
Abstract
The tree-legume Leucaena leucocephala (leucaena) is used as a perennial fodder because of its fast-growing foliage, which is high in protein content. The use of leucaena as a fodder is however restricted due to the presence of the toxin mimosine. Improvements in the nutritional contents as well as other agronomic traits of leucaena can be accomplished through genetic transformation. The objective of this research was to develop a transformation protocol for leucaena using phosphinothricin resistance as the plant selectable marker. Explants obtained from immature zygotic embryos infected with the Agrobacterium tumefaciens strain C58C1 containing the binary plasmid pCAMBIA3201 produced four putative transformed leucaena plants. Transformation was confirmed by PCR, RT-PCR, Southern blot, Western analyses, GUS-specific enzyme activity and herbicide leaf spraying assay. A transformation efficiency of 2% was established using this protocol.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Leucaena leucocephala de Wit. (leucaena) belongs to the family Fabaceae, subfamily Mimosoideae, and is the best known species of the genus Leucaena. Leucaena is a fast-growing, nitrogen-fixing leguminous tree that is grown in many tropical and subtropical countries. It can grow under different soil and environmental conditions, such as alkaline and arid regions, and it is highly resistant to insects and pathogens. Because of its deep root system and ability to fix nitrogen, it is considered to be an ideal plant to control soil erosion and to enhance fertility in nitrogen-depleted soils. In many countries, leucaena is used as a fuelwood, and also for producing pulp and charcoal. Leucaena foliage is especially useful as a fodder for livestock animals because of its high nutritional values (Jones 1979). The protein content of leucaena is much higher (15–18%) compared to tropical grasses and cereal straws (3–5%), making it a valuable protein supplement to low quality forage diets (Shelton and Brewbaker 1994; Soedarjo and Borthakur 1996). Leucaena foliage is highly digestible, favored by animals, and a rich source of macro- and micro-elements (Norton 1995). Although highly nutritious, leucaena has an undesirable attribute; it produces a toxic compound, mimosine, which has harmful effects on animals. Because of the toxic effects of mimosine, leucaena can be fed to animals only in limited amounts (<10% of total diet). Among different methods of plant improvement, genetic engineering may be the fastest alternative to resolve the problem of toxicity in leucaena, however an efficient transformation protocol is necessary. Another possible benefit of genetic engineering of leucaena is in the field of phytoremediation. Leucaena was found to have the natural ability to uptake and detoxify xenobiotic pollutants from the soil and groundwater (Doty et al. 2003). However, it lacks the catabolic pathways for complete breakdown of these compounds compared to microorganisms. Hence, transfer of genes involved in xenobiotic degradation from microbes to leucaena can further enhance its potential for remediation of harmful compounds.
Tissue culture and plant regeneration of leguminous woody trees have been well established, including in leucaena (Saafi and Borthakur 2002), however, there are only a few reports on the genetic transformation of these plants. Only Stylosanthes guianensis (Sarria et al. 1994), Robinia pseudoacacia (Igasaki et al. 2000), Acacia mangium (Xie and Hong 2002) and Acacia crassicarpa (Yang et al. 2008) have been reportedly transformed using Agrobacterium tumefaciens. Although Rastogi and Dwivedi (2006) described a transformation protocol for leucaena using mature nodes and cotyledonary nodes as explant material, we could not replicate their results. One of the main obstacles during genetic transformation of trees is the process of regenerating transformed cells into healthy plantlets. In this paper, we report the development of an Agrobacterium-mediated transformation protocol to produce transgenic leucaena plants resistant to the herbicide phosphinothricin, using immature zygotic embryo segments of green seeds as the explant material.
Materials and methods
Plant regeneration from immature zygotic embryos
Leucaena cv. K-636 seeds were collected from the University of Hawaii farm in Waimanalo, Honolulu, HI. The immature zygotic embryos were obtained from unripe, 10 weeks post-anthesis seeds that were approximately 9 mm long. The green seeds were thoroughly washed with 1% v/v dishwashing detergent before being surface sterilized with 10% v/v sodium hypochlorite for 10 min with shaking, and washed five times with sterile deionized water. After blot-drying the seeds with sterile filter paper, the testa was removed, exposing the zygotic immature embryo and cotyledons. The cotyledons were removed from the immature embryo and discarded. The embryo was cut in half through its main axis, with each half used as the starting explant material for plant regeneration and transformation.
