
Abstract
The coordination compound constructed for nitronyl nitroxide radical NIT-Ph-4-Br and CuII(hfac)2(H2O)2 building blocks (NIT-Ph-4-Br = 2-(4-bromo-phenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide, hfac = hexafluoroacetylacetonato) was successfully synthesized. The single-crystal X-ray diffraction analyses indicated that the complexes {(NIT-Ph-4-Br)2[Cu(hfac)2]3} have centrosymmetric five-spin structures consisting of three Cu(II) ions bridged by two nitroxide ligands and that they consist of two types of copper atoms, one with a heavily Jahn–Teller distorted (4 + 2) octahedral coordination (Cuoct) and hfac in trans-positions and the other with square pyramidal five-coordinated (Cupyr) with three hfac oxygen atoms and N–O oxygen atom at the base and the one hfac oxygen atom at the apex. Different geometries of the copper ions are quite important for magnetochemistry. The magnetic susceptibility study of the coordination compound shows strong antiferromagnetic interactions between the metal center and the organic radical.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
In the past two decades, molecular magnetic materials have attracted scientists’ attention due to their potential applications in high-density magnetic memories, quantum computing devices and molecular spintronics [1,2,3,4,5,6]. Among the variety of molecular magnetic materials with different structures and dimensionalities, the strategy of metal–radical, which is formed by matching paramagnetic organic molecules and transition metal complexes, is still the key way to synthesize and design magnets. Up to now, a variety of organic paramagnetic molecules such as aminoxyl, verdazyl, thiazyl, triazinyl, nindigo, semiquinone and nitroxide radicals have been widely studied in the field of molecular magnetism [7,8,9,10,11,12,13,14,15]. Among them, because of their relatively stable and easy-to-obtain derivatives with substituents containing donor atoms, the nitronyl nitroxide (2-(R)-4,4,5,5-tetramethyl-4,5-dihydro-1H-imidazol-1-oxy-3-oxide, NITR) radicals have received more attention of researchers [16,17,18,19,20,21,22]. Besides, nitronyl nitroxide family of radicals can act as bidentate ligands with identical N–O coordination groups. Compared with regular diamagnetic ligands, the N–O groups of this kind of radical have weak basicity, so strong acidic partners were needed for ligation to the radical. Thus, it is necessary to utilize the strong electron-withdrawing coligands such as hexafluoroacetylacetonate (hfac) and trifluoroacetylacetonate (tfac) in the metal to improve the weak coordination ability. However, the steric demand of hfac or tfac ancillary ligand can restrict the dimensionality of the resulting metal–radical complex.
Furthermore, copper(II)–nitronyl nitroxide coordination complexes are the best documented and characterized on the basis of their coordination geometry toward development of magnetic materials [23, 24]. The copper(II) ion has a well-defined magnetic orbital of 3d2x2-y; in parallel, the N–O group has an unpaired electron in the molecular π* orbital. The magnetic coupling parameter J is typically determined as a singlet–triplet energy gap of 2 J at a > NO–Cu unit. Generally, an equatorial coordination results in a strong antiferromagnetic metal–radical exchange coupling (J < 0), while an axial bonding results in a moderate ferromagnetic coupling (J > 0) [25,26,27,28,29,30,31,32,33,34,35,36].
Inspired by these ideas, we synthesized the nitronyl nitroxide ligands (Scheme 1) which can provide an efficient way for magnetic spin exchange, following a detailed study of the structures and magnetism of the metal–radical compounds. In this contribution, we report the crystal structures and magnetic properties of {(NIT-Ph-4-Br)2[Cu(hfac)2]3} obtained from the nitronyl nitroxide radicals and CuII(hfac)2 building blocks.

Organic radicals NIT-Ph-4-Br and inorganic complexes: CuII(hfac)2
Experimental
Materials and methods
All reagents were obtained from commercial sources and used without further purification.
2-(4-Bromo-phenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide radical (NIT-Ph-4-Br) was prepared as reported previously with only minor modifications (Scheme 2) [37, 38].

Synthesis pathway of NIT-Ph-4-Br radical
Preparation of Cu(hfac)2·2H2O
Hfac (1 g, 7.1 mmol) was added to a suspension of Cu(CH3COO)2·H2O (0.71 g, 3.55 mmol) in H2O (17 mL) at room temperature, and then, NaHCO3 (0.071 g, 0.8 mmol) was added under stirring. After 10 min, a light blue precipitate was obtained and washed three times with n-hexane. Then, the light blue solid was dried at 50 °C for three days, and Cu(hfac)2·2H2O (0.74 g) was obtained. FTIR (KBr, cm−1): 3399(s), 1644(s), 1561(s), 1497(s), 1 210(s), 1151(s), 800(s), 766(w), 743(w), 668(s), 590(s).
