
Abstract
M(II) (M = Cu, Zn) complexes of kojic acid (1, 2) have been synthesised, but characterisation was hampered by their lack of solubility. Related metal derivatives (5, 6) of benzyl-protected comenic acid (4), an oxidised variant of kojic acid, have been prepared, and the structures of 5·H2O·3DMSO (5a) and 6·2H2O (6a) were determined. The menthyl ester of O-benzyl comenic acid (7) has been prepared, but complete de-protection proved difficult. 3-Hydroxy-6-menthoxyacetyloxymethyl-4-pyrone (9) was prepared along with its M(II) derivatives (10, 11), though both were of low solubility; 10 forms soluble 1:1 adducts with pyridine (12) and 2,2′-bipyridine (13). Menthyl-3-oxo-butanoate (14) has been prepared along with its copper complex (15), whose structure has been determined. Included in the report are the structures of 4·DMSO (4a), 4·MeOH (4b), 4·THF (4c) and 9.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
We have an ongoing interest in metal complexes of α-hydroxyketones (I–III) [1–5], particularly those of copper, zinc and tin. These metals, which have antibacterial properties, can be utilised in dental formulations, provided that they can be delivered in stable form and in the correct oxidation state [6]. In this respect, α-hydroxyketones readily complex metals, which can be used to regulate levels of these metals in the body. For example, II
(R = R′ = Me) is known to form a complex with Fe3+ and is used in the treatment of Fe-overload diseases [7], II
(R = Me, R′ = Et) is a promising chelating agent for the treatment of iron overload in transfusion-dependent thalassaemia patients [8], while similar zinc complexes are potential matrix metalloproteinase (MMP) inhibitors [6, 7] and oral therapeutics for type 2 diabetes [9]. Pyridinones have also been used to regulate Fe3+, Cu2+ and Zn2+ overload in the brain, which has implications for Alzheimer’s disease [10–12]. 
While the metal complexation chemistry of ligands such as I–III is well established, that of the related kojic acid (IV) is scarcer. The older literature reports the synthesis [13–18] and anti-fungal activity [13, 14] of some copper and zinc (plus limited other transition metal) derivatives of IV, but characterisation of the resulting complexes is limited. Halogen derivatives of kojic acid e.g. 3-hydroxy-6-chloromethyl-4-pyrone, 3-hydroxy-6-iodomethyl-4-pyrone and 3-hydroxy-6-bromomethyl-4-pyrone [19] and sulphur-containing derivatives [20] have been reported to show significant anti-fungal effects. The bromo- and chloro-derivatives of kojic acid have also been found to have anti-neoplastic effects by significantly inhibiting DNA, RNA and protein synthesis [21]. The oxidised derivative of IV, commonly known as comenic acid (V), has been reported to act on organisms in the same way as vitamins [22] and has both oxidising and reducing activities [23]. It is used in pharmacology (as a component of the drug baliz-2) as an antioxidant [24].
As part of a long-term interest in the use of metal complexes in dental formulations, we now wish to present our findings in this area, which include crystallographic characterisation of copper and zinc complexes of both kojic acid and comenic acid, along with the synthesis of ligands which incorporate a flavourant (menthol) which may help mitigate the metallic taste of these complexes and which is detrimental to their oral delivery.
Experimental
Reactions were carried out in air unless otherwise specified. Elemental analyses were performed on a Carlo-Erba Strumentazione EA model 1106 microanalyser, and the temperature of the furnace was set to 500 °C. The results were duplicated, and the mean of the duplicated measurements was taken as the final result. All 1H and 13C NMR spectra were recorded on either a Bruker Avance (300 MHz) Fourier transform spectrometer or a JEOL JNM-GX270FT instrument. All spectra were recorded in either CDCl3 or d 6-DMSO and are referenced to residual solvent peaks. Peak positions were recorded in δ ppm with abbreviations s, d, t, m denoting singlet, doublet, triplet and multiplet, respectively, with a numbering scheme following that given in either Scheme 2 or Eq. 4 as far as is possible. All coupling constants (J) are quoted in Hertz. Infrared spectra were recorded as thin oil films sandwiched between NaCl plates in the frequency range 4000–400 cm−1 on a Nicolet 510P FT-IR spectrometer. All absorptions are quoted in cm−1.
3-Benzyloxy-6-hydroxymethyl-4-pyrone (3) [25] and menthoxyacetic acid [26] were synthesised using the literature procedures. In the following, menthol refers to the common (−) (1R, 2 S , 5R) stereoisomer.
Synthesis
3-Benzyloxy-6-carboxy-4-pyrone (4)
3-Benzyloxy-6-hydroxymethyl-4-pyrone (3) [25] (2.00 g, 8.62 mmol) was dissolved in acetone (15 mL) and cooled to 0 °C using an ice bath, to which was added Jones reagent (ca. 5 mL) (a mixture of 3.29 g, 32.9 mmol CrO3, 10 mL H2O and 3.29 mL H2SO4). The resulting brown mixture was left to stir for 1 h. The organic material was removed by filtration, and the filtrate was evaporated to dryness to obtain a pale green solid. Recrystallisation from methanol afforded 3-benzyloxy-6-carboxy-4-pyrone (9) as a white powder (1.53 g, 72 %; m.p 195–197, lit 195–197 °C [27]). Analysis, found [calc. for C13H10O5]: C 63.3 (63.4), H 4.13 (4.07) %. 1H NMR (d 6-DMSO): 8.40 [1H, s, C2 H]; 7.30–7.50 [5H, m, C6 H 5 ], 6.80 [1H, s, C5 H]; 5.01 [2H, s, CH 2 Ph]. 13C NMR (d 6-DMSO): 173 [C 4], 161 [C 7], 152 [C 6], 148 [C 3], 136 [C 2], 136–127 [C 6 H5], 117 [C 5], 71 [CH2Ph]. IR data: 3085 ν(O–H); 1731, 1629 ν(C=O); 1604 ν(C=C); 1267, 1216 ν(C–O).