To reduce oxidative browning of the explant material due to leaching of phenolic compounds into the growth medium, the explants were soaked for 30 min at room temperature (RT) in antioxidant solution (50 mg l−1 ascorbic acid and 75 mg l−1 citric acid) prior to in vitro culturing. After antioxidant treatment, the explants were kept for 14 days in the dark at 28°C on solid callus induction medium (CIM) containing full-strength MS salts (Murashige and Skoog 1962) and Gamborg vitamins (Gamborg et al. 1968), supplemented with 1 mg l−1 2,4-dichlorophenoxyacetic acid (2,4-D), 0.5 mg l−1 benzyladenine (BA), 30 g l−1 sucrose, pH adjusted to 5.8 and solidified with 2 g l−1 Phytagel (Sigma-Aldrich, St. Louis, MO). After 14 days, the explants were maintained and cultured in Magenta GA7 boxes (Gibco-BRL, Gaithersburg, MD) with solid shoot induction medium (SIM), containing full-strength MS salts, Gamborg vitamins, supplemented with 3 mg l−1 BA and 0.25 mg l−1 naphthalene acetic acid (NAA), 30 g l−1 sucrose, pH 5.8 under a 16/8-h photoperiod at 60 μmol m−2 s−1 light intensity, until shoots reached 5 cm in length. Shoots developed from these explants were then transferred to Magenta GA7 boxes with solid root induction medium (RIM) containing half-strength MS salts, Gamborg vitamins, supplemented with 2 mg l−1 indole butyric acid (IBA), 0.1 mg l−1 kinetin, and 30 g l−1 sucrose.
Selective agent
To identify an appropriate selective agent and its concentration to be used in the genetic transformation of leucaena, explants were grown for 30 days in SIM, containing either the antibiotic kanamycin or the herbicide phosphinothricin. Kanamycin was used in a range of concentrations starting at 25 mg l−1 up to 250 mg l−1 in increments of 25 mg l−1; while phosphinothricin was used in concentrations starting at 1 mg l−1 up to 10 mg l−1 in increments of 1 mg l−1.
Binary vector and bacterial strains
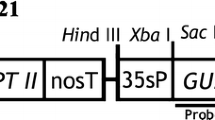
We tested the T-DNA transfer ability of three different Agrobacterium tumefaciens strains, the agropine strain EHA105 (Hood et al. 1993), the octopine strain LBA4404 (Hoekema et al. 1983), and the nopaline strain C58C1 (Ashby et al. 1988) carrying the plant binary vector pCAMBIA3201 (CAMBIA, Canberra, Australia). This binary vector contains the bar selectable marker gene, encoding the herbicide-degrading enzyme phosphinothricin aminotransferase (PAT), and the uidA reporter gene, encoding the enzyme β-glucuronidase (GUS), both under the control of the constitutive cauliflower mosaic 35S virus (35S CaMV) promoter and the nopaline synthase (NOS) terminator (Fig. 1). The binary vector was transferred into the A. tumefaciens strains using the electroporation method described by Lin (1995).

Diagrammatic representation of the binary plasmid vector pCAMBIA3201 T-DNA region indicating the localization of the bar gene, encoding phosphinothricin aminotransferase (PAT) and the uidA gene, encoding β-Glucuronidase (GUS). LB (left border), RB (right border), NOS ter (nopaline synthase terminator), 35S pro (cauliflower mosaic virus 35S promoter)
The A. tumefaciens strains were cultured in a shaker (250 rpm) at 28°C for 12 h in 10 ml liquid YEP medium (Sambrook et al. 1989), containing 10 mg l−1 rifampicin and 25 mg l−1 chloramphenicol. After 12 h, an optical density (OD600) of 0.8–1.0 was obtained, measured with a Beckman Coulter DU530 photometer (Fullerton, CA). The A. tumefaciens cultures were then centrifuged at 10,000 rpm at 4°C for 10 min and the pellet re-suspended in liquid CIM supplemented with 200 μM acetosyringone.