Preparation of {[(NIT-Ph-4-Br)2[Cu(hfac)2]3}
CuII(hfac)2(H2O)2 (1 mmol, 51.5 mg) was dissolved in 10 mL of hot n-heptane, and then, the solution was cooled to about 50 °C. NIT-Ph-4-Br (0.05 mmol, 15 mg), dissolved in 2 mL of CHCl3, was added to it with constant stirring. The mixed solution was stirred for ca. 3 min and then cooled to room temperature. The filtrate was kept under N2 stream at room temperature until solid appeared. A few drops of CHCl3 were added until the solid disappears, and then, the flask was sealed and placed in the refrigerator. After 2–3 days, block-shaped dark blue crystals were obtained, yield 81%. Chemical analyses for C56H38Br2Cu3N4O16 are: calculated (%) C: 32.67, H: 1.85, N: 2.72. Found (%): C: 32.84, H: 1.69, N:2.57. FTIR (KBr, cm −1): 1658(s), 1559(w), 1534(s), 1479(s), 1356(w), 1 221(s), 1145(s), 1084(s), 862(s), 798(w), 663(s), 614(s).
General characterizations
Infrared spectra were recorded on a JASCO FT/IR-660 Plus spectrometer by transmission through KBr pellets in the range 400–4000 cm−1. Elemental analyses for C, H, and N were carried out using a PerkinElmer series II CHNS/O Analyzer 2400. The magnetization measurements on the crystalline solids were taken using Quantum Design MPMS-5S and MPMS-2 SQUID magnetometers. The magnetic field was varied from − 50 to 50 kOe and the temperature from 2 to 300 K. The data were corrected for the sample diamagnetism using Pascal’s constants [39, 40].
Single-Crystal X-ray diffraction studies
Their X-ray diffraction intensities were collected for the selected single crystals individually mounted on glass fibers at different temperature using a Bruker diffractometer SMART APEX II with a CCD and D8-QUEST with a CMOS area detector. Both employed graphite-monochromated Mo Kα (λ = 0.71073 Å) radiation. Data reduction was made using SAINT, and intensities were corrected for absorption by SADABS [41, 42]. The structures were solved by direct methods and refined by full-matrix least squares against F2 using ShelXL [43]. A part of the hydrogen atoms were located in the difference Fourier maps, and those not found were added at theoretical positions using the riding model. The details can be obtained from the cif files deposited at the Cambridge Crystallographic Data Centre. The crystals and refinements data are summarized in Table 1.
Results and discussion
Crystal structure of complex {(NIT-Ph-4-Br)2[Cu(hfac)2]3}
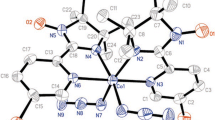
The molecular structures {(NIT-Ph-4-Br)2[Cu(hfac)2]3} were successfully determined by means of X-ray crystallographic analysis at different temperatures. (Table 1 shows selected crystallographic data.) {(NIT-Ph-4-Br)2[Cu(hfac)2]3} crystallizes in the triclinic space group P − 1 with Z = 1. The molecular structure is shown in Fig. 1, and it forms a centrosymmetric dimer (Fig. 1b). As shown in Fig. 1a, the asymmetric unit of complex {(NIT-Ph-4-Br)2[Cu(hfac)2]3} consists of two types of copper atoms, one with a heavily Jahn–Teller distorted (4 + 2) octahedral coordination (Cuoct) and hfac in trans-positions which are occupied by four oxygen atoms (O7, O8) from two hfac units, two oxygen atoms (O1) from two nitroxide ligands, and the other with nearly square pyramidal five-coordinated (Cupyr) with three hfac oxygen atoms (O3, O5, O6) and the N–O (O2) at the base and one hfac oxygen atom (O4) at the apex. The asymmetric unit of {(NIT-Ph-4-Br)2[Cu(hfac)2]3} consists of one radical, half a Cuoct (Cu1) and one Cupyr (Cu2). The whole molecule has five localized S = 1/2 spins, which form a zigzag linear spin system along the copper–radical–copper–radical–copper array. The molecular structure consists of alternating metal–radical units. The radical plays a key role in establishing this zero-dimensional structure, acting as a bidentate ligand to connect the metal centers. In addition, there are two slightly longer intermolecular interactions N2–O2···O2–N2 = 3.540(1) Å and Br1···Br1 = 3.333(1) Å, which bridge the molecular into a layer structure as shown in Fig. 2.

Molecular structure of a asymmetric unit and b trinuclear unit of {(NIT-Ph-4-Br)2[Cu(hfac)2]3} with ellipsoids at 50% probability level. Fluorine and hydrogen atoms are omitted for clarity

Crystal packing of complex {(NIT-Ph-4-Br)2[Cu(hfac)2]3} showing the O···O (blue) and Br···Br (black) interchain short contacts. Fluorine and hydrogen atoms are omitted for clarity
Selected bond lengths and angles are listed in Table 2. For the six-coordinated octahedral copper(II) ions, the apical Cu1–O1 bond lengths are 2.572(1)Å, while the equatorial Cu–O bonds are much shorter than the apical bonds (the Cu1–O distances are ~ 1.92 Å), as shown in Figure 3a. Furthermore, the Cu1–O1 distance was longer compared with typical copper(II)–radical bond lengths (> 2.3 Å) [44,45,46,47]. For the square pyramidal five-coordinated copper(II) ions, the apical Cu2–O4 bond lengths are 2.202(1) Å, while the equatorial Cu2–O bond distances range from 1.923 to 1.961 Å, as shown in Figure 3b. Furthermore, Cu2–O2 bond distance is 1.951(1)Å, which is a typical length for a copper(II)–nitroxide equatorial coordinate bond (1.93–2.03Å) [46,47,48,49]. The bond distances of uncoordinated N–O(1.266(2) Å) are shorter than those of the coordinated ones (1.306(2) Å). In addition, the angles of N1–O1–Cu1 and N2–O2–Cu2 are 161.56(1)° and 118.14(9)°, respectively. The O1–N1–C3–N2–O2 is almost planar. The dihedral angle between nitronyl nitroxide and the benzene ring is 38.7°. The molecular arrangement of complex {(NIT-Ph-4-Br)2[Cu(hfac)2]3} is shown in Fig. 2, the nearest intramolecular metal–metal contacts give a Cu···Cu distance of 7.759(1) Å, while the intermolecular Cu···Cu distance is 6.658(1) Å. The distance of the two corresponding radicals from adjacent molecules is 3.54 Å. No meaningful interdimer interaction, such as a π–π stacking or an interaction through counteranion, was observed in the crystal structures. Thus, the interdimer magnetic interaction is negligible for magnetic analysis.

Octahedral structure of Cu1 (a) and square pyramidal structure of Cu2 (b)
Magnetic Properties
The temperature-dependent magnetic susceptibilities of {(NIT-Ph-4-Br)2[Cu(hfac)2]3} were measured in the temperature range 2–300 K in an external magnetic field of 10 kOe, and the data were corrected for the diamagnetism of the components.
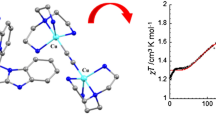
Magnetic data for {(NIT-Ph-4-Br)2[Cu(hfac)2]3} are shown in Fig. 4 as χm versus T and χmT versus T. For the value of χmT (square), at room temperature the value is 0.71 cm3 K mol−1, which is obviously lower than the theoretical value (1.875 cm3 K mol−1) for an uncoupled system of three copper(II) (S = 1/2, χmT = 0.375 cm3 K mol−1) plus two radicals (S = 1/2, χmT = 0.375 cm3 K mol−1), assuming g = 2, indicating the existence of strong antiferromagnetic interactions within the compound [48, 49]. With reduction in temperature, χmT sharply decreases to reach a value of 0.40 cm3 K mol−1, which is close to a spin value for S = 1/2 with g = 2. The effective expression for calculating the susceptibility of the five-spin system has not been found [50, 51]. And, based on the crystal data and the comparison with magnetic properties of other well-understood copper–radical complexes [50,51,52,53,54,55], the χmT − T curves for this complex can be rationalized qualitatively in term of an approximate model shown as follows:

Temperature dependence of χm (circular) and χmT (square) for complex {(NIT-Ph-4-Br)2[Cu(hfac)2]3}
In this model, we presume that the interactions between Cu1 and R1 and Cu1 and R2 are very small in this complex, while the interactions of Cu2–R1 and Cu2–R2 are strongly antiferromagnetic, because it has been found that two spins will have different coupling phenomena in the coordination environment of copper(II): When the N–O occupies an equatorial position, the spins are strongly coupled in an antiferromagnetic way with J > 500 cm−1 (Ĥ = JŜiŜj) [49, 53]; when the nitroxide occupies an axial position, a weak coupling is found [49]. Therefore, the monotonous decrease in χmT with the decrease in temperature should be ascribed to the very strong antiferromagnetic interaction between the Cu2–R1 and the Cu2–R2. The minimum of χmT of 0.40 cm3 K mol−1 at 2.0 K is close to an uncoupled Cu(II) ion (S = 1/2, 0.375 cm3 K mol−1), which may be due to the very strong antiferromagnetic interactions between Cu2 and radicals, which leads to complete spin cancellation. It can also be proved by fitting the susceptibilities (circular) by the Curie–Weiss law (red curve). In the Curie-Weiss fit we obtain C = 0.516 cm3 K mol−1, corresponding to a spin S = 1/2 with g = 2.34, proving the very strong antiferromagnetic interactions in the systems as expected, which is similar to those of other reported complexes [49,50,51].
The magnetization versus field curve obtained at 2 K is depicted in Fig. 5. At a magnetic field of 50 kOe, the magnetization reaches a maximum of 0.56 NμB, which is overall smaller than the expected value.

Isothermal magnetization at 2 K of {(NIT-Ph-4-Br)2[Cu(hfac)2]3}
Conclusion
The centrosymmetric complex {(NIT-Ph-4-Br)2[Cu(hfac)2]3} based on copper(II) and nitronyl nitroxide radicals was successfully prepared. The single-crystal structures show that two radicals are bridges linking three Cu(II) ions to form five-spin systems. The magnetic measurements indicate that the complex exhibited antiferromagnetic interactions between Cu(II) ions and radicals. Their magneto-structural correlations were explained based on the crystal structures.
Supplementary data
CCDC contains the supplementary crystallographic data for this paper (1959819 for 90 K and 1959820 for 173 K). The data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/structures.
References
Itoh K, Kinoshita M (eds) (2000) Molecular magnetism, new magnetic materials. Gordon Breach-Kodansha, Tokyo
Blundell SJ, Pratt FL (2004) J Phys: Condens Matter 16:R771–R828
Gatteschi D, Sessoli R, Villain J (2006) Molecular nanomagnets. Oxford University Press, Oxford
Sorace L, Benelli C, Gatteschi D (2011) Chem Soc Rev 40:3092–3104
Gutlich P, Garcia Y, Goodwin HA (2000) Chem Soc Rev 29:419–427
Kurmoo M (2009) Chem Soc Rev 38:1353–1379
Demir S, Jeon IR, Long JR, Harris TD (2015) Coord Chem Rev 149:289–290
Ratera JV (2012) Chem Soc Rev 41:303–349
Brook DJR (2015) Comments Inorg Chem 35:1–17
Preuss KE (2015) Coord Chem Rev 49:289–290
Morgan IS, Mansikkamäki A, Zissimou GA, Koutentis PA, Rouzières M, Clérac R, Tuononen HM (2015) Chem Eur J 21:15843–15851
Morgan IS, Peuronen A, Hänninen MM, Reed RW, Clérac R, Tuononen HM (2014) Inorg Chem 53:33–35
Fortier S, Le Roy JJ, Chen CH, Vieru V, Murugesu M, Chibotaru LF, Mindiola DJ, Caulton KG (2013) J Am Chem Soc 135:14670–14678
Miller JS (2011) Chem Soc Rev 40:3266–3296
Shultz DA, Miller JS, Drillon M (eds) (2002) Magnetism: molecules to materials II: molecule-based materials. Wiley-VCH, Weinheim, pp 281–304
Witt A, Heinemann FW, Khusniyarov MM (2015) Chem Sci 6:4599–4609
Occharenko V(2010) Stable radicals. Wiely, ch. 13
Miller JS, Drillon M (2003) Magnetism: Molecules to Materials II: Molecule-Based Materials, Wiely-VCH Verlag GmbH & Co, ch. 1
Mckinnon SDJ, Patrick BO, Lever ABP, Hicks RG (2013) Inorg Chem 52:8053–8066
Wu J, MacDonald DJ, Clérac R, Jeon IR, Jennings M, Lough AJ, Britten J, Robertson C, Dube PA, Preuss KE (2012) Inorg Chem 51:3827–3839
Kaszub W, Marino A, Lorenc M, Collet E, Bagryanskaya EG, Tretyakov EV, Ovcharenko VI, Fedin MV (2014) Angew Chem Int Ed 53:10636–10640
Wang J, Li JN, Zhang SL, Zhao XH, Shao D, Wang XY (2016) Chem Commun 52:5033–5036
Caneschi A, Gatteschi D, Sessoli R, Rey P (1989) Acc Chem Res 22:392–398
Caneschi A, Gatteschi D, Rey P (1991) Prog Inorg Chem 39:331–334
Dickman MH, Doedens RJ (1981) Inorg Chem 20:2677–2681
Porter LC, Ickmann MH, Doedens RJ (1983) Inorg Chem 22:1962–1964