Cu(3-benzyloxy-4-pyrone-6-carboxylate)2·2H2O (5)
3-Benzyloxy-6-carboxy-4-pyrone (4) (0.67 g, 2.72 mmol) was reacted with copper(II) acetate (0.27 g, 1.35 mmol) in a mixed H2O/EtOH (1:1) solvent over a 2-h period; on cooling, a light blue precipitate was produced (0.51 g; 49 %). Analysis, found [calc. for C26H22O12Cu]: C 51.9 (52.9), H 3.73 (3.73)%.
Recrystallisation from hot dimethylsulphoxide yielded turquoise crystals, which crystallography established as Cu(3-benzyloxy-4-pyrone-6-carboxylate)2·H2O·3DMSO (5a). Analysis, found [calc. for C32H38O14S3Cu]: C 46.8 (47.6), H 4.67 (4.71)%. IR data: 3100–3150 ν(O–H); 1639, 1589 ν(C=O); 1601 ν(C=C); 1285 ν(C–O).
Zn(3-benzyloxy-4-pyrone-6-carboxylate)2·3·5H2O (6)
The procedure for 5 was followed, using 3-benzyloxy-6-carboxy-4-pyrone (4) (0.71 g, 2.89 mmol) and zinc(II) acetate (0.32 g, 1.46 mmol). On cooling, a white precipitate of Zn(3-benzyloxy-4-pyrone-6-carboxylate)2·3·5H2O (6) was produced. Analysis, found [calc. for C26H25O13.5Zn]: C 50.7 (50.5), H 4.00 (4.04)%.
Colourless crystals of Zn(3-benzyloxy-4-pyrone-6-carboxylate)2·2H2O (6a) were produced by recrystallisation from hot methanol (0.21 g; 25 %). Analysis, found [calc. for C26H22O12Zn]: C 53.7 (52.8), H 3.98 (3.72)%. 1H NMR (d 6-DMSO): 8.20 [1H, s, C2 H], 7.50–7.30 [5H, m, C6 H 5 ], 6.80 [1H, s, C5 H], 4.95 [2H, s, CH 2 Ph]; 13C NMR (d 6-DMSO): 172 [C 4], 160 [C 7], 150 [C 3], 146 [C 6], 141 [C 2], 135–125 [C 6 H5]115 [C 5], 70.6 [CH2Ph]. IR data: 3200–3275 ν(O–H); 1632, 1590 ν(C=O); 1604 ν(C=C); 1287, 1218 ν(C–O).
Menthy-3-benzyloxy-4-pyrone-6-carboxylate (7)
A solution of 3-benzyloxy-6-carboxy-4-pyrone (4) (1.00 g, 4.07 mmol) in dry dichloromethane, under nitrogen, was stirred at room temperature for approximately 10 min. Once it had all dissolved, the solution was treated successively with triethylamine (0.82 g, 8.10 mmol) and thionyl chloride (0.82 mL, 11.2 mmol). After 2 h, the solution was evaporated in vacuo to remove excess thionyl chloride and dichloromethane, leaving a pink powder. This was re-dissolved in dichloromethane, and it was added to menthol (15) (0.95 g, 6.09 mmol) and dimethylaminopyridine (DMAP) in dichloromethane (20 %). This mixture was allowed to stir for 24 h. When esterification was complete, the reaction mixture was partitioned between diethyl ether and sodium bicarbonate (10 %), and then the organic phase was washed with brine (2 × 20 mL) (50 % sat.), dried over magnesium sulphate, filtered and dried under rotary evaporation to yield a yellow oil. A dark brown solid was produced from hot hexane, which was further purified by column chromatography (silica gel: hexane) to yield a yellow solid (1.32 g; 56 %). Analysis, found [calc. C23H28O5]: C 70.2 (71.9), H 7.30 (7.29)%. 1H NMR (CDCl3): 7.55 [1H, s, C2 H], 7.30–7.20 [5H, m, C6 H 5 ], 7.15 [1H, s, C5 H], 4.80 [1H, ddd, 3J 10.5, 3J 10.5, 3J 4.5 Hz, C8 H], 1.05–2.00 [2H, m, C9 H 2 ; 1H, m, C10 H; 2H, m, C11 H 2 ; 2H, m, C12 H 2 ; 1H, m C13 H; 1H, m, C14 H], 0.78, 0.71 [2 × 3H, d, 3J 3.6, 4.2 Hz, C15 H 3 ,/C16 H 3 ], 0.79 [3H, d, 3J 3.3 Hz, C17 H 3 ]; 5.10 [2H, s, CH 2 Ph]. 13C NMR(CDCl3): 173 [C 4], 158 [C 7], 151 [C 6], 141 [C 3], 134 [C 2], 134–127 [C 6 H5],118 [C 5], 76.6 [C 8], 71.0 [CH2Ph], 46.0 [C 13], 39.5 [C 9], 33.0 [C 11], 30.4 [C 10], 25.4 [C 14], 22.4 [C 17], 20.9 [C 12], 19.7, 15.3 [C 15 /C 16]. IR data: 1745, 1631 ν(C=O); 1595 ν(C=C); 1278, 1215 ν(C–O).
