to be present [12, 13]. The PepMV-PK contained five ORFs coding for a putative replicase of 163 kDa (1,438 aa residues), for TGB1 26 kDa (234 aa), for TGB2 14 (123 aa), for TGB3 9 kDa (82 aa) and 25 kDa (237 aa) for CP. The 5′ UTR consists of 85 nt up to the first AUG codon. These 5′ non–coding regions start with the 5′ GAAAACAAA 3′ sequence, the putative promoter sequence found within all potexviruses. The organization of reading frames was identical for PepMV-PK and Ch2 isolates. We also observed the same insertions of two nucleotides at the same positions as in the Ch2 genome in comparison to US2, i.e., at position 24 in the 5′UTR and at position 4,411 in the intergenic region between the ORF1 and TGB1. ORF1 is the RNA-dependent RNA polymerase, as it has three well-conserved domains found in the replicases of the other potexviruses: (i) the putative methyltransferase domain, (ii) the NTPase/helicase domain with the NTP-binding motifs, and (iii) the RNA-dependent-RNA polymerase domain. The TGB genes are juxtaposed between the replicase gene and the viral CP gene. There is a response to systemic movement requiring cell–to-cell spread of a plant. In TGB1 gene we found conserved regions including the ATP-GTP binding domain (P-Loop) consensus GXXGKTSTS. After a 38-base long intercistronic region the 3′ proximal ORF 5 is found, which encodes for the CP and contains the core motif sequence KFAAFDFFDGVT. The 3′ UTR consists of 69 residues downstream of the termination codon of ORF5, followed by poly A. The polyadenylation signal AAUAAA is the same as in the Ch2 isolate, and it starts at position 6,409 with the last AAA being a part of the polyA.
The variations in the genome were mainly found to be concentrated in two regions: the 5′ UTR, and between the second half of ORF1 replicase and the first half of ORF2 (TGB1). In comparisons with Ch2 we found several point mutations in the PepMV-PK genome. Most of them were silent and did not change the amino acid sequence although we found 14 different amino acids in the RdRp gene, nine in TGB1, two in TGB2, and only one different amino acid in CP and TGB3 genes. The presence of the variable regions was confirmed by a SNP analysis using the DNAsp program (Fig. 2).

Profile of Single Nucleotide Polymorphism of the US2 group. The Pi diversity, the average number of nucleotide differences per site between two sequences was generated based on the alignment of full-genome-length PepMV-PK, Ch2 and US2 isolates. Sliding windows site 100 nt and step size 25 nt
Phylogenetic analysis
PepMV-PK shows a very high nucleotide sequence identity of 98% with the Ch2 isolate from Chile. In contrast, PepMV-PK shares only 78% similarity with the European tomato strains, while the identity with the US2 strain is 91%. The amino acid sequences of all RT-PCR products showed that PepMV-PK shares the following identities with the Ch2 isolate: 99.5% for the CP genes, 99% for the RdRp, 96, 98.3, and 98.7% for the TGB1, TGB2, and TBG3, respectively. Comparison with US2 revealed more distinct values in the RdRp gene: 91% and 97% nucleotide and amino acids identity, respectively (Table 2). In the non-coding regions we found the similarity: for the 5′UTR to be 99% with Ch2 and 80% with US2, while for 3′ UTR: both US2 and Ch2 isolates showed 99% identity. The variability between PepMV-PK and US2 in the part of RdRp and 5′ UTR was confirmed by SNP analysis (Fig. 2). This analysis showed that more SNPs occurred in the 5′ end than in 3′ end and in the middle section that encodes RdRp and TGB1. These relationships and the distinct relationships with other published PepMV sequences were also clearly demonstrated by phylogenetic trees, constructed on the basis of each gene product (Fig. 3).