Plant transformation
The immature embryo-derived explants were pre-cultured for 4 days in CIM at 28°C in the dark, then vacuum-infiltrated at 400 mm Hg pressure for 30 min with the A. tumefaciens inoculums, washed five times in sterile deionized water, blotted dry on sterile filter-paper and placed on solid CIM supplemented with 200 μM acetosyringone for 10 days at 28°C in the dark and 5 days at 28°C with a 16/8-h photoperiod (60 μmol m−2 s−1). This co-cultivation period allowed for the formation of callus tissues and multiplication of the agrobacteria. Following co-cultivation, the explants were cultured for 90 days (transferred to fresh medium every 15 days) in SIM, supplemented with 250 mg l−1 cefotaxime, for the elimination of A. tumefaciens, and 3 mg l−1 phosphinothricin, for the selection of transformed tissues. The surviving explants were then subcultured for another 60 days (transferred to fresh medium every 15 days) on SIM supplemented with 3 mg l−1 phosphinothricin for continued selective pressure, without cefotaxime, under a 16/8-h photoperiod at 60 μmol m−2 s−1 light radiation from cool-white fluorescent tubes. Regenerating putative transgenic shoots were cut off the callus and rooted on RIM supplemented with 3 mg l−1 phosphinothricin until the formation of well developed roots, usually within 30 days. Rooted putative transgenic plants were then transferred to pots and grown in a controlled environmental chamber (R. W. Smith & Co., San Diego, CA).
PCR analysis
Using the method of Lin et al. (2001), 100 mg of leaves from young transformed leucaena plants were used for isolation of total genomic DNA. PCR reaction mixture was composed of 5 μl 10× buffer, 3 μl 25 mM MgCl2, 1 μl 10 mM dNTP mixture, 1 μl 20 μM each of both forward and reverse primers, 1 unit of Taq-DNA Polymerase (Promega, Madison, WI) and 100 ng of DNA template for a 50 μl reaction. PCR was carried out in a GeneAmp PCR system 2700 (Applied Biosystems, Foster City, CA) starting with a denaturing step at 95°C for 10 min, followed by 30 cycles at 94°C for 1 min, 55°C for 1 min, 72°C for 1 min 30 s, and finishing with an extension step at 72°C for 5 min. PCR products were separated on a 1% agarose gel using electrophoresis, stained with ethidium bromide and photographed. The primer set used for DNA amplification of a 452 bp segment of the bar gene was 5′-ATGAGCCCAGAACGACGCC-3′ and 5′-TCAAATCTCGGTGACGGGCAG-3′.
RT-PCR analysis
A QIAGEN RNeasy plant kit (Valencia, CA) was used for the isolation of total RNA, which was treated with RNase-free DNase (Promega) to remove contaminating DNA before reverse transcriptase PCR (RT-PCR) analysis. The RT-PCR reaction was done according to Promega’s Reverse Transcription System protocol. For the synthesis of first-strand cDNA, 1 μg of total RNA was incubated at 70°C for 10 min, and used in a reaction containing 4 μl 25 mM MgCl2, 2 μl 10× RT-buffer, 2 μl 10 mM dNTP mixture, 0.5 μl ribonuclease inhibitor, 15 units of AMV reverse transcriptase, 0.5 μg random primers, and nuclease-free water to a final volume of 20 μl. The reaction was incubated at room temperature for 10 min, 42°C for 15 min, 95°C for 5 min, and kept on ice for 5 min. From the first-strand cDNA reaction, 2 μl was used as the template for further PCR amplification of double-stranded DNA. The set of primers used for RT-PCR was the same used for PCR analysis.