Synthesis of 3-hydroxy-6-menthoxyacetyloxymethyl-4-pyrone (9)
Menthoxyacetylchloride (8) was produced by dissolving menthoxyacetic acid [26] (2.60 g, 12.1 mmol) in thionyl chloride (4.32 mL, 59.3 mmol) and warming at 50 °C for 3 h. The excess thionyl chloride was removed by warming in a water bath under reduced pressure to produce a colourless oil (2.49 g; 88 %). Kojic acid (0.41 g, 2.89 mmol) was dissolved in dichloromethane (5 mL) and cooled to 0 °C using an ice bath. Pyridine (1.16 mL, 14.3 mmol) was added followed by a dichloromethane solution of the acetyl chloride 8 (1.00 g, 4.30 mmol). This was slowly warmed to room temperature, and stirring was continued for another 24 h. After completion of the reaction, the mixture was quenched with water (2 mL) and further stirred for 10 min. The aqueous phase was extracted with dichloromethane (2 × 10 mL). The organic fractions were combined and washed with 10 % HCl (25 mL), water (10 mL) and brine (25 mL), dried over magnesium sulphate, filtered and rotary-evaporated to produce a sticky white solid. Colourless crystals were obtained from hot ethanol (1.60 g, 39 %; m.p. 100 °C). Analysis, found [calc. C18H26O6]: C 63.6 (63.9), H 7.73 (7.69)%. 1H NMR (CDCl3): 7.85 [ 1H, s, C2 H], 6.30 [1H, s, C5 H], 6.02 [1H, br s, C3OH], 4.95 [2H, s, C7 H 2 ], 4.18 [1H, d, 2J 15.0 Hz, C9 H a], 4.02 [1H, d, 2J 15.0 Hz, C9 H b], 3.10 [1H, ddd, 3J 11.1, 3J 11.1, 3J 5.1 Hz, C10 H], 2.20–1.25 [2H, m, C11 H 2 ; 1H, m, C12 H; 2H, m, C13 H 2 ; 2H, m, C14 H 2 ; 1H, m, C15 H; 1H, m, C16 H], 0.87 [3H, d, J19–12 = 5.10 Hz, C19 H 3 ], 0.83, 0.72 [2 × 3H, d, 3J 7.50, 6.00 Hz, C17 H 3 /C18 H 3 ]. 13C NMR (CDCl3): 173 [C 8], 169 [C 4], 162 [C 6], 145 [C 3], 137 [C 2], 111 [C 5], 79.5 [C 10], 64.7 [C 9], 60.4 [C 7], 47.0 [C 15], 38.9 [C 11], 33.3 [C 13], 30.4 [C 12], 24.6 [C 16], 22.3 [C 19], 21.2 [C 14], 19.9, 15.3 [C 17/ C 18]. IR data: 1756, 1655, 1623 ν(C=O); 1558 ν(C=C); 1230, 1170 ν(C–O).
Cu(3-hydroxy-6-menthoxyacetyloxymethyl-4-pyronate)2 (10)
3-Hydroxy-6-menthoxyacetyloxymethyl-4-pyrone (9) (0.16 g, 0.47 mmol) was reacted with copper(II) acetate (0.05 g, 0.25 mmol) in a mixed H2O/EtOH (1:1) solvent over a 2-h period, and on cooling, a light green precipitate was produced (0.30 g; 86 %). Analysis, found [calc. C36H50O12Cu]: C 58.9 (58.6), H 6.62 (6.78)%. IR data: 1756, 1621, 1564 ν(C=O); 1506 ν(C=C); 1252, 1169 ν(C–O).
Zn(3-hydroxy-6-menthoxyacetyloxymethyl-4-pyronate)2 (11)
3-Hydroxy-6-menthoxyacetyloxymethyl-4-pyrone (9) (0.16 g, 0.49 mmol) was reacted with zinc(II) acetate (0.05 g, 0.27 mmol) in a manner similar to 10; on cooling, a cream precipitate was produced (0.32 g; 91 %). Analysis, found [calc. C36H50O12Zn]: C 58.4 (58.4), H 6.81 (6.76)%. IR data: 1762, 1611, 1592 ν(C=O); 1533 ν(C=C); 1267, 1155 ν(C–O).
Cu(3-hydroxy-6-menthoxyacetyloxymethyl-4-pyronate)2·py (12)
Pyridine (5 mL) was added to a suspension of Cu(3-hydroxy-6-menthoxyacetyloxymethyl-4-pyronate)2 (10) (0.50 g, 0.68 mmol) and toluene (30 mL). The dark green solution formed was stirred for 30 min and cooled to −20 °C. A dark green precipitate of 12 was produced on standing (0.32 g; 58 %). Analysis, found [calc. C41H55O12NCu]: C 60.5 (60.3), H 6.80 (6.74), N 1.75 (1.71)%. IR data: 1760, 1649, 1525 ν(C=O); 1501 ν(C=C); 1261, 1155 ν(C–O).
Cu(3-hydroxy-6-menthoxylaetyloxymethyl-4-pyronate)2·bipy (13)
Bipyridine (0.80 g, 5.13 mmol) was added to a mixture of toluene (30 mL) and Cu(3-hydroxy-6-menthoxyacetyloxymethyl-4-pyronate)2 (10) (0.60 g, 0.81 mmol). The reaction mixture was stirred for 30 min at room temperature and then left for several days, yielding a brown solid (0.52 g; 72 %). Analysis, found [calc. C46H58O12N2Cu]: C 60.9 (61.7), H 6.50 (6.49), N 3.20 (3.13)%. IR data: 1768, 1657, 1523 ν(C=O); 1509 ν(C=C); 1265, 1145 ν(C–O).