Neighbor-joining trees based on the amino acids alignments for five genomic regions of Polish isolates of Pepino mosaic virus (PepMV): RNA-dependent RNA polymerase gene (RdRp), triple gene block—TGB1, TGB2, TGB3, and coat protein gene (CP). Sequences for other PepMV isolates obtained from public data bases were included. The scale bar represents, for the horizontal branch lengths, genetic distances of 0.01
Discussion
In this study, we have determined the complete genome sequence and amino acid sequences of the PepMV-PK isolate. In comparison with the available, fully sequenced PepMV isolates from different geographic regions, we found a very high similarity between Chilean, USA and PepMV-PK isolates. Our analysis of the entire coding sequences revealed that PepMV-PK, Ch2, and US2 always cluster together. As was reported before by Ling, the complete sequences of both Chilean variants were determined from the virus isolates obtained from commercial tomato seed produced in Chile [2]. We postulate that PepMV-PK originated from Chile and was brought to Poland via seeds, tomato plants, or fruits. The same genomic organization and very high amino acid similarities between PepMV-PK and Ch2 seem to confirm this hypothesis. We noticed that US2 differed in some partial regions from the above-mentioned isolates, and showed only 91% amino acids similarity with them. Both SNP and phylogenetic analyses showed two variable regions in this group: the 5′UTR and the second covering the area between the second half of RdRp and the first half of TGB1. The study of genetic relationships based on these parts of genome should be considered with caution, because it can lead to false inference. We also observed a number of non-synonymous mutations in PepMV–PK genome. This may indicate that the virus searches for the best variants to survive in a host organism. Besides the PepMV-PK we have found recently (end of 2006 and in 2007) other isolates of PepMV. We sequenced 20 clones of TGB1 gene (as it contains both conserved and variable regions) for each isolate (until now 7) and we noticed that all these clones shared identical sequences with some nucleotide substitutions resulted in amino acids changes. These could indicate the presence of genetic heterogeneity in the population of these isolates. RNA viruses do not form a homogenous population, but circulate in a host organism as a pool of genetically distinct variants. To describe such a complex structure of a virus population, the concept of quasi-species was introduced. The virus quasi-species can be defined as a set of phylogenetically related variants, which are present in a single infected organism. We assume that the US2 strain population can create new variants more suitable for a host organism. This may indicate that more similar genetic variants have been introduced into Poland. Very recently, both the Peruvian as well as the US strains were detected in Spain. Recombinant isolates between the European tomato and US2 strains also occurred in the Spanish population of PepMV isolates. The introduction of new strains and the appearance of new genetic types by interstrain recombinants represent a high potential risk and should be considered by developing efficient control strategies. So far, no specific vectors have been identified for PepMV, but its efficient mechanical transmission and high stability may contribute to its rapid spread.
In summary, we have genetically characterized the PepMV-PK isolate by analyzing its complete nucleotide and amino acid sequences, and comparing the majority of the available fully sequenced PepMV genomes. Interestingly, the highest nucleotide and amino acids identities PepMV-PK showed with Chilean variant isolated from a commercial tomato seed lot produced in Chile. These findings will facilitate the determination of viral gene product functions and pathogenesis at the molecular level. Furthermore, better understanding of genetic relationships among PepMV strains worldwide will help to understand their molecular evolution and origin. With the genome sequences obtained from the present studies, we are now developing an infectious transcript of the PepMV-PK variant.
References
R.A.C. Jones, R. Koenig, D.E. Lesemann, Ann. Appl. Biol. 94, 61–68 (1980)
K.S. Ling, Virus Genes 34, 1–8 (2006)
R.A. Mumford, E.J. Metcalfe, Arch. Virol. 146, 2455–2460 (2001)
R.A.A. Vlugt van der, C.C.M.M. Stijger, J.Th.J. Verhoeven, D.E. Lesemann, Plant Dis. 84, 103 (2000)
I. Pagan, M.C. Cordoba-Selles, L. Martinez-Priego, A. Fraile, J.M. Malpica, C. Jorda, F. Garcia-Arenal, Phytopathology 96, 274–279 (2006)
H. Pospieszny, N. Borodynko, Plant Dis. 90, 1106 (2006)
P. Chomczyński, Biotechniques 15, 532–537 (1993)
T.A. Hall, Nucleic Acids Symp. Ser. 41, 95–98 (1999)
S. Kumar, K. Tamura, M. Nei, Brief Bioinform. 5, 150–163 (2004)
J.D. Thompson, D.G. Higgins, T.J. Gibson, Nucleic Acids Res. 22, 4673–4680 (1994)
J. Rozas, J.C. Sanches DelBarrio, X. Messeguer, R. Rozas, Bioinformatics 19, 2496–2497 (2003)
J.M. Aquilar, M.D. Hernandez-Gallasrdo, J.L. Cenis, A. Lacasa, M.A. Aranda, Arch. Virol. 147, 2009–2015 (2002)
C.J. Maroon-Lango, M.A. Guaragna, R.L. Jordan, J. Hammond, M. Bandla, S.K. Marquardt, Arch. Virol. 150, 1187–1201 (2005)
Acknowledgment
The authors would like to thank Prof. Dr. Jack A.M. Leunissen of Wageningen University, The Netherlands, for his comments on the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hasiów, B., Borodynko, N. & Pospieszny, H. Complete genomic RNA sequence of the Polish Pepino mosaic virus isolate belonging to the US2 strain. Virus Genes 36, 209–214 (2008). https://doi.org/10.1007/s11262-007-0171-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-007-0171-3