Southern blot analysis
Ten μg of plant genomic DNA were digested with EcoRI (Promega), separated on an 1% agarose gel, and transferred overnight with salt transfer buffer (20× SSC) to a positively charged nylon membrane (GeneScreen Plus NEN, Boston, MA) by capillary blot according to manufacture’s instruction. The probe was obtained by labeling a PCR-amplified 452 bp fragment of the bar gene from pCAMBIA3201 with Digoxigenin-11-dUTP using the DIG High Prime DNA Labeling and Detection Starter Kit I from Roche Applied Science (Mannheim, Germany). The probe was denatured by heating in boiling water for 10 min and quickly cooling in ice-water. The denatured probe was hybridized to the membrane overnight at 42°C with gentle agitation in DIG Easy Hyb buffer (Roche Applied Science). The membrane was then washed twice for 5 min at RT in 2× SSC, 0.1% (w/v) SDS, and twice for 15 min at 68°C in 0.5× SSC, 0.1% SDS with constant agitation. Immunological detection was performed using anti-digoxigenin-AP conjugate and visualized with the colorimetric substrates NBT/BCIP according to manufacture’s instruction.
Western analysis
Leaf tissue (100 mg) was frozen in liquid nitrogen and ground with mortar and pestle. The resulting powder was immediately added to 1.0 ml of ice-cold extraction buffer (150 mM NaPO4 (pH 7.0), 10 mM β-mercaptoethanol, 0.1% Triton X-100, 10 mM Na2 EDTA (pH 8.0), 0.1% Sarkosyl, 100 mg/ml PVPP). After homogenization of the tissue, extracts were centrifuged at 12,000 rpm for 20 min at 4°C, and the supernatants were placed in Eppendorf tubes and stored at 4°C. Protein concentrations of the crude extracts were estimated by the method of Bradford (1976) with a kit supplied by Bio-Rad laboratories (Richmond, CA). Fifty μg of total proteins isolated from leaves of transformed leucaena were separated in 10% polyacrylamide gels supplemented with sodium dodecyl sulfate (SDS-PAGE), and transferred to a 0.45 μm nitrocellulose membrane (Bio-Rad). Immunodetection was carried out with anti-PAT polyclonal antibody (Sigma) at a 1:3,000 dilution in TBS (100 mM Tris–Cl (pH 7.4) and 0.9% (w/v) NaCl). Western analysis was performed with the ‘WesternBreeze’ blotting system from Invitrogen (Carlsbad, CA) according to manufacturer’s instructions.
Fluorometric GUS assay
The fluorometric assay for specific GUS enzyme activity of transformed leucaena plants was conducted using 4-methyl umbelliferyl glucuronide (MUG) as the substrate, according to protocols established by Jefferson et al. (1987) with some modifications (Cote and Rutledge 2002). The MUG assays were conducted in triplicates for each crude extract (including an extract from a wild-type leucaena for negative control), using microtiter plates. Fifty microliters of crude extract were added to 450 μl of assay buffer (1 mM MUG), incubated at 37°C in the dark, and 100 μl aliquots were removed at time zero and at 30-min intervals for 120 min and added to 900 μl stop buffer (0.2 M Na2CO3) to terminate the reaction. Aliquots (200 μl) from each sample were placed in Eppendorf tubes and visualized under UV light. The same volume was placed into the wells of a microtiter plate and the fluorescence was read by using excitation at 360 nm and emission at 460 nm with a FLX800 Microplate Fluorescence Reader (BioTek Instruments, Winooski, VT). To calibrate the fluorometer, six 4-methyl umbelliferone (MU) standards (0 nM, 100 nM, 250 nM, 500 nM, 750 nM and 1,000 nM) were prepared in stop buffer and measured for the establishment of a standard curve. The GUS activity of each transformant was calculated from the slope of the line showing the increase in fluorescence per minute and was expressed in nanomoles of MU produced per minute per milligram of protein by extrapolating from the MU standard curve.