Synthesis of menthy-3-oxo-butanoate (14) [28]
Diketene (4.44 mL, 57.6 mmol) in acetonitrile (6 mL) was added dropwise to an acetonitrile (25 mL) solution of menthol (4.50 g, 28.8 mmol) and sodium acetate (0.15 g, 1.83 mmol) and refluxed for 2 h. The reaction mixture was cooled to 0 °C and treated with water (6 mL) and extracted with diethyl ether (2 × 20 mL). The combined organic layer was washed with brine (3 × 10 mL), dried over magnesium sulphate and rotary-evaporated to yield an orange oil. Further purification using column chromatography (petroleum ether/ethyl acetate, 95:5) gave a colourless oil (6.20 g; 89 %). Analysis, found [calc. for C14H24O3]: C 69.5 (70.0), H 10.2 (10.0)%. 1H NMR (CDCl3): 4.59 [1H, ddd, 3J 10.5, 3J 10.5, 3J 4.6 Hz, C5 H], 3.28 [2H, s, C3 H 2 ], 2.10 [3H, s, C1 H 3 ], 1.88–1.20-[2H, m, C6 H 2 ; 1H, m, C7 H; 2H, m, C8 H 2 ; 2H, m, C9 H 2 ; 1H, m, C10 H; 1H, m, C11 H], 0.73, 0.61 [2 × 3H, d, 3J 5.40, 6.30 Hz, C12 H 3 /C13 H 3 ], 0.76 [3H, d, 3J 4.2 Hz, C14 H 3 ]. 13C NMR (CDCl3): 201 [C 4], 167 [C 2], 75.9 [C 5], 51.0 [C 10], 47.3 [C 1], 41.1 [C 6], 34.6 [C 8], 31.8 [C 7], 30.4 [C 3], 26.5 [C 11], 23.7 [C 14], 22.4 [C 9], 21.1, 16.5 [C 12 /C 13]. IR data: 1625, 1710 ν(C=O).
Cu(menthyl 3-hydroxy-but-2-enoate)2·MeOH (15)
A minimum amount of dilute ammonia (ca. 3 mL) was added to menthyl 3-oxo-butanoate (14) (2.89 g, 12.0 mmol) in order to dissolve the ligand and give a homogeneous solution. This solution was added slowly to CuSO4·5H2O (1.50 g, 6.01 mmol) dissolved in cold water (10 mL). A blue precipitate was produced and filtered off. The precipitate was washed slowly with cold acetone and finally with diethyl ether. The solid was dried in vacuo and recrystallised from warm methanol to produce dark blue crystals of 15 (3.21 g; 46 %). Analysis, found [calc. for C29H50O7Cu]: C 59.9 (60.7), H 8.75 (8.72)%. IR data: 1590, 1555 ν(C=O).
X-ray crystallography
Experimental details relating to the single-crystal X-ray crystallographic studies are summarised in Table 1. For all structures, data were collected on a Nonius Kappa CCD diffractometer at 150(2) K using Mo-Kα radiation (λ = 0.71073 Å). Structure solution followed by full-matrix least squares refinement was performed using the WinGX-1.80 suite of programmes [29]. Specific details are 4a: the asymmetric unit consists of one molecule of 4 and one molecule of DMSO. H2 was located in the difference Fourier map and was freely refined. 4b: the asymmetric unit consists of one molecule of 4 and one MeOH. All O–H hydrogen atoms were located in the difference Fourier map and were refined with bond lengths restraints and additionally bond angle restraint for H6A. 4c: the asymmetric unit consists of one molecule of 4 and one THF. The O–H hydrogen atom H1 was located in the difference Fourier map and was refined freely. 5a: the asymmetric unit consists of one molecule of 5 (half of the dimer), one water and three molecules of DMSO. The water hydrogen atoms H11a and H11b were located in the difference Fourier map and were freely refined; the largest residual in the electron density map (1.5 eÅ3) is 1.25 Å from sulphur in a non-chemically significant location. 6a: the asymmetric unit consists of one molecule of 5 (half of the dimer), the other half being generated via an inversion centre. Hydrogen atoms for the water molecules (H11A/B, H12A/B) were located in the difference Fourier map and were refined using bond length and bond angle restraints. 9: the asymmetric unit consists of two molecules of 9 connected via hydrogen bonds. The COH hydrogens H1 and H7 were located in the difference Fourier map and were refined freely. 15: the asymmetric unit consists of two molecules of 15 and two molecules of MeOH. O–H hydrogen atoms (H7, H14) were located in the difference Fourier map and were refined with bond length restraints. Data completeness of 92 % reflects sample quality, which was optimal despite several recrystallisations.
Results and discussion
Initial attempts to prepare copper and zinc derivatives of kojic acid proved largely ineffective. The products (1, 2; Eq. 1) proved to be completely insoluble and defied full characterisation. While 2 was analytically pure as precipitated, 1 was impure but could not be purified further. Presumably the lack of solubility in the complexes, in contrast to similar derivatives of related α-hydroxyketones (maltol, pyridinione, tropolone), is due to extensive intermolecular hydrogen bonding involving the pendant CH2OH/C=O, COH functions.

In order to introduce solubility and/or functionality, IV can be elaborated via either the hydroxyketone or hydroxyalkyl positions. We initially prepared the related carboxylic acid (4; O-benzylcomenic acid) by protection of the hydroxyketone (3) then oxidation of the remaining primary alcohol using Jones’ reagent (CrO3, H2O and H2SO4), as shown in Scheme 1.

Synthesis of O-benzylcomenic acid (4)
The identities of 3 and 4 were confirmed by 1H, 13C NMR, IR and elemental analysis. In 1H NMR Spectrum of 4, the disappearance of the C7 H 2 peak is observed confirming the replacement of the CH2 in 3 by a C=O group in 4. The infrared spectra of 4 show the appearance of a second C=O band (1731 cm−1) typical of a carboxylic acid, in addition to the C=O of C4 (1629 cm−1).
