
Abstract
Plant MADS box transcription factors play key roles in many developmental processes, including the transition to reproductive phase and determination of floral meristem and organs identity. Here we describe the obtaining and characterization of transgenic Nicotiana tabacum L. plants with constitutive expression of Asteraceae MADS box genes CDM111, CDM41, CDM8, CDM77, CDM44 (Chrysanthemum morifolium L.) and HAM92, HAM75 (Helianthus annuus L.). Phylogenetic analysis confirmed that CDM111, HAM75 and HAM92 belong to APETALA1 (AP1), CDM41 and CDM8—FRUITFULL (FUL), CDM44—SEPALLATA3 (SEP3), and CDM77—ASTERACEAE.SEP3 (AST.SEP3) clades. Overexpression of Chrysanthemum and Helianthus AP1/FUL-like genes in tobacco plants resulted in early flowering, shortened stem and decreased number of leaves, which confirmed the functional similarity of Asteraceae AP1/FUL-like factors to AP1 and FUL. This observation testified the conservatism of processes taking place in different plants including Asteraceae. The yeast GAL4 two- and three-hybrid analysis of interactions between CDM77 and other CDM proteins revealed that CDM77 shares similar interaction map with Gerbera SEP-proteins GRCD1 and GRCD2. Overexpression of CDM44 in tobacco caused early flowering without any alterations in vegetative tissues, while overexpression of CDM77 did not reveal any visible developmental changes, which verified the functional similarity between CDM44 and SEP3, and assumed the unique role of CDM77 as whorl- and flower-type specific C-function partner.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Extensive studies of flowering plants show that the genetic functions controlling switch to flowering, inflorescence and flower development are highly conserved among different species (Theissen et al. 1996). In the well-investigated model plant Arabidopsis thaliana, one of the key players in the transition from the vegetative to the reproductive growth is the MADS domain transcription factor APETALA1 (AP1) (Mandel et al. 1992; Kaufmann et al. 2010). Mutation in AP1 gene produces phenotypes in which inflorescence meristems replace floral meristems, while overexpression of AP1 converts shoots into flowers (Mandel and Yanofsky 1995b). Kaufmann et al. (2010) demonstrated that AP1 directs floral initiation by integrating growth, patterning, and hormonal pathways, in particular, down-regulating flowering repressors genes SVP, AGL24, SOC1 and TFL1 and activating the transcription of floral organ identity genes APETALA2, APETALA3 (AP3) and SEPALLATA3 (SEP3). The AP1 subfamily includes MADS box genes from many other plant species (Theissen et al. 2000) and has been divided on AP1 and FRUITFULL (FUL) (Gu et al. 1998) clades. In Arabidopsis, MADS domain transcription factor FUL redundantly with AP1 and CAULIFLOWER (CAL) regulates the floral meristem identity and inflorescence architecture and controls the transcription of genes required for cellular differentiation during fruit and leaf development (Mandel and Yanofsky 1995a; Gu et al. 1998; Ferrándiz et al. 2000).
Taking into account the conservatism of the developmental processes in flowering plants, a model was proposed, according to which A, B, C, D and E homeotic activities mostly presented by MADS domain transcription factors specify the identity of flower organs (Coen and Meyerowitz 1991; Angenent et al. 1994; Pnueli et al. 1994; Colombo et al. 1995; Theissen 2001). The so called ‘floral quartets’, that is tetrameric complexes between mentioned proteins in different combinations are responsible for appropriate floral whorl initiation (Honma and Goto 2001). It has been shown that the class E proteins belonging to the SEP subfamily act redundantly for the specification of floral meristem and all type floral organs identity (Pelaz et al. 2000; Goto et al. 2001; Ditta et al. 2004). In Arabidopsis, SEP1, SEP2 and SEP3 in combination with other MADS box proteins determine petal, stamen, carpel and ovule identity, while protein complex with SEP4 protein specifies sepal formation. Quadruple mutant lacking all SEP factors results in complete conversion of flower organs into leaves (Pelaz et al. 2000; Honma and Goto 2001; Ditta et al. 2004). Transgenic plants with expression of chimeric repressor version of SEP3 demonstrate late flowering, decrease in number and size of flower organs, defects of differentiation and organ identity, and sterility (Kaufmann et al. 2009). It was proved that SEP3 is one of the key players in different growth-related and hormonal pathways (Kaufmann et al. 2009) and mediates the formation of the floral quartets responsible for floral induction, floral development and seed production (Honma and Goto 2001; Immink et al. 2009; Melzer et al. 2009). Namely, mentioned multimeric complexes suppress flowering repressors genes SOC1, AGL24 and SVP, activate flower meristem and flower organs identity genes AP1, AG, SHP1, SHP2, AP3 and SEP3, and control hormonal signal pathways (Immink et al. 2009; Kaufmann et al. 2009).
The AP1, FUL and SEP3-like genes are isolated from many different plant species. The members of each MADS genes clade share similar expression patterns and have highly related activity models. At the same time, morphological diversity in the plant kingdom indicates that there are certain features of the regulatory genes network in each individual plant species (Ruokolainen et al. 2010a, b; Guo et al. 2010; Li et al. 2011; Zhao et al. 2011). Chrysanthemum (Chrysanthemum morifolium) and sunflower (Helianthus annuus) are attractive systems for comparative studies on the molecular genetics of flowering. Although Asteraceae members are also eudicot plants like Arabidopsis, the development of inflorescences and flowers in compositae significantly differs from these processes in the model species. The Asteraceae indeterminate inflorescence (capitulum or head) is considered to be condensed raceme composed of hundreds or thousands florets of two or more distinct types, which may vary in symmetry and morphology, and where flower calyx either is absent or turns into a pappus bristles (Yu et al. 1999; Fambrini et al. 2003; Shchennikova et al. 2004). Comparative investigations of MADS box transcription factors of different plant species can clarify the genetic regulation of flower morphology diversity between species and within single genotype.