Herbicide leaf spraying assay
Eight-month-old transgenic leucaena plants expressing the PAT protein and a non-transformed (control) plant were analyzed for their resistance to the herbicide phosphinothricin by the leaf spraying assay (Zaragoza et al. 2004). The leaves were sprayed to run-off (approximately 3 ml) with an aqueous solution of the commercial herbicide formulation (Finale, Bayer) diluted to contain 200 mg l−1 of the active compound phosphinothricin. The tolerance of plants to the herbicide was evaluated visually 5 days after application.
Results
Selective agent
Although the higher concentrations of kanamycin slowed down the development and regeneration of immature embryos, leucaena K-636 was found to be highly tolerant to kanamycin, and the regeneration of shoots from the explants was not completely arrested even at a kanamycin concentration of 250 mg l−1. For this reason, we used the herbicide phosphinothricin as an alternative selective agent. Based on physical observations of the explants, including overall health, formation of tissue necrosis, and arrest of shoot and callus development, it was determined that 3 mg l−1 would be the ideal concentration of herbicide to be used as a selective agent for our protocol.
Plant regeneration and transformation
Our observations showed that the combined effects of the plant growth regulators BA and NAA were essential for shoot regeneration from immature embryo-derived explants, with higher concentrations of BA (3 mg l−1) and lower concentrations of NAA (0.25 mg l−1) giving the highest frequency of shoot formation (Table 1). Root development from the immature embryo-derived shoots was tested under different concentrations of IBA and kinetin. After the regeneration and establishment of primary shoots, they were removed from the explants and transferred to RIM supplemented with 2 mg l−1 IBA plus 0.1 mg l−1 kinetin. At this hormonal concentrations, 78% of the shoots produced roots within 4 weeks (Table 2). Our experiments also revealed that the pre-culture of the explants on CIM prior to Agrobacterium co-cultivation and their maintenance on this medium in the dark for another 10 days allowed for the formation of phosphinothricin-resistant shoots. No phosphinothricin-resistant shoots developed in the absence of the pre-culture and callus induction steps, when the explants were placed directly on SIM after Agrobacterium co-cultivation. It seems however that the transgenic shoots originated from the cells of the immature zygotic embryo, and not from the cells of the callus. The callus formed during this stage may have played a role in enhancing Agrobacterium transformation by providing a protective layer to the emerging phosphinothricin-resistant shoots.
Three different A. tumefaciens strains, EHA105, LBA4404, and C58C1, were used to determine the best-suited strain for generating transgenic leucaena from immature zygotic embryos. A total of 300 explants in one experiment (100 explants per Agrobacterium strain) were co-cultivated with the three A. tumefaciens strains, and this experiment was repeated twice. After A. tumefaciens infection and selection in phosphinothricin-supplemented medium, no shoots were developed from the explants infected with either LBA4404 or EHA105; however, a number of explants infected with the strain C58C1 survived under selective pressure and developed shoots. After multiple transfers, a total of four phosphinothricin-resistant shoots were obtained from immature zygotic embryos infected with strain C58C1. These shoots survived multiple passages in selective medium and were also able to develop roots and be successfully transplanted to pots containing soil (Fig. 2). After 8 months of growth in a controlled environmental chamber, four stable transformants (one from the first experiment and three from the second experiment) were obtained from a total of 200 explants, giving on average, two transgenic lines from 100 immature embryo-derived explants. The overall transformation efficiency of leucaena using the described method was therefore established at 2%. All four transformants showed normal growth and phenotype.

Transformation and regeneration of leucaena. a Excision of zygotic immature embryos from green seeds (bar = 3 mm). b Embryos were cut through main axis (bar = 2 mm). c Elongation step of split embryos (bar = 2 mm). d Callus initiation from dissected embryos in CIM supplemented with 1 mg l−1 2,4-D and 0.5 mg l−1 BA (bar = 2 mm). e Shoot formation from callus tissue in SIM containing 3 mg l−1 BA, 0.1 mg l−1 NAA and 3 mg l−1 phosphinothricin (bar = 15 mm). f Shoots excised from callus tissue and placed in selective medium (bar = 15 mm). g Root development from herbicide-resistant shoots (bar = 15 mm). h Eight-month-old transformed plants (bar = 0.2 mm)
Analysis of transformants
The four putative leucaena transformants were analyzed for the presence and expression of the bar gene through PCR, RT-PCR and Western blotting. PCR amplification of DNA extracted from leaves of the transgenic plants using bar-specific primers resulted in a 452-bp band that indicates the presence of the transgene (Fig. 3a). Wild-type leucaena plants were used as a control and did not produce bands when analyzed by PCR. RT-PCR analyses using total RNA extracted from leaves of the transformed plants confirmed the presence of the bar transcripts (Fig. 3b). The expression of the bar genes in the RT-PCR positive transformed leucaena plants was further confirmed by Western blot analyses using polyclonal antisera raised against the PAT protein. Western analyses revealed the presence of a single band with a molecular mass of 21 kDa in all four transformed plants (Fig. 3c). Southern hybridization confirmed stable integration of the T-DNA in the genome of the four transgenic plants, showing at least one copy of the bar gene. The unique banding pattern shown on the Southern blot also suggests that each transgenic plant originated from a separate transformation event (Fig. 4). Long-term, stable expression of the bar gene was also analyzed by applying the herbicide phosphinothricin to the leaves of the 8-month-old greenhouse-grown transgenic leucaena plants. Within 2 days, necrotic spots appeared on the non-transformed leaves. Five days after application, the transgenic leaflets showed much higher resistance to the herbicide spraying than the leaflets from the non-transformed plant (Fig. 5). In addition to the bar gene for resistance to phosphinothricin, the T-DNA of the binary vector pCAMBIA3201 contained the E. coli uidA gene, which encodes GUS. Quantitative analyses of the four independently transformed leucaena plants, confirmed the expression of the uidA gene in the transgenic plants after 8 months of growth in a greenhouse (Fig. 6).

Molecular analyses of four transgenic leucaena plants expressing the bar gene. a PCR analyses show that the four herbicide-resistant transformants contained the bar gene (452 bp) b RT-PCR analyses show that all four plants produced the bar transcript c Western analyses show that the transgenic plants produced a 21-kDa PAT protein. The lanes 1, 2, 3 and 4 represent four independently transformed lines. WT—wild-type (non-transformant negative control)

Southern blot hybridization analysis of genomic DNA extracted from leaves of a non-transformed and four putative leucaena transformants. EcoRI-digested DNA was hybridized with a DIG-labeled bar probe. M: molecular weight marker; C−: negative control (wild-type); L-1, L-2, L-3 and L-4: four independently transformed lines of leucaena; C+: positive control (PCR-amplified bar probe from the pCAMBIA3201 plasmid)

Leaf spraying assay. Leaves from 8-month-old non-transformed (WT) and transformed (L-1, L-2, L-3, L-4) leucaena plants 5 days after herbicide application (200 mg l−1 phosphinothricin)

Fluorometric GUS specific activity in extracts prepared from four 8-month-old leucaena transformants, L-1, L-2, L-3 and L-4. Values represent the mean ± SD of triplicates. WT—wild-type
Discussion
We report here the development of an Agrobacterium-mediated transformation protocol for leucaena, in which immature zygotic embryos were used as the explant material for the generation of transgenic leucaena. During the development of our transformation protocol, we found leucaena to be recalcitrant to genetic transformation by Agrobacterium. One of the main obstacles during in vitro culture and plant regeneration of leucaena was the exudation of phenolic compounds by wounded tissues. This caused oxidative browning and subsequent necrosis of the tissues, hindering organogenesis and inhibiting further development of the explants into plantlets.
A number of different explant materials have been previously used in the development of Agrobacterium-mediated transformation protocols for woody plants (Archilletti et al. 1995; Le et al. 1996; Franche et al. 1997; Gartland et al. 2000; Bishop-Hurley et al. 2001; Corredoira et al. 2004; Polin et al. 2006; Li et al. 2007). More specifically, for the transformation of leguminous trees, stems from rejuvenated shoots and leaves were used as explants for A. mangium and R. pseudoacacia, respectively (Xie and Hong 2002; Igasaki et al. 2000). