
Abstract
The reaction of M(OAc)2 with 2,2′-bis(2-hydroxybenzylideneamino)-4,4′-dimethyl-1,1′-biphenyl (H2L1) allows the synthesis of 2,2′-bis(2-oxidobenzylideneamino)-4,4′-dimethyl-1,1′-biphenyl complexes of Cu(II) (CuL1), Co(II) (CoL1) and Ni(II) (NiL1) that were characterized by elemental analysis, FTIR spectroscopy and for CuL1 also by X-ray crystallography verifying a tetradentate binding mode of L1 via an (ONNO) motif of the two phenolic oxygen atoms and two azomethine nitrogen atoms. Recrystallization from a solvent mixture of dichloromethane and methanol promotes the formation of methanol adducts. Different binding modes of the methanol–complex were investigated using density functional theory calculations and binding energies, and thermodynamic data of the interaction are reported. The results show that the favored interaction occurs when the methanol molecule acts as a Lewis acid weakly binding via an O–H···O hydrogen bridge to a phenoxide moiety leading to an elongation of the respective M–O bond.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Tetradentate Schiff bases, especially with two N and two O donor sites, resulting from the condensation of aliphatic and aromatic diamines and their derivatives with salicylaldehyde have been studied intensely [1,2,3,4,5,6,7,8,9,10,11,12]. Schiff bases and their metal complexes have played a seminal role in the development of coordination chemistry. This class of compounds has been extensively utilized in many fields, such as industrial applications [13], as intermediates in organic chemistry [14], polymers [15, 16], catalytic reactions [17,18,19,20,21,22] and in medicinal chemistry [23,24,25,26,27,28,29]. In addition, complexes of Schiff base ligands with different transition metal ions have been investigated in detail with considerable attention by inorganic biochemists because of their remarkable biological activities. Some Schiff bases show antifungal activities, while others can act as antibacterial agents [30,31,32,33]. Nair et al. [34] reported that Cu(II), Zn(II), Co(II) and Ni(II) complexes of Schiff bases derived from the condensation of indole-3-carboxaldehyde with 3-aminobenzoic acid exhibited excellent antifungal and antibacterial activities. Antimicrobial activity studies verify that Schiff bases are significantly less potent than their metal complexes [35].
In our previous investigations, we have chosen tetradentate ligands with a 2,2′-diimino-4,4′-dimethyl-1,1′-biphenyl backbone. These Schiff bases are easily accessible by the condensation reaction of 2,2′-diamino-4,4′-dimethyl-1,1′-biphenyl with two equivalents of 2-hydroxybenzaldehyde yielding 2,2′-bis(2-hydroxybenzylideneamino)-4,4′-dimethyl-1,1′-biphenyl (H2L1) [36]. The rotational flexibility of the doubly deprotonated congeners, namely 2,2′-bis(2-oxidobenzylideneamino)-4,4′-dimethyl-1,1′-biphenyl (L1), allows the formation of cis- and trans-isomers of octahedrally coordinated metal ions. These diastereomers can be easily distinguished by NMR spectroscopy as demonstrated for the isomers of the titanium complex [(L1)TiCl2] [36].
Recently, we have reported the deprotonation of 2,2′-bis(salicylideneamino)-4,4′-dimethyl-6,6′-dibromo-1,1′-biphenyls (with and without the methoxy substituents at the salicylidene) with diethylzinc yielding diamagnetic zinc(II) complexes. The choice of this metal ensured straightforward preparative protocols via metalation of the 2,2′-bis(salicylideneamino)-4,4′-dimethyl-6,6′-dibromo-1,1′-biphenyls with commercially available diethylzinc without any purification procedures. In addition, reliable NMR spectroscopic experiments were performed on these diamagnetic compounds [37].
For the synthesis of the complexes of Cu(II), Co(II) and Ni(II) with the Schiff base ligand L1 derived from the condensation reaction of 2,2′-diamino-4,4′-dimethyl-1,1′-biphenyl with 2-hydroxybenzaldehyde (Scheme 1), another procedure had to be chosen due to the missing availability of simple organometallic compounds. The X-ray structure of the Cu(II) complex was determined, and molecular geometries of the Cu(II) complex along with its methanol adduct were theoretically modeled using density functional theory (DFT) calculations and correlated with experimental XRD data.

Synthesis of the Schiff base 2,2′-bis(2-hydroxybenzylideneamino)-4,4′-dimethyl-1,1′-biphenyl H2L1
Experimental
All manipulations of air- and moisture-sensitive compounds were carried out in an inert nitrogen atmosphere using Schlenk techniques. Solvents were purified and dried prior to use according to standard procedures. Deuterated solvents were dried over sodium and saturated with nitrogen. Elemental analyses (C, H, N) were performed using a PerkinElmer 2400 instrument. 1H and 13C{1H} NMR spectra of H2L1 were measured with Bruker AC 400 and AC 500 spectrometers. All commercially available substrates were purchased from Sigma-Aldrich, Merck or Alfa Aesar and used as received without further purification. Starting materials 2,2′-diamino-4,4′-dimethyl-1,1′-biphenyl and 2,2-bis(salicylideneamino)-4,4′-dimethyl-1,1′-biphenyl were prepared according to literature procedures [36, 38].
Synthesis of CuL1
A solution of Cu(OAc)2·H2O (0.05 g, 0.24 mmol) in methanol was added to a solution of 2,2′-bis(salicylideneamino)-4,4′-dimethyl-1,1′-biphenyl (H2L1, 0.1 g, 0.24 mmol) in 10 ml of anhydrous methanol. The reaction mixture was stirred and refluxed for 3 h. The solvent was removed under vacuum, and CuL1 was obtained as a dark green solid. The crude product was collected, washed with methanol and recrystallized from a mixture of dichloromethane and methanol yielding green needles. Yield: 94%. CuL1·MeOH (C29H26CuN2O3, 514.08): calcd. C 67.76, H 5.10, N 5.45%; found C 67.68, H 4.91, N 5.46%. MS (EI, m/z): 481 [M]+. IR: \(\tilde{\nu }\) = 1601, 1591, 1522, 1461, 1433, 1395, 1376, 1358, 1345, 1328, 1313, 1245, 1191, 1149, 1123, 1112, 1007, 978, 907, 874, 851, 813, 760, 739, 659, 615, 640, 577, 548, 526, 518, 495, 481, 455 cm–1.
Synthesis of CoL1
Co(OAc)2 (0.075 g, 0.3 mmol) was added to a solution of 2,2′-bis(salicylideneamino)-4,4′-dimethyl-1,1′-biphenyl (H2L1, 0.13 g, 0.3 mmol) with sodium methoxide (0.032 g, 0.6 mmol) under nitrogen in 15 ml of anhydrous ethanol. The reaction mixture was refluxed for 6 h. After cooling to room temperature, the precipitated red solid was collected on a frit and dried in vacuo. Yield: 88%. CoL1·MeOH (C29H26CoN2O3, 509.47): calcd. C 68.37, H 5.14, N 5.50%; found C 68.29, H 4.44, N 5.08%. MS (EI, m/z): 477 [M]+. IR: \(\tilde{\nu }\) = 1602, 1581, 1549, 1491, 1439, 1379, 1353, 1327, 1290, 1247, 1190, 1152, 1136, 1123, 1113, 1031, 1006, 980, 957, 876, 855, 813, 753, 738, 668, 613, 584, 572, 557, 539, 518, 488, 453, 434 cm–1.
Synthesis of NiL1
A solution of 0.075 g (0.3 mmol) of Ni(OAc)2·4H2O in 15 ml of methanol was added to 0.13 g (0.3 mmol) of 2,2′-bis(salicylideneamino)-4,4′-dimethyl-1,1′-biphenyl (H2L1) in 10 ml of methanol. The mixture was stirred and refluxed for 5 h. The obtained solution was cooled to room temperature. The solvent was removed in vacuo leaving a yellow residue of NiL1. This crude product was collected by filtration, washed with methanol and dried in vacuo yielding yellow NiL1. Yield: 94%. NiL1·MeOH (C29H26NiN2O3, 509.23): calcd. C 68.40, H 5.15, N 5.50%; found C 67.84, H 4.94, N 5.34%. MS (EI, m/z): 476 [M]+. IR: \(\tilde{\nu }\) = 1604, 1561, 1541, 1466, 1442, 1382, 1337, 1300, 1277, 1192, 1150, 1137, 1124, 1111, 1089, 1040, 1006, 978, 957, 846, 908, 874, 812, 751, 658, 614, 579, 561, 528, 593, 515, 493, 455, 433 cm–1.
Crystal structure determination of CuL1
The intensity data were collected on a Nonius Kappa CCD diffractometer, using graphite-monochromated Mo-Kα radiation. Data were corrected for Lorentz and polarization effects; absorption was taken into account on a semi-empirical basis using multiple scans [39,40,41]. The structure was solved by direct methods (SHELXS [42]) and refined by full-matrix least squares techniques against Fo2 (SHELXL-97 [42]). The hydrogen atoms (with exception of the methyl-groups C26 and C27) were located by difference Fourier synthesis and refined isotropically. The hydrogen atoms bonded to the methyl-groups C26 and C27 were included at calculated positions with fixed thermal parameters. All non-hydrogen atoms were refined anisotropically [42]. XP was used for structure representations [43]. Crystal Data for CuL1: C29H26CuN2O3, M = 514.06 g mol−1, dark green prism, size 0.132 × 0.132 × 0.122 mm3, orthorhombic, space group P212121, a = 13.3090(3), b = 17.0197(4), c = 10.4536(2) Å, V = 2367.90(9) Å3, T = − 140 °C, Z = 4, ρcalcd. = 1.442 g cm−3, µ (Mo-Kα) = 9.57 cm−1, multi-scan, transmin: 0.6971, transmax: 0.7456, F(000) = 1068, 16,351 reflections in h(− 17/17), k(− 22/22), l(− 13/13), measured in the range 2.286° ≤ Θ ≤ 27.459°, completeness Θmax = 99.9%, 5362 independent reflections, Rint = 0.0356, 5164 reflections with Fo > 4σ(Fo), 399 parameters, 0 restraints, R1obs = 0.0270, wR2obs = 0.0639, R1all = 0.0287, wR2all = 0.0650, GOOF = 1.061, Flack parameter 0.038(12), largest difference peak and hole: 0.233/− 0.215 e Å−3.
Computational method
All electronic structure calculations were performed using the Spartan 18 package [44]. The initial molecular geometry is extracted from the experimental crystal structure. The ground-state geometries of CuL1 complex and its methanol adduct CuL1·MeOH were fully optimized without any geometry or symmetry constraints in the gas phase utilizing the B3LYP hybrid functional and the polarized 6-31G(d) basis set [45,46,47,48,49].
Examination of the output from the vibrational analysis for the optimized complex ensured the absence of imaginary frequencies in the vibrational mode calculations indicating minimal-energy structures.
Results and discussion
The Schiff base H2(L1) was efficiently prepared via the condensation reaction of 2,2′-iamino-4,4′-dimethyl-1,1′-biphenyl with 2-hydroxybenzaldehyde [36] in a 1:2 stoichiometric ratio in anhydrous ethanol yielding 2,2′-bis(2-hydroxybenzylideneamino)-4,4′-dimethyl-1,1′-biphenyl as shown in Scheme 1.
The isolated Schiff base H2L1 is stable at room temperature and soluble in common organic solvents. The Cu(II), Co(II) and Ni(II) metal complexes of the doubly deprotonated Schiff base ligand L1 were obtained from the reaction of H2L1 with the metal(II) acetates (M = Cu, Co, Ni) with high yields and purity (Scheme 2). For the synthesis of the cobalt complex sodium methoxide was added to ensure a quantitative conversion. Analytical and elemental analyses verified a 1:1 ratio of the ligand L1 and the metal ions. The crystalline complexes were stable in air and could safely be handled without Schlenk techniques.