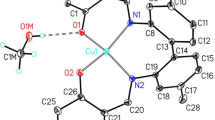
Crystals of solvated 4 (DMSO, 4a; MeOH, 4b; THF, 4c), obtained fortuitously as unreacted material from reactions with metal acetates in various solvents to form M(II) derivatives, confirm the identity of 4. As an example, the structure of 4a is shown in Fig. 1; structures of 4b and 4c, both of which show extensive hydrogen-bonded networks, can be found in the Supplementary data (Figures S1, S2 and Tables T1, T2) and will not be discussed further here. In 4a, the oxygen of the DMSO solvent [O(6)] is hydrogen bonded to the hydrogen of the carboxylic group, H(2) [H(2)···O(6) 1.69(5); O(2)···O(6) 2.490(3) Å; ∠O(2)–H(2)···O(6) 169(5)°]. In addition, there is another weak H(6)···O(2′) interaction to the adjacent molecule [H(6)···O(2′) 2.41, C(6)···O(2′) 3.347(3) Å; ∠C(6)–H(6)···O(2′) 168.9°; symmetry operation: 2 −x, −y, −z], which creates a dimeric structure. The structure is further stabilised by much weaker bifurcated hydrogen bonds involving hydrogen atoms of the DMSO methyl groups on the adjacent solvent molecule [H(14A), H(15B)] with carbonyl oxygen of the pyronic ring [O(4)···C(14) 3.192 (3), H(14B)···O(4): 2.50 Å; ∠C(14)–H(14B)···O(4) 127.2°; (4)···C(15) 3.261(3), H(15B)···O(4): 2.59 Å; ∠C(140–H(14B)···O(4) 125.9°; symmetry operation: −x + 3/2, y − 1/2, −z + 1/2] to produce an extended lattice network.

Asymmetric unit of 4a, showing the labelling scheme used; ellipsoids are at the 50 % level. Symmetry operations: ′2 − x, −y, −z; ″3/2 − x, ½ + y, ½ − z
Reaction of 4 with the appropriate metal acetate in a water/alcohol mix successfully yielded the copper (5) and zinc (6) complexes (Eq. 2):

Microanalysis suggests that these complexes are hydrated i.e. 5·2H2O, 6·3·5H2O. The 1H NMR spectrum of 6 shows the presence of the C2 H (8.20 ppm) and C5 H (6.80 ppm) from the ligand and the presence of the protecting group (7.30–7.50 ppm).
Recrystallisation of 5 from hot DMSO produced Cu(3-benzyloxy-4-pyrone-6-carboxylate)2·H2O·3DMSO (5a). The asymmetric unit of 5a (Fig. 2) consists of one half of a dimer in which each copper atom is coordinated by carboxylate groups, DMSO, one water molecule and a pyronic carbonyl. The carboxylate ligands are monodentate with respect to copper, and the [C=O] involves itself in hydrogen bonding. There are a further two molecules of DMSO incorporated in the structure, but these are not interacting with the metal centre. The remainder of the structure is generated via an inversion centre at the heart of the dimer (symmetry operation 1 −x, −y, 2 −z) creating a head-to-tail configuration. The coordination sphere around each copper atom is square-pyramidal CuO5 (τ = 0.10) [30], two of which are from the carboxylate ligands, one from the water molecule, one from the DMSO molecule and one from the pyronic carbonyl oxygen; the pyronic carbonyl is in the axial position [Cu–O(9′) 2.2690(17) Å] and the carboxylate ligands [Cu–O(1) 1.9635(18), Cu–O(6) 1.9762(15) Å], and solvent molecules, water [Cu–O(11) 1.9352(18) Å] and DMSO [Cu–O(12) 1.9511(17) Å], in the equatorial positions. The angles subtended by the atoms in the equatorial plane at the centre are close to the ideal for a square-planar geometry [O(6)–Cu–O(11): 88.50(7)°; (1)–Cu–O(12): 91.26(7)°; (6)–Cu–O(12): 89.64(7)°; (1)–Cu–O(11): 90.54(7)°]. In addition, the trans pair of atom angles also are close to the ideal value of 180° [O(1)–Cu–O(6); 171.84(7)°; (11)–Cu–O(12): 178.10(7)°]. Two other molecules of DMSO are included in the structure (Fig. 2), which are linked to the dimer by hydrogen bonds [H(7a)···O(14) 2.51 Å, C(7)···O(14) 3.453(4) Å, ∠C(7)–H(7a)···O(14) 160.1°; H(19)···O(13) 2.42 Å, C(19)···O(13) 3.371(3) Å, ∠C(19)–H(19)···O(13) 174.2°].

Asymmetric unit of 5a, showing the labelling scheme used; ellipsoids are at the 50 % level. Selected geometric data: Cu–O(1) 1.9352(18), Cu–O(6) 1.9762(15), Cu–O(9′) 2.2689(16), Cu–O(11) 1.9352(18), Cu–O(12) 1.9511(17), (1)–C(1) 1.271(3), (2)–C(1) 1.241(3), (4)–C(4) 1.236(3), (6)–C(14) 1.275(3), (7)–C(14) 1.234(3), (9)–C(17) 1.242(3) Å; (1)–Cu–O(6) 171.84(7), (1)–Cu–O(9′) 86.42(6), (1)–Cu–O(11) 90.54(7), (1)–Cu–O(12) 91.26(7), (6)–Cu–O(9′) 101.72(6), (6)–Cu–O(11) 88.50(7), (6)–Cu–O(12) 89.64(7), (9′)–Cu–O(11) 92.74(7), (9′)–Cu–O(12) 87.98(7), (11)–Cu–O(12) 178.10(7)°. Symmetry operations: ′1 − x, −y, 2 − z; ″−x, −y, 2 − z
In addition, there is lattice association through a network of hydrogen bonds (Fig. 3), primarily involving the coordinated water molecule [H(11a)···O(2′) 1.88(4), (11)···O(2′)··· 2.631(2), ∠O(11)–H(11a)···O(2′) 167(3); H(11b)···O(7′) 1.85(4), (11)···O(7′).2.622(2), ∠O(11)–H(11b)···O(7′) 176(3)°], supported by a weaker CH···O interaction [H(3)···O(14) 2.56 Å, C(3)···O(14) 3.441(3) Å, ∠C(3)–H(3)···O(14) 153.9°; symmetry operation: −x, −y, −z + 1]. The DMSO based on S(3) thus forms bifurcated hydrogen bonds in the same way as seen in 4a.