All that is known today about the Asteraceae MADS domain proteins belonging to AP1/FUL and SEP subfamilies assembled in a few papers devoted to isolation and characterization of appropriate genes from Chrysanthemum (Shchennikova et al. 2003, 2004), Helianthus (Shulga et al. 2008), and Gerbera (Yu et al. 1999; Kotilainen et al. 2000; Ruokolainen et al. 2010a, b). Expression patterns of the Chrysanthemum AP1/FUL-like MADS-box cDNAs, corresponding protein sequence alignment and the results of yeast two- and three-hybrid studies suggested that CDM8 and CDM41 belong to the FUL clade, CDM111 is the functional equivalent to AP1, and CDM44 is a SEP3 functional equivalent (Shchennikova et al. 2004). Eight full-length cDNAs of MADS genes have been isolated from Helianthus, among which the HAM75 and HAM92 genes are homologous to AP1, and the HAM137 gene is homologous to SEP3 (Shulga et al. 2008). Ruokolainen et al. (2010a) described six Gerbera AP1/FUL-like genes among which GSQUA1 and GSQUA3 are members of the AP1 clade, while GSQUA2, GSQUA4, GSQUA5, and GSQUA6 are co-orthologs of the Arabidopsis FUL gene. Based on expression patterns, none of the Gerbera AP1-like genes are likely to control flower organ identity in the sense of the floral A function. However, it was shown that the FUL-like gene GSQUA2 plays a vital role in meristem transition. The roles of other AP1-genes in Gerbera floral development require further study. Ruokolainen et al. (2010b) performed yeast two- and three-hybrid analysis with fourteen Gerbera MADS domain proteins and demonstrated that these proteins exhibit both conserved and derived behavior in multimeric complex formation. It has been shown that GRCD4 and GRCD5 act redundantly and perform general E function in Gerbera, comparable to the SEP proteins in Arabidopsis, while GRCD1 and GRCD2 are specific regulators involved in female flower staminode and pistil development, respectively.
Little is known about the effect caused by the ectopic expression of AP1/FUL- and SEP-like MADS-genes in the Asteraceae plants. The constitutive expression of three Asteraceae AP1-like genes CDM111, HAM75 and HAM92 in transgenic Chrysanthemum morifolium under short-day conditions trigger bud initiation 2 week earlier than non-transgenic Chrysanthemum plants and transgenic flowers showed color earlier and resulted in full opening of inflorescences 3 week prior to non-transgenic plants (Shulga et al. 2011). Lack of GRCD1 activity in Gerbera caused homeotic changes in one whorl only: sterile staminodes, which normally develop in whorl 3 of marginal female florets, converted into petals (Kotilainen et al. 2000). Down-regulation of gene GRCD2 gives rise to Gerbera plants in which carpel development affected (Uimari et al. 2004). Finally, overexpression of GSQUA2 in Gerbera led to accelerated flowering, dwarfism and vegetative abnormalities (Ruokolainen et al. 2010a).
The important differences observed in Asteraceae flower and inflorescence development, compared to other model species, raise the question of how orthologous genes operate in such different contexts. To understand this, we focused on roles of Asteraceae AP1-, FUL- and SEP3-like genes from Chrysanthemum morifolium L. (CDM111, CDM41, CDM8, CDM77, CDM44) and H. annuus L. (HAM92, HAM75) (Shchennikova et al. 2003, 2004; Shulga et al. 2008) in heterologous environment. In order to uncover possible special activities of these genes, we studied the phenotypes produced by overexpression of CDM and HAM genes under the control of the Cauliflower mosaic virus (CaMV) 35S RNA promoter in Nicotiana tabacum L. background. Transgene’s integration and expression were analyzed using molecular methods. The transgenic plant phenotype was compared with the non-transgenic control, and developmental changes were treated using statistical methods. These experiments allowed us to verify functional similarity between Asteraceae AP1/FUL- and SEP3-like factors and other members of those subfamilies, and to assume the unique role of CDM77.
Materials and methods
Plant material and transformation
For functional analysis of CDM and HAM genes, tobacco plants (N. tabacum L. cv. Samsun NN) have been used. Tobacco plants grew under the standard greenhouse conditions (day/night—16/8 h, 21–24°C, 5,000 lux, 65–70% humidity). Tobacco seeds were sterilized as described by Zia et al. (2010). Leaf disks were used as explants for Agrobacterium-mediated transformation according to Horsch et al. (1984) and Kim et al. (2011) with some modifications. The infection of tobacco explants was carried out in liquid MS (Murashige and Skoog 1962) with Agrobacterium for 40 min instead of 10 min (Kim et al. 2011). For selection, we used 100 mg/l kanamycin and 500 mg/l carbenicillin instead of 50 mg/l kanamycin and 300 mg/l cefotaxim (Kim et al. 2011). A co-cultivation period of 2 days was used, as previously found to be ideal for many species, including N. tabacum (Uranbey et al. 2005; Shilpa et al. 2010).
Bacterial strains and vector/gene constructs
The disarmed Agrobacterium strain LBA 4404 was used for transformation. The overexpression constructs were made by cloning of full-length cDNAs of CDM111, CDM41, CDM8, CDM44, CDM77, HAM75, and HAM92 in sense orientation into the pBin19 plasmid under the control of the double CaMV 35S RNA promoter (Shchennikova et al. 2004). The gene constructs have nptII gene as plant selection marker. The Agrobacterium culture was grown overnight in liquid LB medium (5 g/l Bacto yeast extract, 10 g/l peptone, 10 g/l NaCl) containing 50 mg/l kanamycin and 25 mg/l rifampicin at 28°C on a shaker at 180 rpm.
Phenotypic analysis of transgenic plants
T1 progeny (10 kanamycin-resistant seedlings of each independent PCR-positive transgenic plants) was grown in greenhouse simultaneously with 10 control non-transgenic plants. The effect of gene expression was estimated by the number of days taken from bedding to greenhouse until terminal flower opening, the number of leaves, and stem length from the base to the terminal flower. For the plants with AP1-like genes overexpression, number of flowers and fruits was calculated.
Genomic DNA isolation and polymerase chain reaction
PCR was performed to detect presence of the expression cassette of the introduced compositae genes in putative transgenic tobacco plants rooted on Kanamycin selection medium. Total genomic DNA was extracted from leaves of transgenic and wild type (WT) plants by the method described by Dellaporta et al. (1983). The PCR reactions were carried out in 25 μl reaction mixture prepared according to Banerjee and Chattopadhyay (2010). Primers specific to 35S CaMV promoter (5′-CAA TCC CAC TAT CCT TCG CAA GAC CC-3′) and nopalyne synthase gene terminator region (5′-CGA ATT CCC GGG ATC TAG TAA CAT AGA TGA C-3′) were used. Amplification was performed with the following cycling conditions: initial denaturation step at 95°C for 4 min followed by 40 cycles of denaturation at 94°C for 30 s, annealing at 55°C for 30 s, extension at 72°C for 1 min and a final extension step at 72°C for 7 min. Amplicons were separated on a 1% agarose gel containing ethidium bromide and visualized under a UV light.
RNA isolation and reverse transcriptase-polymerase chain reaction
Total RNA was isolated from 100 mg frozen leaf tissue of T1 transgenic and WT plants using the RNeasy Plant Mini Kit (QIAGEN Sciences). The RNA was then treated with RNase-free DNase Set (QIAGEN Sciences). RT-PCR was performed with the OneStep RT-PCR Kit (QIAGEN Sciences) according to the manufacturer’s instructions. Expression of the integrated transgenes was analyzed by RT-PCR with genes-specific primers. The CDM41 gene was amplified with the following primers: forward, 5′-ATG GGT AGA GGA AGA GTT-3′, and reverse, 5′-TTA TTG GTT AAG GTG GCG-3′. The CDM111 gene was amplified with the following primers: forward, 5′-GGA ATT CAT GGG AAG AGG TAA GGT ACA G-3′, and reverse, 5′-CCA GCT GTT AAG ATG GAA AGC ACC TCA TGT GGC-3′. The CDM8 gene was amplified with the following primers: forward, 5′-TCA TGA GAT CTC CGT TCT GT-3′, and reverse, 5′-TCA CTT GCT CAT GTG TTG-3′. The HAM92 gene was amplified with the following primers: forward, 5′-GAA CCA ACT CCT GCA TGA AT-3′, and reverse, 5′-TTT ACG GAA AGC ACC TT-3′. The HAM75 gene was amplified with the following primers: forward, 5′-GGA ATT CGG GAT GGG GAG AGG AAA GG-3′, and reverse, 5′-GGT CGA CTT TAG GAA GGA AAG CAC CTC-3′. The CDM77 gene was amplified with the following primers: forward, 5′-GAA AGG AAT TAC TAT GGT-3′, and reverse, 5′-TCA TGC TGG CCA ACC CTG-3′. The CDM44 gene was amplified with the following primers: forward, 5′-GAA AGG CAA CTC GAT ACA-3′, and reverse, 5′-TCA CTG ATA CCA TCC TGG-3′. Reverse transcription was carried out at 50°C. PCR amplification was carried out with initial PCR activation step at 95°C for 15 min, followed by 35 cycles of 95°C for 30 s, 53°C for 30 s, and 72°C for 1 min, and a final extension step at 72°C for 10 min. Amplicons were separated on a 1% agarose gel containing ethidium bromide and visualized under a UV light.
Statistical analysis
A statistical analysis was carried out by the one-way ANOVA (Microsoft Office Excel) with given reliability value of 95%. Each replication consisted of 10 plants. Each experiment was repeated at least three times. The absolute value of actual difference ∆ avrg = Xc − Xtr, where Xc is the average value of 10 control plants data, Xtr is the average value of 10 transgenic plants data. If ∆avrg is more or equal to LSD05 criterion, it is significant (Table 1).
Phylogenetic analysis of protein sequences
In order to better understand the phylogenetic relationships between Chrysanthemum and Helianthus MADS-box genes used in this research, phylogenetic tree was constructed using the complete amino acid sequences of the AP1/FUL- and SEP-like MADS-box proteins from Chrysanthemum, Helianthus, Arabidopsis and other plant species. Protein sequences were analyzed with the BLAST search program (Altschul et al. 1997) at the National Center for Biotechnology Information (www.ncbi.nlm.nih.gov). Asteraceae MADS box proteins were aligned with known MADS box proteins from other plants using the ClustalX program (Larkin et al. 2007).
Analysis of protein–protein interactions with the yeast two-hybrid GAL4 system
Yeast two- and three-hybrid GAL4 analyses were performed according to HybriZAP-2.1 Two-Hybrid cDNA Synthesis kit protocol (Stratagene) at room temperature and 30°C. Two constructs were generated by cloning full-length cDNA of the CDM77 gene into pAD-GAL4 and pBD-GAL4-Cam vectors (Stratagene). The bait and prey constructs with full-length cDNAs of other CDM genes were described previously (Shchennikova et al. 2004). For three-hybrid analysis (Egea-Cortines et al. 1999) the construct consisting of pRED-NLSa vector (Ferrario et al. 2003) with cloned CDM86 coding sequence in frame with a nuclear localization signal was used (Shchennikova et al. 2004). The two- and three- hybrid experiments were carried out as described previously (Immink et al. 2003; Ferrario et al. 2003).
Results
Constitutive expression of AP1/FUL and SEP3 Asteraceae homologs in tobacco influence flowering time
The homology of sequence and expression pattern similarity suggested a possible functional relationship between CDM111, HAM75, HAM92 and AP1-like proteins, between CDM41, CDM8, and FUL-like proteins, and between CDM44, CDM77 and SEP3-like proteins (Shchennikova et al. 2003, 2004; Shulga et al. 2008).
Propagation of Chrysanthemum, in most cases, carried out by vegetative methods (seeds do not preserve the plant variety), using the bushes dividing or cuttings. Therefore, it is not possible to obtain correct further generations and to get the homozygous state of the transgene. The Helianthus belongs to species with a low competence to genetic transformation. Because of the simplicity and high efficiency of Agrobacterium-mediated transformation techniques with tobacco, it is common practice to use this plant for functional analysis of heterologous genes (Lemmetyinen et al. 2004; Shin et al. 2011; An et al. 2011). To all the above, we note that the flower structure in tobacco is different from Arabidopsis one, but rather similar to Asteraceae disk floret structure. Therefore, the effect of Asteraceae genes constitutive expression has been studied in a heterologous tobacco system. That is, we analyzed functions of CDM111, CDM41, CDM8, HAM75, HAM92, CDM44 and CDM77 in transgenic N. tabacum plants ectopically expressing these genes under the control of the double CaMV 35S promoter.
Eighteen transgenic tobacco plants were generated with the 35S::HAM75 construct. Six transgenic tobacco plants were generated with the 35S::CDM111 construct; twenty—with 35S::HAM92; twenty—with 35S::CDM44; twenty—with 35S::CDM77; eleven—with 35S::CDM41; eight—with 35S::CDM8. The flowering time was scored in the progeny of some of the transgenic lines in which the transgene segregated as a single locus (Table 1). Analysis of T1 progeny of these plants on kanamycin selection medium identified in total twenty-two lines with 3:1 (kanamycin-resistant : kanamycin-sensitive) segregation, indicating that the transgene was stably inherited in progeny plants and that there is one transgene integration locus in plant genome. For the following experiment sixteen lines with 3:1 segregation and one line (92–3) with 15:1 segregation (that supposed independent two locus integration 35S::HAM92) were used (Table 1).