Synthesis of the Cu(II), Co(II) and Ni(II) complexes containing the Schiff base ligands L1
The IR spectrum of H2L1 shows characteristic bands at 3050 cm−1 and 1614 cm−1 attributed to ν(OH) and ν(C=N), respectively [35]. The IR spectra of the Cu, Co and Ni complexes show absorption bands at 1601, 1602 and 1604 cm−1, respectively, which are assigned to ν(C=N) stretching vibrations. These ν(C=N) bands are shifted to lower wavenumbers by around 10 cm−1 in the complexes indicating complexation from the two azomethine nitrogen atoms. Moreover, the absence of the OH stretching vibration in the metal complexes verifies that the Schiff base ligand L1 is also coordinated to the metal atoms via phenolic oxygen atoms resulting in a (ONNO)-tetradentate coordination to the metal atoms. The spectroscopic parameters of these complexes verify very similar coordination environments of the metal atoms and explain the negligible spectroscopic variations of these complexes. NMR spectroscopy was not utilized to characterize any of the prepared complexes since they were inactive in this technique, implying that all complexes are paramagnetic. In order to shed light on the coordination environment of the copper(II) ion, we have determined the crystal structure of CuL1. Our attempts to grow single crystals of the Ni and Co complexes with different solvents and methods were unsuccessful. Although single crystals could not be isolated for the Co and Ni complexes, the analytical and spectroscopic data and the DFT calculations along with having the same coordination environment around the Co and Ni metal atoms enabled us to predict geometries similar to that of the Cu complex.
X-ray structure analysis
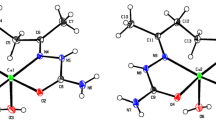
The molecular structure and atom labeling scheme of CuL1 are depicted in Fig. 1. The copper(II) center Cu1 is in an intermediate coordination sphere between tetrahedral and planar arrangement of the donor atoms with an angle between the O1–Cu1–N1 and O2–Cu1–N2 planes of only 40.8°. The bond angles between neighboring donor sites in the CuN2O2 core lie in the range between 90.26(6)° (O1–Cu–O2) and 96.74(7)° (N1–Cu–N2) and are quite similar to the values found in unsubstituted copper(II) 2,2′-(2-oxidobenzylideneamino)-1,1′-biphenyl (89°–96°) [50].

Molecular structure and atom labeling scheme of CuL1. The ellipsoids represent a probability of 30%, H atoms are omitted for the sake of clarity with the exception of those of the methanol molecule. The hydrogen bridge is drawn with a dashed line. Selected bond lengths (pm): Cu1–O1 191.10(14), Cu1–O2 189.66(15), Cu1–N1 194.82(17), Cu1–N2 196.84(17), O1–C1 131.5(2), O2–C26 130.4(2), N1–C7 129.6(3), N1–C8 144.2(3), N2–C19 144.3(3), N2–C20 130.2(3), C6–C7 143.9(3), C13–C14 148.2(3), C20–C21 143.6(3), O1M–H1M 70(4), O1M–C1M 139.5(3), O1···H1M 213(4); selected bond angles (°): O1–Cu1–O2 90.26(6), O1–Cu1–N1 93.82(7), O1–Cu1–N2 150.25(7), O2–Cu1–N1 150.31(7), O2–Cu1–N2 94.08(7), N1–Cu1–N2 96.74(7), C7–N1–C8 117.37(17), C19–N2–C20 115.46(18), O1···H1M–O1M 176(5), C1M–O1M-H1M 110(3)
The methanol molecule can in principle act as a Lewis base via formation of a metal–oxygen bond (leading to an enhanced coordination number of 5 for the metal center) or as a Lewis acid forming an O–H···O hydrogen bridge to the phenoxide functionality. In the crystalline state, the coordination of the methanol molecule at a phenoxide subunit is preferred and leads to a weak asymmetric hydrogen bridge with an O1···O1M distance of 282.1(3) pm (O1M-H1M 70(4), O1···H1M 213(4) pm). In agreement with a weak hydrogen bridge, the methanol molecules can easily be removed in vacuo at room temperature. The coordination of methanol at O1 leads to a slight elongation of the Cu1–O1 bond by 1.5 pm. The coordination number of 3 for O1 also enhances the O1–C1 bond length by 1.1 pm compared to the unaffected O2–C26 distance. Furthermore, the Cu1–N1/2 bonds are also affected in the same order of magnitude, but now the Cu1–N1 bond length is smaller by 2.0 pm than the Cu1–N2 value. These structural parameters are in agreement with those of unsubstituted copper(II) 2,2′-(2-oxidobenzylideneamino)-1,1′-biphenyl [50] verifying an only small influence of the biphenyl-bound methyl groups (C27, C28) and the O1-bound methanol molecule on the structure of the inner core.
The C13–C14 bond length between the aryl units of the biphenyl backbone features a typical single bond value of 148.2(3) pm ruling out interaction between the π-systems of these moieties. This result is analogous to the reported value of 149.1 Å for a comparable zinc complex containing ligated methanol molecules [37]. In that system, the biphenyl backbone is twisted with an angle of 56.5° between the aryl planes which is comparable to the value of 50.85° observed here in CuL1·MeOH. Such systems can be relatively flexible due to the absence of bulky groups at the biphenyl backbone to enforce larger torsion angles as observed for the binaphthyl congeners.
Copper(II) complexes with (substituted) 2,2′-bis(2-oxidobenzylideneamino)-1,1′-biphenyl ligands and their dinaphthyl congeners [50,51,52,53,54,55,56] are known for several decades justified by their importance in organic and coordination chemistry. Regardless of the substitution patterns, mononuclear copper(II) complexes are formed. Additional donor sites are required to stabilize dinuclear copper(II) derivatives with (ONNO) binding pockets for both metal atoms with bridging phenoxide subunits [57, 58]. Despite the tremendous interest in this type of complexes, not many structures of copper(II) complexes have been reported as of yet which offer the opportunity to elucidate the influence of substitution patterns at the phenoxide and biphenyl subunits as well as of methanol coordination at a phenoxide moiety on the molecular structure.
In order to clarify the influence of methanol coordination on the molecular structure of CuL1, we performed computational calculations for CuL1 and its methanol adduct because suitable crystallographically authenticated complexes for a reliable comparison of structural data are not available.
Computational study
The molecular structures of the copper complex CuL1 and its methanol adducts were theoretically modeled and examined in correlation with the experimentally solved XRD structure. Intensive DFT calculations verified the ground-state optimized geometry of the copper complex which is depicted in Fig. 2. Additionally, selected calculated and experimental bond lengths and angles are compared in Table 1. In order to examine the interaction of the methanol ligand with open coordination sites of the copper complex, two binding modes were considered by allowing a methanol molecule to either approach the metal atom and bind as a Lewis base or by approaching a phenoxide moiety and binding as a Lewis acid. Since all calculations were carried out in the gas phase without any geometry or symmetry constraints, the output of geometry optimization always revealed that the most-plausible and the minimal-energy structure is a complex with the methanol ligand acting as a Lewis acid weakly binding via an O–H···O hydrogen bridge to a phenoxide moiety.