Intermolecular association through hydrogen bonds in 5a
Recrystallisation of the zinc complex 6 from hot methanol resulted in the formation of Zn(3-benzyloxy-4-pyrone-6-carboxylate)2·2H2O (6a). The structure of 6a (Fig. 4) is similar to that of 5a, whereby it consists of a dimer generated via the inversion operation 1 − x, 1 − y, −z, but with H2O coordinated to zinc instead of DMSO. The zinc(II) ion is coordinated to five oxygen atoms, of which two oxygens are from the carboxylate residue [Zn–O(1) 2.012(3), Zn–O(9) 2.063(3) Å], one from the carbonyl oxygen atom of the pyranone ring [Zn–O(10′) 1.995(3) Å] and two from the water molecules [Zn–O(11) 2.039(4), Zn–O(12) 2.019(3) Å]. 6a thus exhibits a five-coordinated ZnO5 motif and a square-pyramidal geometry (τ = 0.098) [30] with two carboxylate atoms [O(1)–Zn–O(9): 157.04(16) and two water molecules in the basal plane [O(11)–Zn–O(12): 163.80(17)°] and the pyranone oxygen (10) axial; this geometry is notably more distorted than that in 5a.

Asymmetric unit of 6a, showing the labelling scheme used (H11a is not labelled); ellipsoids are at the 50 % level. Selected geometric data: Zn–O(1) 2.012(3), Zn–O(9) 2.063(3), Zn–O(10′) 1.995(3), Zn–O(11) 2.039(4), Zn–O(12) 2.019(3), (1)–C(1) 1.261(6), (2)–C(1) 1.245(7), (3)–C(4) 1.235(7), (8)–C(24) 1.240(6), (9)–C(24) 1.261(6), (10)–C(26) 1.267(5) Å; (1)–Zn–O(9) 157.04(16), (1)–Zn–O(10′) 94.33(15), (1)–Zn–O(11) 92.36(15), (1)–Zn–O(12) 89.77(14), (9)–Zn–O(10′) 108.60(13), (9)–Zn–O(11) 84.08(14), (9)–Zn–O(12) 87.32(13), (10′)–Zn–O(11) 96.45(15), (10′)–Zn–O(12) 100.41(14), (11)–Zn–O(12) 163.80(17)°. Symmetry operation: 1 − x, 1 − y, z
As with 5a, 6a forms an associated lattice through a network of hydrogen bonds (Fig. 5). The carbonyl oxygen of each carboxylate group, which are themselves monodentate with respect to zinc, forms a pair of bifurcated hydrogen bonds involving, in total, all the water hydrogen atoms, to form hydrogen-bonded molecular stacks [H(11b)···O(2) 1.95(6), (11)···O(2) 2.710(5) Å, ∠O(11b)–H(11)···(O2) 142(9)°; H(11a)···O(8) 1.88(4), (11)···O(8) 2.699(5) Å, ∠O(11b)–H(11)···(O2) 154(8)°; H(12a)···O(8) 2.03(8), (12)···O(8) 2.691(4) Å, ∠O(11b)–H(11)···(O2) 131(9)°; H(12b)···O(2) 1.89(3), (12)···O(2) 2.737(5) Å, ∠O(12)–H(12b)···(O2) 159(6)°].

Intermolecular association through hydrogen bonds in 6a
Our initial attempt to introduce a flavourant as a ligand component involved the initial conversion of 4 to its acid chloride, followed by reaction with menthol in the presence of base to yield 7 (Scheme 2). 1H NMR of 7 shows new peaks due to the menthyl group at δ 0.71–2.00 ppm, while the C8 H proton is shifted from 3.12 ppm in menthol to 4.80 ppm in 7 and the alkene protons on the pyronic ring shift from 8.40 ppm [C2 H] and 6.80 ppm [C5 H] in 4 to 7.55 ppm [C2 H] and 7.15 ppm [C5 H] in 7. However, despite numerous attempts to de-protect 7 using an atmosphere of hydrogen gas in the presence of a catalytic amount of Pd/C, with varying temperatures (25, 50 °C), different reaction times (4, 16, 24 h) and different solvents (THF, EtOH, MeOH, aq EtOH), 1H NMR showed that there was still the presence of the protecting group (5.10 ppm, CH 2 Ph; 7.20–7.30 ppm, C6 H 5 ).

Synthetic routes to flavourant-functionalised derivatives of kojic acid
However, the related species 9 was obtained, albeit fortuitously. Menthoxyacetic acid was converted to its acyl chloride (8) which was directly reacted with kojic acid in the presence of a pyridine [31]. However, from spectroscopic data and X-ray analysis, it was found that the base deprotonated the C7-OH on kojic acid instead of the anticipated C3-OH to produce 3-hydroxy-6-menthoxyacetyloxymethyl-4-pyrone (9) (Scheme 2). X-ray analysis of 9 (Fig. 6) confirmed the coupling site as C7-OH rather than the C3-OH, leaving a free chelating hydroxyketone for zinc and copper chelation. The asymmetric unit of 9 consists of a dimer, involving two ligands that are intermolecularly hydrogen bonded to each other via two complimentary C=O···OH hydrogen bonds [H(7)···O(2) 1.91(8), (7)···O(2): 2.718(5) Å, ∠O(7)-H(7)···O(2) 142(8)°; H(1) (8) 2.00(6), (1)···O(8): 2.744(6) Å, ∠O(1)-H(1)···O(8) 150(6)°]. This dimeric arrangement is common among the family of α-hydroxyketones I–IV and a pattern which we and others have encountered elsewhere in these studies [4, 32]. However, the two menthyl groups are disposed in a trans manner with respect to the two pyranone rings, unlike the related dimer of 1-menthoxyethyl-2-methyl-3-hydroxypyridin-4-one, which incorporates the same flavorant but attached to a pyridinone ring, where the two menthyl groups are cis to each other [5].