Porter LC, Doedens RJ (1985) Inorg Chem 24:1006–1010
Lim YY, Drago RS (1972) Inorg Chem 11:1334–1338
de Panthou FL, Belorizky E, Calemczuk R, Luneau D, Marcenat C, Ressouche E, Turek P, Rey P (1995) J Am Chem Soc 117:11247–11253
Guedes GP, Zorzanelli RG, Comerlato NM, Speziali NL, Santos-Jr S, Vaz MGF (2012) Inorg Chem Commun 23:59–62
Grand A, Rey P, Subra R (1983) Inorg Chem 22:391–394
Caneschi A, Gatteschi D, Grand A, Laugier J, Pardi L, Rey P (1988) Inorg Chem 27:1031–1035
Caneschi A, Gatteschi D, Laugier J, Rey P (1987) J Am Chem Soc 109:2191–2192
Gatteschi D, Laugier J, Rey P, Zanchini C (1987) Inorg Chem 26:938–943
Luneau D, Romero FM, Ziessel R (1998) Inorg Chem 37:5078–5087
Porter LC, Dickman MH, Doedens RJ (1986) Inorg Chem 25:678–684
Ullman FE, Osiecki JH, Boocock DGB, Darcy R (1972) J Am Chem Soc 94:7049–7059
Hirel C, Vostrikova KE, Pécaut J, Ovcharenko VI, Rey P (2001) Chem Eur J 7:2007–2014
Boudreaux EA, Mulay LN (1976) Theory and application of molecular paramagnetism. Wiley, New York
Gordon AB, John FB (2008) J Chem Edu 85:532–536
SAINT-Plus, version 6.02, Bruker Analytical X-ray System, Madison, WI (1999)
Sheldrick GM (1996) SADABS—an empirical absorption correction program; Bruker Analytical X-ray Systems, Madison, WI
Sheldrick GM (2015) SHELXTL refinement program version 2016/6. Acta Crystallogr Sect C 71:3–8
Osanai K, Okazawa A, Nogami T, Ishida T (2006) J Am Chem Soc 128:14008–14009
Okazawa A, Nogami T, Ishida T (2007) Chem Mater 19:2733–2735
Okazawa A, Nagaichi Y, Nogami T, Ishida T (2008) Inorg Chem 47:8859–8868
Okazawa A, Nogami T, Ishida T (2009) Polyhedron 28:1917–1921
Jiang ZH, Yi Q, Liao DZ, Huan ZW, Yan SP, Wang GL, Yao XK, Wang RJ (1995) Trans Met Chem 20:136–137
Caneschi A, Gatteschi D, Sessoli R, Hoffmann SK (1988) Inorg Chem 27:2390–2392
Wang XL, Li YX, Yang SL, Zhang CX, Wang QL (2017) J Coord Chem 70:1–10
Wang YL, Gao YY, Yang MF, Gao T, Ma Y, Wang QL, Liao DZ (2013) Polyhedron 61:105–111
Caneschi A, Ferraro F, Gatteschi D, Rey P, Sessoli R (1991) Inorg Chem 30:3162–3166
Fokin S, Ovcharenko V, Romanenko G, Ikorskii V (2004) Inorg Chem 43:969–977
Tretyakov E, Fokin S, Romanenko G, Ikorskii V, Vasilevsky S, Ovcharenko V (2006) Inorg Chem 45:3671–3678
Musin RN, Schastnev PV, Malinovskaya SA (1992) Inorg Chem 31:4118–4121
Acknowledgment
This work was supported by (a) a Grant-in-Aid for Scientific Research (S) (No. 25220803) “Toward a New Class Magnetism by Chemically-controlled Chirality,” (b) a National Nature Science Foundation of China (No. 51762042), (c) Shaanxi Provincial Science and Technology Department Innovative Talent Promotion Plan Project of China (No. 2018KJXX-078) and (d) Doctoral Scientific Research Foundation of Yulin university (18GK24).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Gao, YL., Inoue, K. Synthesis, crystal structures and magnetic properties of nitronyl nitroxide radical-coordinated copper(II) complexes. Transit Met Chem 45, 195–201 (2020). https://doi.org/10.1007/s11243-019-00370-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11243-019-00370-y