The optimized ground-state geometry of CuL1 (top) and the methanol-bound complex (bottom) at the B3LYP/6-31G(d) level of theory (O red, N blue, C gray). (Color figure online)
The structural parameters of the monoligated optimized complex feature a tetradentate coordination environment around the central metal atom that exhibits a tetrahedral-based geometry. The computational results are in good agreement with the experimental data. For example, the bond angles in the CuN2O2 core are comparable to the experimental values as for O1–Cu–O2 angle of 92.98° (exp. 90.26°) and N1–Cu–N2 angle of 98.65° (exp. 96.74°). The calculated angle between the O1–Cu1–N1 and O2–Cu1–N2 planes is 48.54° (exp. 40.77°) suggesting a severely distorted tetrahedral geometry. The C13–C14 bond length between the aryl units of the biphenyl backbone is calculated at 1.489 Å (exp. 1.482 Å) verifying a typical single bond value. Additionally, the calculated twist angle between the aryl planes of the biphenyl backbone is slightly larger (57.26°) than observed experimentally (50.85°).
The computed MeOH···O hydrogen bridge (O···H 1.870 Å) is 26 pm shorter than the experimentally determined value of 213(3) pm. Such difference between experimental and theoretical data is attributable to the fact that the experimental data were obtained at crystalline materials inducing lattice interactions such as packing effects and intermolecular van der Waals forces, while the calculated values refer to an isolated molecule in the gas phase. Generally, percentage error (Δ%) in bond lengths around the metal atom ranged from 0 to 1.6% (average 0.75%), and in bond angles from 0.01 to 4.1% (average 1.2%). This high degree of accordance points out that the selected computational method yields reliable results for the prediction of structural parameters of such compounds. Selected calculated bond lengths and angles of the copper complex CuL1 compared with its methanol adducts are listed in Table 1.
It is obvious that there are only small structural differences between the complex CuL1 and its methanol adduct. This can be attributed to the weak binding strengths of the methanol bridge. However, the effect of methanol coordination is evident for the elongated Cu–O1 bond of 1.910 Å (exp. 1.911 Å) compared to 1.900 Å in the methanol-free complex and for the shortened Cu–O2 bond of 1.895 Å (exp. 1.897 Å) compared to 1.899 Å in the methanol-free complex. Computed IR spectrum of the Cu complex shows a strong absorption band at 1651 cm−1, which is assigned to ν(C=N). Other weaker bands that appear at 3211 cm−1 are attributed to ν(O–H) of the aromatic rings. In order to get a better understanding on the stabilizing effect of methanol on the copper complex, binding energy and thermodynamic data of the interaction were calculated. The methanol–complex interaction energy ΔEint was calculated using this relation:
where ECuL1 and EMeOH are the electronic energies of the methanol-free complex CuL1 and isolated methanol, respectively, and ECuL1·MeOH is the energy of the methanol adduct CuL1·MeOH.
The thermal energies of the methanol–complex molecular binding, shown in Table 2, exhibit negative energies of ΔEint and ∆H, which reveals that the interaction is attractive and results in a stable association. The Gibb’s energy has a small positive value of ~ 1 kJ/mol indicating that the CuL1·MeOH formation is non-spontaneous (but very close to spontaneity) at ambient temperature. Using the reported thermodynamic data (∆H, ∆G and ∆S), we can conclude that upon cooling, the interaction becomes spontaneous which explains how the methanol molecule is embedded in the complex during the recrystallization process. These calculations verify the weak bonding of methanol which can easily be removed in vacuo at room temperature even in the solid state.
Conclusions
The reaction of M(OAc)2 (M = Co, Ni, Cu) with tetradentate 2,2′-bis(2-hydroxybenzylideneamino)-4,4′-dimethyl-1,1′-biphenyl H2L1 yields the red cobalt(II) (CoL1), yellow nickel(II) (NiL1) and green copper(II) (CuL1) 2,2′-bis(2-oxidobenzylideneamino)-4,4′-dimethyl-1,1′-biphenyl complexes. The molecular structure of CuL1·MeOH with a tetracoordinate metal atom features an environment between a tetrahedral and planar arrangement of the oxygen and nitrogen donor atoms as revealed by X-ray crystallography.
The optimized ground-state geometry of the prepared CuL1 complex and its methanol adduct were elucidated using DFT calculations at the B3LYP level of theory using the 6-31G(d) basis set, and the results verify the XRD data. The results indicate that the methanol-containing complex is thermodynamically favored and forms more spontaneously upon cooling of the solution. Since azomethine derivatives, especially when metal complexed, exhibit many useful biological and catalytic activities, it may be of interest for future research to investigate this series of complexes with the potential of biological activity and as catalysts such as in the catalytic oxidation of organic compounds.
Supporting Information
Crystallographic data deposited at the Cambridge Crystallographic Data Centre under CCDC-1976020 for CuL1 contain the supplementary crystallographic data excluding structure factors; these data can be obtained free of charge via www.ccdc.cam.ac.uk/conts/retrieving.html (or from the Cambridge Crystallographic Data Centre, 12, Union Road, Cambridge CB2 1EZ, UK; fax: (+ 44) 1223-336-033; or deposit@ccdc.cam.ac.uk).
References
Abd-Elzaher MM (2001) J Chin Chem Soc 48:153–158
Ramesh R, Suganthy PK, Natarajan K (1996) Synth Inorg Met Org Chem 26:47–60
Xinde Z, Chenggang W, Zhiping L, Zhifeng L, Zhshen W (1996) Synth Inorg Met Org Chem 26:955–966
Siddiqui RA, Raj P, Saxena AK (1996) Synth Inorg Met Org Chem 26:1189–1203
Ghose BN, Lasisi KM (1986) Synth Inorg Met Org Chem 16:1121–1125
Yuan R, Chai Y, Liu D, Gao D, Li J, Yu R (1993) Anal Chem 65:2572–2575
Jha NK, Joshi DM (1984) Synth Inorg Met Org Chem 14:455–465
Ohashi Y (1997) Bull Chem Soc Jpn 70:1319–1324
Wong YL, Ma JF, Law WF, Yan Y, Wong WT, Zhang ZY, Mak TC, Ng DK (1999) Eur J Inorg Chem 1999:313–321
Koksal H, Dolaz M, Tumer M, Serin S (2001) Synth Inorg Met Org Chem 31:1141–1162
Boucher LJ, Coe CG (1976) Inorg Chem 15:1334–1340
Atkins R, Bewer G, Kokot E, Mockler GM, Sinn E (1985) Inorg Chem 24:127–134
Pui A, Policar C, Mahy JP (2007) Inorg Chim Acta 360:2139–2144
Jia HP, Li W, Ju ZF, Zhang J (2007) J Mol Struct 833:49–52
El-Saeed SM, Farag RK, Abdul-Raouf ME, Abdel-Azim AAA (2008) Int J Polym Mater 57:860–877
Nishat N, Parveen S, Dhyani S, Asma A, Ahamad T (2009) J Appl Polym Sci 113:1671–1679
Singh DP, Kumar R, Mehani R, Verma SK (2006) J Serb Chem Soc 71:939–944
Deepa NT, Madhu PK, Krishnan R (2005) Synth React Inorg Met Org Chem 35:883–888
Chohan ZH, Pervez H, Rauf A, Khan KM, Supwern CT (2002) J Enzyme Inhib Med Chem 17:117–122
Karvembu R, Natarajan K (2002) Polyhedron 21:219–223
Ali SA, Soliman AA, Aboaly MM, Ramadan RM (2002) J Coord Chem 55:1161–1170
Chatterjee D, Mitra A, Roy BC (2000) J Mol Catal 161:17–21
Katsuki T (1995) Coord Chem Rev 140:189–214
Kleij AW (2009) Eur J Inorg Chem 2009:193–205
Kleij AW (2009) Dalton Trans 24:4635–4639
Cort AD, De Bernardin P, Forte G, Mihan FY (2010) Chem Soc Rev 39:3863–3874
Whiteoak CJ, Salassa G, Kleij AW (2012) Chem Soc Rev 41:622–631
Yin HY, Tang J, Zhang JL (2017) Eur J Inorg Chem 2017:5085–5093
Erxleben A (2018) Inorg Chim Acta 472:40–57
Ranga SP, Sharma S, Chowdhary V, Parihar M, Mehta RK (1988) J Curr Bio Sci 5:98–100
Chohan ZH (1999) Met Based Drug 6:187–192
Chohan ZH (1999) Met Based Drugs 6:75–80
Chohan ZH, Kausar S (2001) J Chem Soc Pak 23:163–167
Nair MS, Arish D, Joseyphus RS (2012) J Saudi Chem Soc 16:83–88
Gopalakrishnan S, Joseph J (2009) Mycobiology 37:141–146
Al-Shboul TMA, Ziemann S, Görls H, Jazzazi TMA, Krieck S, Westerhausen M (2018) Eur J Inorg Chem 2018:1563–1570
Al-Shboul TMA, Ziemann S, Görls H, Krieck S, Westerhausen M (2019) Z Anorg Allg Chem 645:292–300
Carlin RB, Foltz GE (1956) J Am Chem Soc 78:1997–2000
Hooft R, Nonius BV (1998) Collect data collection software. Nonius BV, Delft
Otwinowski Z, Minor W (1997) Methods in enzymology. Academic Press, New York
Krause L, Herbst-Irmer R, Sheldrick GM, Stalke D (2015) J Appl Cryst 48:3–10
Sheldrick GM (2015) Acta Cryst C 71:3–8
XP Siemens Analytical X-Ray Instruments Inc (1990) Karlsruhe, Germany (1994) Madison
Spartan’18 Wavefunction. Inc. Irvine, CA
Becke AD (1993) J Chem Phys 98:5648–5652
Becke AD (1996) J Chem Phys 104:1040–1046
Lee C, Yang W, Parr RG (1988) Phys Rev B 37:785–789
Petersson GA, Bennett A, Tensfeldt TG, Al-Laham MA, Shirley WA, Mantzaris JA (1988) J Chem Phys 89:2193–2218
Petersson GA, Tensfeldt TG, Montgomery JA Jr (1991) J Chem Phys 94:6091–6101
Cheeseman TP, Hall D, Waters TN (1966) J Chem Soc A 1966:1396–1406
Wang Y, Stack TDP (1996) J Am Chem Soc 118:13097–13098
Ho CW, Cheng WC, Cheng MC, Peng SM, Cheng KF, Che CM (1996) J Chem Soc Dalton Trans 4:405–414
Che CM, Kwong HL, Cheung CW, Cheng KF, Lee WS, Yu HS, Yeung CT, Cheung KK (2002) Eur J Inorg Chem 2002:1456–1463
Heo J, Jeon Y, Mirkin CA (2007) J Am Chem Soc 129:7712–7713
Mariko S, Hisako S, Yukie M, Yutaka F (2009) Bull Chem Soc Jpn 82:1266–1273
Chu Z, Ding LQ, Long Y, Chen LL, Lu XQ, Song JR, Fan DD, Bao F, Ma R (2010) J Inorg Organomet Polym Mater 20:235–241
Brychcy K, Drager K, Jens KJ, Tilset M, Behrens U (1994) Chem Ber 127:1817–1826
Panther T, Baumann U, Behrens U (2001) Z Anorg Allg Chem 627:238–243
Acknowledgements
We acknowledge the valuable support of the NMR (www.nmr.uni-jena.de) and mass spectrometry service platforms (www.ms.uni-jena.de) of the Faculty of Chemistry and Earth Sciences of the Friedrich Schiller University Jena, Germany.
Funding
We highly appreciate the generous financial support of the Project We1561/21 by the German Research Foundation (DFG, Bonn, Germany).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there is no conflict of interests regarding the publication of this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ababneh, T.S., Al-Shboul, T.M.A., Jazzazi, T.M.A. et al. Crystallographic and computational study of the structure of copper(II) 2,2′-bis(2-oxidobenzylideneamino)-4,4′-dimethyl-1,1′-biphenyl. Transit Met Chem 45, 435–442 (2020). https://doi.org/10.1007/s11243-020-00395-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11243-020-00395-8