Asymmetric unit of 9, showing the labelling scheme used; ellipsoids are at the 50 % level
Copper (10) and zinc (11) complexes have been prepared from 9 in a similar manner to that described above for 5, 6 (Eq. 3):

10, 11 were obtained as highly insoluble precipitates and characterised by elemental analysis and IR spectroscopy. The elemental analysis suggested that the two complexes are both anhydrous. The absence of the υ(O–H) mode and the decrease in the υ(C=O) mode by ca. 34–44 cm−1 in the IR spectra of the metal complexes (10: υ(C=O) 1621 cm−1; 11: υ(C=O) 1611 cm−1) as compared to the ligand (9: 1655 cm−1) confirm the expected O,O κ 2 chelation by the ligand, while ν(C=O) of the ester group in the spectrum of 9 (1756 cm−1) is similar to the metal complexes (10: 1756 cm−1; 11:1762 cm−1), suggesting that no coordination occurs via this carbonyl group.
In order to obtain more soluble species for detailed structural analysis, 10 was suspended in toluene and either pyridine or 2,2′-bipyridyl was added. 10 was thus solubilised, and on standing these solutions produced dark precipitates of Cu(3-hydroxy-6-menthoxyacetyloxymethyl-4-pyronate)2·py (12, green) and Cu(3-hydroxy-6-menthoxyacetyloxymethyl-4-pyronate)2·bipy (13, brown). 12 and 13 were characterised by elemental analysis, and the results confirmed that 1:1 copper complexes with either bipyridine or pyridine coordinated to the metal complex has been formed. Unfortunately, crystals suitable for definitive structural elucidation were not forthcoming, but 12 presumably contains a five-coordinate copper and the bipyridine adduct 13 a six-coordinate metal.
Finally, to gain some insight into the structures of insoluble 10, 11, a new flavoured diketone ligand, menthyl-3-oxo-butanoate (14) was synthesised following a literature procedure (Eq. 4) [21], involving base-catalysed acylation reaction between menthol, anhydrous sodium acetate and diketene:

Two bands were observed in the IR spectrum of 14 (1710, 1625 cm−1) which correspond to the υ(C=O) bands. 1H NMR showed the presence of the menthol component of 14, with C5 H resonating at a δ 4.59 ppm, shifted from 3.12 ppm in menthol. In addition, the coupling pattern of the C5 H in 1H NMR spectrum confirms the retention of the menthol configuration at C5 (ddd, J8–9ax = J8–13 = 10.5, J5–8eq = 4.6 Hz; see Scheme 2 for menthyl group numbering].
14 was reacted with copper acetate following the reaction conditions employed in the synthesis of bis(acetylacetonato)copper(II) [33]. A blue precipitate was formed which was crystallised from hot methanol to produce Cu(menthyl-3-hydroxy-but-2-enoate)2·MeOH (15; Eq. 4). The υ(C=O) bands in the starting material are shifted to lower wavenumber in 15 (1590; 1555 cm−1), suggesting that the metal chelates via the two oxygen atoms. X-ray analysis shows that the asymmetric unit of 15 contains two independent molecules that are hydrogen bonded, generating a dimer (Fig. 7). A molecule of methanol is coordinated to the metal centre in each molecule to generate a five-coordinate species. The hydrogen atom of each methanol forms an intermolecular hydrogen bond to the hydroxyl oxygen on the next molecule [H(7)···O(8) 2.00(2), (7)···O(8): 2.857(4) Å, ∠O(7)–H(7)···O(8) 164(6)°; H(14)···O(3) 1.893(19), (14)···O(3): 2.745(4) Å), ∠O(14)–H(14)···O(3) 164(5)°]. The coordination sphere around the copper metal is square-pyramidal in both molecules (τ = 0.01, 0.13 for Cu(1), Cu(2), respectively) [30], in which the solvent molecule occupies the apical position and involves the longest of the Cu–O bonds present in the molecule [Cu1)–O(7) 2.261(4); Cu(2)–O(14) 2.296(4) Å]. The pairs of chelate rings on the basal plane of both molecules are oriented cis with respect to the two menthyl groups, a situation dictated by the formation of the dimer via hydrogen bonds. Within each chelate ring, there is a short Cu–O bond associated with a longer C–O interaction [e.g. Cu(1)–O(1) 1.910(3), (1)–C(1) 1.283(5) Å] and one long Cu–O associated with a shorter C=O [e.g. Cu(1)–O(2) 1.923(3), (2)–C(4) 1.253(5) Å]. However, in the chelate rings which are also involved in hydrogen bonding, the C–O bond is lengthened further [e.g. (3)–C(15) 1.291(5) Å]. The structure of 15 is similar to those bis(2,3,3,4,4-pentamethyl-8, 8, 8-trifluoro-4-silaoctane-5,7-dionato)copper(II) (methanol)) [34] and bis(pivaloyltrifluoroacetonate)copper(II)·EtOH [35], both of which incorporate two O,O-chelate rings and a solvent molecule in a square-pyramidal environment (Fig. 7).

Asymmetric unit of 15, showing the labelling scheme used in the text; thermal ellipsoids are at the 40 % level. C(5) is hidden behind C(22) and is not labelled. Selected geometric data for the molecule containing Cu(1): Cu(1)–O(1) 1.910(3), Cu(1)–O(2) 1.923(3), Cu(1)–O(3) 1.919(3),Cu(1)–O(4) 1.938(2), Cu(1)–O(7) 2.261(4), O(1)–C(1) 1.283(5), O(2)–C(4) 1.253(5), O(3)–C(15) 1.291(5), O(4)–C(18) 1.257(4) Å; O(1)–Cu(1)–O(2) 93.47(12), O(1)–Cu(1)–O(3) 88.14(12), O(1)–Cu(1)–O(4) 171.12(16), O(1)–Cu(1)–O(7) 96.04(15), O(2)–Cu(1)–O(3) 171.45(16), O(2)–Cu(1)–O(4) 84.19(10), O(2)–Cu(1)–O(7) 95.19(15), O(3)–Cu(1)–O(4) 92.94(11), O(3)–Cu(1)–O(7) 92.99(14), O(4)–Cu(1)–O(7) 92.70(15)°
Conclusions
Simple copper(II) and zinc(II) derivatives of kojic acid (1, 2) are insoluble, presumably due to intermolecular hydrogen bonds involving the exo-cyclic CH2OH groups. Unfortunately, elaboration of this functionality in the form of 6-menthoxyacetyloxymethyl-4-pyrone (9), which incorporates a flavouring component, failed to enhance the solubility of the corresponding metal complexes, although addition of a Lewis base (pyridine, bipyridyl) does cause dissolution. Similarly, the two metals form complexes with comenic acid (5, 6) after oxidation of kojic acid, though again these are only soluble in strongly coordination solvents (DMSO, MeOH). Consequently, these metal complexes collectively seem less attractive as potential antibacterial agents’ dental formulations in comparison with their previously reported pyranone (maltol, ethyl maltol) and pyridinone analogues [5].
References
Barret MC, Mahon MF, Molloy KC, Steed JW, Wright P (2001) Inorg Chem 40:4384
Barret MC, Mahon MF, Molloy KC, Wright P (2000) Main Group Metal Chem 23:663
Barret MC, Mahon MF, Molloy KC, Wright P, Creeth JE (2002) Polyhedron 21:1761
Barret MC, Bhatia PH, Kociok-Köhn G, Molloy KC (2014) Trans Met Chem 39:543
Bhatia PH, Kociok-Köhn G, Molloy KC (2014) Trans Met Chem 40:241
Creeth J, Molloy KC, Wright P (2000) Oral Care Compos Int. Patent No. WO 00/16736
Faller B, Nick H (1994) J Am Chem Soc 116:3860
Weatherall DJ, Pippard MJ, Callender ST (1983) New Eng J Med 308:456
Adachi Y, Yoshikawa Y, Yoshida J, Kodera Y, Katoh A, Kojima Y, Sakurai H (2006) Biomed Res Trace Elem 17:17
Scott LE, Telpoukhovskaia M, Rodrıguez-Rodrıguez C, Merkel M, Bowen ML, Page BDG, Green DE, Storr T, Thomas F, Allen DD, Lockman PR, Patrick BO, Adam MJ, Orvig C (2011) Chem Sci 2:642
Scott LE, Page BDG, Patrick BO, Orvig C (2008) Dalton Trans 45:6364
Green DE, Bowen ML, Scott LE, Storr T, Merkel M, Böhmerle K, Thompson KH, Patrick BO, Schugar HJ, Orvig C (2010) Dalton Trans 39:1604
Melnik M, Uher M, Brtko J, Mrozinska D, Mrozinski J (1993) Pol J Chem 67:1219
Balaz S, Uher M, Brtko J, Veverka M, Bransova J, Dobias J, Podova M, Buchvald J (1993) Folia Microbiol 38:387
Gerard C (1979) Bull Soc Chim Fr 11–12:451
Murakami Y, Mera K (1966) Bull Chem Soc Jpn 39:396
Sharma S, Ramani J, Bhalodia J, Patel N, Thakkar K, Patel R (2011) Adv Appl Sci Res 2:37482
Emami S, Hosseinimehr SJ, Taghdisi SM, Akhlaghpoor S (2007) Bioorg Med Chem Lett 17:45
Hudecova D, Uher M, Brtko J (1992) Biologia 47:483
Uher M, Kyselicova L, Rajniakova O, Hudecova D, Bransova J (1997) Chem Pap 51:421
Bransova J, Uher M, Novtny L, Brtko J (1998) Anticancer Res 18:4423
Derbenev AV, Krylov BV, Shurygin AY (2000) Membrane Cell Biol 13:379
Grishina EP, Shurygin AY, Alekseeva AN, Ramenskaya LM (2001) Russ J Electrochem 37:976
Shurygin, AY, Yugai GA (2000) Izv. Vyss. Uchebn. Zaved. Sev-Kavk. Re. Estestv. Nauki, 71
Hare LE, Lu MC, Sullivan CB, SullivanP T, Counsell RE (1974) J Med Chem 17:1
Cochran TG, Huitric AC (1971) J Org Chem 36:3046
Thomas AF (1962) J Chem Soc 439
Landais Y, Planchenault D (1997) Tetrahedron 53:2855
Farrugia LJ (1999) J Appl Crystallogr 32:837
Addison AW, Rao TN, Reedijk J, van Rijn J, Verschoor GC (1984) J Chem Soc Dalton Trans 1349
Campbell AL, Miyano M (1986) Eur. Pat. EP 0171814 A1
Burgess J, Fawcett J, Russell DR, Waltham E (1998) Acta Crystallogr Sect C 54:2011
Peacock RD (1971) J Chem Ed 48:199
Higashiya S, Banger KK, Ngo SC, Lim PN, Toscano PJ, Welch JT (2003) Inorg. Chim Acta 351:291
DelaRosa MJ, Banger KK, Higashiya S, Ngo SC, Hunt DH, Bousman KS, Toscano P, Welch JT (2003) J Fluor Chem 123:109
Acknowledgments
We thank M B Barret (University of Bath) for the synthesis of compounds 1 and 2.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
11243_2015_9935_MOESM1_ESM.docx
CCDC 1045954-1045960 contains the supplementary crystallographic data for this paper (Table 1). These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif (DOCX 400 kb)
Rights and permissions
About this article

Cite this article
Bhatia, P.H., Kociok-Köhn, G. & Molloy, K.C. Copper and zinc complexes of kojic acid and related ligands. Transition Met Chem 40, 459–470 (2015). https://doi.org/10.1007/s11243-015-9935-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11243-015-9935-0