Flowering of transgenic plants was monitored under the controlled greenhouse conditions in comparison to control plants. According to statistic analysis, 13 transgenic lines reliably differed from the control by the number of leaves, flowering time and stem length (Table 1; Fig. 1a). On the average, lines 75–1, 75–2 and 75–16 flowered 32 days earlier, were 52 cm shorter and generated 11 leaves less in comparison to control plants. Lines 75–4 and 75–8 did not differ from the control. Plants with 35S::CDM111, 35S::CDM41, 35S::CDM8, 35S::HAM92, and 35S::CDM44 constructs flowered 24, 20, 16, 14 and 14 days earlier than control plants, respectively. On the average, 35S::CDM41, 35S::CDM8, and 35S::CDM111 plants produced 16, 9 and 9 leaves less, respectively, and their stems were 30 cm shorter in comparison to the control. 35S::HAM92 plants were 29 cm shorter than control plants on the average, but the number of leaves was not changed. The number of leaves and stem length of 35S::CDM44 plants did not differ from the control. 35S::CDM77 plants had wild type tobacco phenotype. RT-PCR analysis of the lines listed in Table 1 confirmed transcription of the corresponding genes everywhere except 75–4 and 75–8 transgenic lines with WT tobacco phenotype (Fig. 1b, c). Ectopic expression of Asteraceae MADS-box genes did not affect morphology of tobacco plants. We could not detect any abnormality in the structure of the inflorescence or in the flowers of the early flowering transgenic tobacco plants.

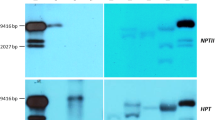
a Phenotypes comparison of the transgenic line 75-16 with constitutive expression of HAM75 gene (on the left) and wild type N. tabacum plant the same age (on the right). Scale bar = 0.05 m. b RT-PCR analysis of introduced HAM75 gene expression in transgenic tobacco plants. 1—non-transgenic plant; 2–4, 8–10—plants of 75-1 and 75-16 lines; 5–7—plants of 75-4 line; 11—the control of plant RNA for DNA contamination. c RT-PCR analysis of introduced genes transcription in transgenic tobacco plants (T1). 10—molecular weight marker (850 bp, 400 bp, Fermentas); 1—non-transgenic plant; 2—the control of plant RNA for DNA contamination; 3, 4—35S::CDM111 plants; 5–8—35S::HAM92 plants; 9—35S::CDM41 plant; 11–13—35S::CDM44 plants; 14, 15—35S::CDM77 plants; 16, 17—35S::CDM8 plants
To check the influence of copy number and transgene allelic state on plant ontogenesis we analyzed 35S::HAM92 plants with 15:1 segregation and homozygotic 35S:HAM75 plants in comparison with appropriate controls. T1 plants of 92–3 line with 15:1 segregation flowered 36 and 22 days earlier than the control and plants of 92–12/92–20 lines with 3:1 segregation, respectively, had 18 leaves less than control plants, and stem of 92–3 plants was 59 and 30 cm shorter than control and 92–12/92–20 stems, respectively (Table 1). Homozygotic state of HAM75 transgene enhanced the effect of early flowering as compared with the heterozygote. Homozygotic plants of 75–131 line (T3 progeny) flowered 36, 6 and 6 days earlier than control, heterozygotic parental 75–1 and sister 75–135 plants, respectively, and formed 31, 20 and 4 leaves less than control, 75–1 and 75–135 plants, respectively (Table 1).
Phylogenetic analysis of Chrysanthemum and Helianthus MADS box proteins
It is considered that plant transcription MADS box factors evolved, primarily, due to the changes in cis-regulatory elements that altered their expression patterns and functions (Litt and Irish 2003). Therefore, the duplication and diversification of ancestral MADS box genes may be the reason of morphological reorganizations, which resulted in inflorescence and flower shape variety. For instance, the AP1/FUL lineage consists of closely related euAP1 and FUL clades, where the euAP1 clade presumably evolved due to the frame shift mutation in C-terminal paleoAP1 motif of euFUL/FUL proteins. SEP clade proteins also share the conservative C-terminal motif closely related to paleoAP1 motif (Litt and Irish 2003). In confirmation of common origin, all reported AP1/FUL- and SEP-like genes maintain their ancestral roles, such as the meristem identity determination via the regulation of flowering time genes, and possess their own specific functions.
Figure 2 shows the phylogenetic tree, which illustrates relationships between CDM, HAM and MADS-box proteins belonging to AP1/FUL and SEP3 subfamilies from different angiosperms. Sequence alignment of the entire CDM and HAM protein sequences and other known MADS-box proteins indicates that CDM111, HAM92, HAM75, CDM41, and CDM8 are the members of the AP1/FUL subfamily, CDM44 and CDM77 match most with members of the SEP3 subfamily (Purugganan et al. 1995; Theissen et al. 1996). The putative protein products of CDM111, HAM92 and HAM75 contain the conserved euAP1 motif YSC(H)HM(L)RCFPS at the C terminus, which is typical for the AP1-like proteins. Similarly, CDM41 and CDM8 share a conserved paleoAP1 motif MPL(P)WMI(Y)R(Q)HL(M) with the FUL protein, while CDM44 and CDM77 share a conserved motifs AGPSCSNYMPGWYQ and HQMQGWPA with the SEP3 and ASTERACEAE.SEP3 (AST.SEP3) clade members (Shchennikova et al. 2004; Litt and Irish 2003; Wanderbussche et al. 2003; Malcomber and Kellogg 2005; Shulga et al. 2008).

Dendrogram based on comparative structural-phylogenetic analysis of complete amino acid sequences of AP1/FUL- and SEP-like MADS-box transcription factors of from H. annuus (HAM75, HAM92, HAM137), C. morifolium (CDM111, CDM8, CDM41, CDM77, CDM44), A. thaliana (AP1, FUL, SEP1, SEP2, SEP3, SEP4, AG), A. majus (SQUA), G. hybrida (GSQUA1, GSQUA3, GSQUA2, GRCD1, GRCD2, GRCD4, GRCD5), P. hybrida (FBP2), N. tabacum (NtFUL), M. domestica (MdMADS2), P. sativum (PEAM4), and B. pendula (BpMADS3). The AG protein from Arabidopsis was used as an outgroup. Vertical line indicates AST.SEP3 clade
Protein–protein interactions between CDM proteins
MADS-box proteins form specific heterodimers and higher order complexes between different members of the MADS-box family (Egea-Cortines et al. 1999). In most cases, the specificity of such interactions has been conserved throughout angiosperm evolution. Thus, the identification of protein interactions offers additional possibilities to determine the functional homologs among species. Therefore, the interactions between CDM77 and other CDM proteins were analyzed with the yeast GAL4 two- and three-hybrid system and are presented in Table 2 and Fig. 3, which also includes published data on interactions between SEP3 homologs and other MADS proteins from Arabidopsis, Chrysanthemum and Gerbera (Honma and Goto 2001; Shchennikova et al. 2004; Ruokolainen et al. 2010b). The CDM86, CDM115 (and its close homolog CDM19), CDM37, and CDM36 proteins are putative homologs of Arabidopsis MADS-box proteins PI, AP3, AG (Bowman et al. 1989), and SOC1 (Lee et al. 2000), respectively (Shchennikova et al. 2003). The two-hybrid analysis revealed that CDM77 does not display auto-activation of the yeast reporter gene in the absence of a bait containing the GAL4 activation domain, does not form homodimer, but interacts strongly with CDM37 at room temperature and at 30°C and weakly with CDM44 at 30°C. From studies of Arabidopsis, Antirrhinum, Petunia, Chrysanthemum and Gerbera class B proteins it is known that they form heterodimers between each other and specific ternary complexes with proteins representing the A, C, and E homeotic functions (Egea-Cortines et al. 1999; Honma and Goto 2001; Ferrario et al. 2003; Shchennikova et al. 2004; Ruokolainen et al. 2010b). Our three-hybrid studies demonstrated that CDM77 forms ternary complexes with B-heterodimer CDM86-CDM115 at both room temperature and 30°C, which is in agreement with interactions observed between the E-type protein and a B-dimer in other species, and does not interact with CDM86-CDM19.

Comparative scheme of the interactions of Chrysanthemum, Gerbera and Arabidopsis SEP-like proteins (CDM44, CDM77, GRCD1, GRCD2, GRCD5, SEP3) with AP1-(CDM111, GSQUA1 and GSQUA3), FUL-(CDM8, CDM41 and GSQUA2), and AG-like (CDM37, GAGA1 and GAGA2) proteins, and also with AP3-PI-like heterodimers (CDM115-CDM86, CDM19-CDM86, GDEF1/GDEF2-GGLO1) (Shchennikova et al. 2003; Shchennikova et al. 2004; Ruokolainen et al. 2010b; Honma and Goto 2001)
Discussion
The developmental process is considered to be conservative among different plant species. Functional investigation of Asteraceae MADS box transcription factors is especially interesting as the morphology of the representatives of this plant family differ strongly from the well-studied model plants (Yu et al. 1999; Fambrini et al. 2003; Shchennikova et al. 2004). The goal of the study was to determine whether Asteraceae AP1, FUL and SEP3 homologs function similarly to known AP1/FUL- and SEP3-like proteins, or they have different models of activity with some features peculiar to Asteraceae genes network. Since the members of major plant MADS box genes clades share highly related functions (Becker and Theissen 2003), we assumed that ectopic expression of CDM111, HAM75, HAM92, CDM41, CDM8, CDM44 and CDM77 genes may affect plant ontogeny like ectopic expression of reported AP1-, FUL- and SEP3-like genes, respectively.
Arabidopsis 35S::AP1 plants were shown to demonstrate early flowering, conversion of inflorescence meristem into flower meristem and terminal composite flower formation (Mandel and Yanofsky 1995b) via premature suppression of flowering repressors SVP, AGL24, SOC1 and TFL1 and transcriptional activation of LEAFY and flower organs identity genes AP2, AP3 and SEP3 (Kaufmann et al. 2010). Constitutive expression of heterologous AP1-like gene PEAM4 (Pisum sativum) in Arabidopsis reproduced the phenotype caused by the constitutive expression of AP1 (Berbel et al. 2001). Heterologous AP1 overexpression in tomato (Lycopersicon esculentum Mill.) reduced plant vegetative phase without affecting fruit number and morphology (Ellul et al. 2004). In transgenic Fortunella crassifolia, ectopic expression of AP1 also caused precocious flowering (Duan et al. 2010). In tobacco, constitutive expression of AP1-like genes BpMADS3 (Betula pendula) and PEAM4 (Pisum sativum) accelerated flowering without changes in inflorescence and flower (Berbel et al. 2001; Elo et al. 2001).
To assess whether Asteraceae proteins can trigger floral initiation similarly to AP1 and FUL, previously we generated transgenic Arabidopsis plants where constitutive expression of CDM111 and HAM75 affected Arabidopsis ontogeny similarly to ectopic expression of AP1 (Shchennikova et al. 2004; Shulga O.A. not published). In addition, CDM111 was able to partially complement the ap1-1 mutant Arabidopsis flower, illustrating that CDM111 is the functional equivalent to AP1 (Shchennikova et al. 2004). In transgenic Chrysanthemum plants, 35S::CDM111, 35S::HAM75, and 35S::HAM92 expression caused precocious flowering without affecting of inflorescence structure and morphology (Shulga et al. 2011).
In this study, early flowering of transgenic tobacco plants with constitutive expression of HAM75, HAM92, and CDM111 genes testifies functional homology of corresponding proteins to AP1/FUL factors. Whereas the model of AP1, we can assume that CDM111, HAM75 and HAM92 prematurely suppress tobacco flowering repressors genes, activate tobacco flowering activators genes transcription, and function similarly in host plants Chrysanthemum (CDM111) and Helianthus (HAM75 and HAM92).
It is known that the function of some MADS box proteins depends on protein expression level. For instance, according to the quantitative model of AGAMOUS (AG) activity, the two AG functions require different levels of AG transcription (Sieburth et al. 1995; Mizukami and Ma 1995). Considerably precocious flowering of transgenic tobacco plants with the higher gene dosage of HAM92 and HAM75 indicates that gene expression level is also very important for the efficiency of AP1 activity (Table 1). However, we observed no morphological alterations in flower. Thus, we concluded that Asteraceae AP1-like genes are the key players in flowering initiation but their possible homeotic function ‘A’ (specification of perianth identity) remains in question.
With respect to the FUL-like genes, except for early flowering, overexpression of FUL and FUL-like genes MADSB (Brassica napus) and DEFH28 (Antirrhinum majus) ensured pod shattering resistance in A. thaliana (Ferrándiz et al. 2000; Müller et al. 2001; Liljegren et al. 2004; Chandler et al. 2005; Østergaard et al. 2006). Conversely, heterologous constitutive expression of FUL in B. juncea did not affect flowering time but helped to maintain pod shattering resistance by inhibiting the expression of the valve margin identity genes in the valves (Østergaard et al. 2006). Ectopic expression of FUL-like BpMADS5 (B. pendula), MdMADS2 (Majus domestica), and NtFUL (N. tabacum) genes caused only early flowering in A. thaliana (Sung et al. 1999; Elo et al. 2001; Smykal et al. 2007). In Gerbera, GSQUA2 overexpression led to accelerated flowering, dwarfism and vegetative abnormalities without any changes in fruit development (Ruokolainen et al. 2010a). In our study, early flowering of transgenic 35S::CDM41 and 35S::CDM8 tobacco plants testifies these genes participation in flowering initiation and, hence, functional homology of CDM41 and CDM8 to the members of FUL clade.
The involvement of the SEP3-like genes in the meristem transition and floral initiation has been proved by the effect of ectopic expression of these genes in transgenic plants. For instance, constitutive SEP3 expression in Arabidopsis caused extremely early flowering, a single terminal flower, and curled rosette and cauline leaves (Pelaz et al. 2001). Overexpression of Petunia hybrida SEP3-like gene FBP2 in Arabidopsis caused similar phenotype (Ferrario et al. 2003). Constitutive expression of FBP2 gene showed a partial to almost complete complementation of the sep1 sep2 sep3 Arabidopsis mutant phenotype (Ferrario et al. 2003). Overexpression of NsMADS3 and NtMADS4 genes revealed to extremely early flowering and dwarfism of tobacco plants (Jang et al. 1999, 2002). In all the cases, decrease in leaves number and stem length was observed.
In our work, the overexpression of two Chrysanthemum SEP3-like genes CDM44 and CDM77 caused different effects in transgenic tobacco: early flowering without affecting vegetative characteristics and no effect, respectively (Table 1). To understand whether these proteins function like SEP3 or not, we compared their protein–protein interaction maps.
In Chrysanthemum, the CDM44 gene is the closest homolog of SEP3 (Mandel and Yanofsky 1998; Shchennikova et al. 2004). It was previously shown by Shchennikova et al. (2004) that CDM44 activates transcription in vitro, interacts with CDM proteins of the AP1/FUL and AG subfamilies, and with the heterodimer between the presumed B-type CDM proteins (Fig. 3). Honma and Goto (2001) examined the protein interactions of SEP3, and found that SEP3 has transcriptional activation domain, interacts with AP1, AG and B-heterodimer PI-AP3. Later on, Ruokolainen et al. (2010b) demonstrated that only GRCD4 and GRCD5 are able to activate transcription in vitro, all Gerbera SEP factors interact with C-function MADS box proteins and, except for GRCD1, with B-function proteins heterodimer (Figs. 2, 3).
The effect of CDM44 on flowering time, its belonging to the SEP3 clade (Fig. 2), as well as the similarity of protein–protein interaction map of CDM44 and other SEP3 homologs (Shchennikova et al. 2004; Ferrario et al. 2003; Ruokolainen et al. 2010b) indicate that CDM44 plays SEP3 function in plant development.
As mentioned above, the ectopic expression of the other member of Chrysanthemum SEP3 subfamily CDM77 did not reveal any alteration in transgenic tobacco. Compared to other SEP3 proteins, the C-terminus of CDM77 is divergent and common for AST.SEP3 clade members, which might indicate modified function, likely, not detectable in heterologous tobacco system. In the yeast interaction assay, we demonstrated CDM77-CDM37 heterodimer formation, which is in agreement with observed interactions between SEP3-AG, CDM44-CDM37, and GRCD1/2-GAGA1/2 (Honma and Goto 2001; Shchennikova et al. 2004; Ruokolainen et al. 2010b; Table 2, Fig. 3). Unlike CDM44, GRCD4, GRCD5 and SEP3, CDM77 does not activate transcription in vitro, but interacts with B-heterodimer (Table 2). Such interaction map is similar to ones of GRCD1 and GRCD2 with some exceptions (Fig. 3; Ruokolainen et al. 2010b). Taking into account the structural homology of CDM77 and GRCD1 (Fig. 2), we suggest that CDM77, similar to GRCD1, could play the unique role specific for the certain Chrysanthemum floret type and organs.
Our results allow us to conclude that Chrysanthemum and Helianthus AP1/FUL-like transcription factors play the key role in flowering promotion, like all described members of AP1/FUL subfamily. The early flowering of transgenic tobacco is attended by the conservation of productivity (bolls number) that suggests the possibility of the Asteraceae AP1/FUL homologues application in plant biotechnology. The early flowering caused by the ectopic CDM44 expression confirms its orthology with SEP3 and shows that, apparently, this phenomenon is common for all SEP3 orthologs. The exception is the members of AST.SEP3 clade, in particular CDM77. We assume the unique role of CDM77 as whorl- and flower-type specific C-function partner. Thus, the presented results confirm that the established plant ontogeny scheme is conservative for Asteraceae members. We found that compositae genes operate in other plant similar to homologous genes from the model plants and do not affect the inflorescence architecture and flower type identity.
References
Altschul SF, Thomas LM, Alejandro AS, Jinghui Z, Zheng Z, Webb M, David JL (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
An X, Ye M, Wang D, Wang Z, Cao G, Zheng H, Zhang Z (2011) Ectopic expression of a poplar APETALA3-like gene in tobacco causes early flowering and fast growth. Biotechnol Lett 33:1239–1247
Angenent GC, Franken J, Busscher M, Weiss D, Van Tunen AJ (1994) Co-suppression of the petunia homeotic gene fbp2 affects the identity of the generative meristem. Plant J 5:33–44
Banerjee A, Chattopadhyay S (2010) Effect of over-expression of Linum usitatissimum PINORESINOL LARICIRESINOL REDUCTASE (LuPLR) gene in transgenic Phyllanthus amarus. Plant Cell Tiss Organ Cult 103:315–323
Becker A, Theissen G (2003) The major clades of MADS-box genes and their role in the development and evolution of flowering plants. Mol Phylogenet Evol 29:464–489
Berbel A, Navarro C, Ferrandiz C, Canas LA, Madueno F, Beltran J-P (2001) Analysis of PEAM4, the pea AP1 functional homologue, supports a model for AP1-like genes controlling both floral meristem and floral organ identity in different plant species. Plant J 25:441–451
Bowman JL, Smyth DR, Meyerowitz EM (1989) Genes directing flower development in Arabidopsis. Plant Cell 1:37–52
Chandler J, Corbesier L, Spielmann P, Dettendorfer J, Stahl D, Apel K, Melzer S (2005) Modulating flowering time and prevention of pod shatter in oilseed rape. Mol Breed 15:87–94
Coen ES, Meyerowitz EM (1991) The war of the whorls: genetic interactions controlling flower development. Nature 353:31–37
Colombo L, Franken J, Koetje E, Van Went J, Dons HJM (1995) The petunia MADS box gene FBP11 determines ovule identity. Plant Cell 7:1859–1868
Dellaporta SL, Wood J, Hicks JB (1983) A plant DNA minipreparation: version II. Plant Mol Biol Rep 1:19–21
Ditta G, Pinyopich A, Robles P, Pelaz S, Yanofsky MF (2004) The SEP4 gene of Arabidopsis thaliana functions in floral organ and meristem identity. Curr Biol 14:1935–1940
Duan Y-X, Fan J, Guo W–W (2010) Regeneration and characterization of transgenic kumquat plants containing the Arabidopsis APETALA1 gene. Plant Cell Tiss Organ Cult 100:273–281
Egea-Cortines M, Saedler H, Sommer H (1999) Ternary complex formation between the MADS-box proteins SQUAMOSA, DEFICIENS and GLOBOSA is involved in the control of floral architecture in Antirrhinum majus. EMBO J 18:5370–5379
Ellul P, Angosto T, García-Sogo B, García-Hurtado N, Martín-Trillo M, Salinas M, Moreno V, Losano R, Martínez-Zapater JM (2004) Expression of Arabidopsis APETALA1 in tomato reduces its vegetative cycle without affecting plant production. Mol Breed 13:155–163
Elo A, Lemmetyinen J, Turunen ML, Tikka L, Sopanen T (2001) Three MADS-box genes similar to APETALA1 and FRUITFULL from silver birch (Betula pendula). Physiol Plant 112:95–103
Fambrini M, Cionini G, Bertini D, Michelotti V, Conti A, Pugliesi C (2003) MISSING FLOWERS gene controls axillary meristems initiation in sunflower. Genesis 36:25–33
Ferrándiz C, Liljegren SJ, Yanofsky MF (2000) Negative regulation of the SHATTERPROOF genes by FRUITFULL during Arabidopsis fruit development. Science 289:436–438
Ferrario S, Immink RGH, Shchennikova A, Busscher-Lange J, Angenent GC (2003) The MADS Box Gene FBP2 Is Required for SEPALLATA Function in Petunia. Plant Cell 15:914–925
Goto K, Kyozuka J, Bowman JL (2001) Turning floral organs into leaves, leaves into floral organs. Curr Opin Genet Dev 11:449–456
Gu Q, Ferrándiz C, Yanofsky MF, Martienssen F (1998) The FRUITFULL MADS-box gene mediates cell differentiation during Arabidopsis fruit development. Development 125:1509–1517
Guo J-L, Yu C-L, Fan C-Y, Lu Q-N, Yin J-M, Zhang Y-F, Yang Q (2010) Cloning and characterization of a potato TFL1 gene involved in tuberization regulation. Plant Cell Tiss Organ Cult 103:103–109
Honma T, Goto K (2001) Complexes of MADS box proteins are sufficient to convert leaves into floral organs. Nature 409:525–529
Horsch RB, Fraley RT, Rogers SG, Sanders PR, Lloyd A, Hoffman N (1984) Inheritance of functional foreign genes in plants. Science 223:496–498
Immink RGH, Ferrario S, Busscher-Lange J, Kooiker M, Busscher M, Angenent GC (2003) Analysis of the petunia MADS-box transcription factor family. Mol Genet Genomics 268:598–606
Immink RGH, Tonako IAN, de Folter S, Shchennikova A, van Dijk ADJ, Busscher-Lange J, Borst JW, Angenent GC (2009) SEPALLATA3: the ‘glue’ for MADS box transcription factor complex formation. Genome Biol 10:R24
Jang S, Hong MY, Chung YY, An G (1999) Ectopic expression of tobacco MADS genes modulates flowering time and plant architecture. Mol Cells 9:576–586
Jang S, An K, Lee S, An G (2002) Characterization of tobacco MADS-box genes involved in floral initiation. Plant Cell Physiol 43(2):230–238
Kaufmann K, Muiño JM, Jauregui R, Airoldi CA, Smaczniak C, Krajewski P, Angenent GC (2009) Target genes of the MADS transcription factor SEPALLATA3: integration of developmental and hormonal pathways in the Arabidopsis flower. PLoS Biol 7:854–875
Kaufmann R, Wellmer F, Muiño JM, Ferrier T, Wuest SE, Kumar V, Serrano-Mislata A, Madueño F, Krajewski P, Meyerowitz EM, Angenent GC, Riechmann JL (2010) Orchestration of floral initiation by APETALA1. Science 328:85–89
Kim M-Y, Kim T-G, Yoo H-S, Yang M-S (2011) Expression and assembly of ApxIIA toxin of Actinobacillus pleuropneumoniae fused with the enterotoxigenic E. coli heat-labile toxin B subunit in transgenic tobacco. Plant Cell Tiss Organ Cult 105:375–382
Kotilainen M, Elomaa P, Uimari A, Albert VA, Yu D, Teeri TH (2000) GRCD1, an AGL2-like MADS box gene, participates in the C function during stamen development in Gerbera hybrida. Plant Cell 12:1893–1902
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948
Lee H, Suh S–S, Park E, Cho E, Ahn JH, Kim S-G, Lee JS, Kwon YM, Lee I (2000) The AGAMOUS-LIKE 20 MADS domain protein integrates floral inductive pathways in Arabidopsis. Genes Dev 14:2366–2376
Lemmetyinen J, Hassinen M, Elo A, Porali I, Keinonen K, Makela H, Sopanen T (2004) Functional characterization of SEPALLATA3 and AGAMOUS orthologues in silver birch. Physiol Plant 121:149–162
Li M, Li H, Hu X, Pan X, Wu G (2011) Genetic transformation and overexpression of a rice Hd3a induces early flowering in Saussurea involucrata Kar. et Kir. ex Maxim. Plant Cell Tiss Organ Cult 106:363–371
Liljegren SJ, Roeder AH, Kempin SA, Gremski K, Østergaard L, Guimil S, Reyes DK, Yanofsky MF (2004) Control of fruit patterning in Arabidopsis by INDEHISCENT. Cell 116:843–853
Litt A, Irish VH (2003) Duplication and Diversification in the APETALA1/FRUITFULL floral homeotic gene lineage: implications for the evolution of floral development. Genetics 165:821–833
Malcomber ST, Kellogg EA (2005) SEPALLATA gene diversification: brave new whorls. Trends Plant Sci 10:427–435
Mandel MA, Yanofsky MF (1995a) The Arabidopsis AGL8 MADS box gene is expressed in inflorescence meristems and is negatively regulated by APETALA1. Plant Cell 7:1763–1771
Mandel MA, Yanofsky MF (1995b) A gene triggering flower formation in Arabidopsis. Nature 377:522–524
Mandel MA, Yanofsky MF (1998) The Arabidopsis SEP3 MADS box gene is expressed in young flower primordia. Sex Plant Reprod 11:22–28
Mandel MA, Gustafson-Brown C, Savidge B, Yanofsky MF (1992) Molecular characterisation of the Arabidopsis floral homeotic gene apetala-1. Nature 360:273–277
Melzer R, Verelst W, Theissen G (2009) The class E floral homeotic protein SEPALLATA3 is sufficient to loop DNA in ‘floral quartet’-like complexes in vitro. Nucleic Acids Res 37(1):144–157
Mizukami Y, Ma H (1995) Separation of AG function in floral meristem determinacy from that in reproductive organ identity by expressing antisense AG RNA. Plant Mol Biol 28:767–784
Müller BM, Saedler H, Zachgo S (2001) The MADS-box gene DEFH28 from Antirrhinum is involved in the regulation of floral meristem identity and fruit development. Plant J 28:169–179
Murashige T, Skoog F (1962) A revised medium for rapid growth and bioassay with tobacco tissue cultures. Physiol Plant 15:473–497
Østergaard L, Kempin SA, Bies D, Klee HJ, Yanofsky MF (2006) Pod shatter-resistant Brassica fruit produced by ectopic expression of the FRUITFULL gene. Plant Biotechnol J 4:45–51
Pelaz S, Ditta GS, Baumann E, Wisman E, Yanofsky MF (2000) B and C floral organ identity functions require SEPALLATA MADS box genes. Nature 405:200–203
Pelaz S, Gustafson-Brown C, Kohlami SE, Crosby WL, Yanofsky MF (2001) APETALA1 and SEPALLATA3 interact to promote flower development. Plant J 26:385–394
Pnueli L, Hareven D, Broday L, Hurwitz C, Lifschitz E (1994) The TM5 MADS box gene mediates organ differentiation in the three inner whorls of tomato flowers. Plant Cell 6:175–186
Purugganan MD, Rounsley SD, Schmidt RJ, Yanofsky MF (1995) Molecular evolution of flower development: diversification of the plant MADS-box regulatory gene family. Genetics 140:345–356
Ruokolainen S, Ng YP, Broholm SK, Albert VA, Elomaa P, Teeri TH (2010a) Characterization of SQUAMOSA-like genes in Gerbera hybrida, including one involved in reproductive transition. BMC Plant Biol 10:1–11
Ruokolainen S, Ng YP, Albert VA, Elomaa P, Teeri TH (2010b) Large scale interaction analysis predicts that the Gerbera hybrida floral E function is provided both by general and specialized proteins. BMC Plant Biol 10:1–13
Shchennikova AV, Shulga OA, Angenent GC, Skryabin KG (2003) Genetic regulation of inflorescence development in Chrysanthemum. Dokl Biol Sci 391:368–370
Shchennikova AV, Shulga OA, Immink R, Skryabin KG, Angenent GC (2004) Identification and characterization of four chrysanthemum MADS-box genes, belonging to the APETALA1/FRUITFULL and SEPALLATA3 subfamilies. Plant Physiol 134:1632–1641
Shilpa KS, Kumar VD, Sujatha M (2010) Agrobacterium-mediated genetic transformation of safflower (Carthamus tinctorius L.). Plant Cell Tiss Organ Cult 103:387–401
Shin M-R, Seo S-G, Kim J-S, Joen S-B, Kang S-W, Lee G-P, Kwon S-Y, Kim S-H (2011) Alteration of floral organ identity by over-expression of IbMADS3–1 in tobacco. Transgenic Res 20:365–376
Shulga OA, Shchennikova AV, Angenent GC, Skryabin KG (2008) MADS-box genes controlling inflorescence morphogenesis in sunflower. Russian J Dev Biol 39:2–5
Shulga OA, Mitiouchkina TYu, Shchennikova AV, Skryabin KG, Dolgov SV (2011) Overexpression of AP1-like genes from Asteraceae induces early-flowering in transgenic Chrysanthemum plants. In Vitro Cell Dev Biol Plant. doi:10.1007/s11627-011-9393-0
Sieburth LE, Running MP, Meyerowitz EM (1995) Genetic separation of third and fourth whorl functions of AGAMOUS. Plant Cell 7:1249–1258
Smykal P, Gennen J, De Bodt S, Ranganath V, Melzer S (2007) Flowering of strict photoperiodic Nicotiana varieties in non-inductive conditions by transgenic approaches. Plant Mol Biol 65:233–242
Sung SK, Yu GH, An G (1999) Characterization of MdMADS2, a member of the SQUAMOSA subfamily of genes, in apple. Plant Physiol 120:969–978
Theissen G (2001) Development of floral organ identity: stories from the MADS house. Curr Opin Plant Biol 4:75–85
Theissen G, Kim JT, Saedler H (1996) Classification and phylogeny of the MADS-box gene families in the morphological evolution of eukaryotes. J Mol Evol 43:484–516
Theissen G, Becker A, Di Rosa A, Kanno A, Kim JT, Münster T, Winter K-U, Saedler H (2000) A short history of MADS-box genes in plants. Plant Mol Biol 42:115–149
Uimari A, Kotilainen M, Elomaa P, Yu D, Albert VA, Teeri TH (2004) Integration of reproductive meristem fates by a SEPALLATA-like MADS-box gene. PNAS 101(44):15817–15822
Uranbey S, Sevimay CS, Kaya MD, Ipek A, Sancak C, Basalma D, Er C, Ozcan S (2005) Influence of different cocultivation temperatures, periods and media on Agrobacterium tumefaciens-mediated gene transfer. Biol Plant 49:53–57
Wandenbussche M, Theissen G, de Peer YV, Gerats T (2003) Structural diversification and neo-functionalization during floral MADS-box gene evolution by C-terminal frameshift mutations. Nucleic Acids Res 31:4401–4409
Yu D, Kotilainen M, Pollanen E, Mehto M, Elomaa P, Helariutta Y, Albert VA, Teeri TH (1999) Organ identity genes and modified patterns of flower development in Gerbera hybrida (Asteraceae). The Plant J 17(1):51–62
Zhao Y, Li X, Chen W, Peng X, Cheng X, Zhu S, Cheng B (2011) Whole-genome survey and characterization of MADS-box gene family in maize and sorghum. Plant Cell Tiss Organ Cult 105:159–173
Zia M, Mirza B, Malik SA, Chaudhary MF (2010) Expression of rol genes in transgenic soybean (Glycine max L.) leads to changes in plant phenotype, leaf morphology, and flowering time. Plant Cell Tiss Organ Cult 103:227–236
Acknowledgments
The research was supported by the SC No. 02.518.11.7148 and the fundamental investigations program “Molecular and Cell Biology” of Presidium of Russian Academy of Sciences.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Goloveshkina, E.N., Shchennikova, A.V., Kamionskaya, A.M. et al. Influence of ectopic expression of Asteraceae MADS box genes on plant ontogeny in tobacco. Plant Cell Tiss Organ Cult 109, 61–71 (2012). https://doi.org/10.1007/s11240-011-0074-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11240-011-0074-9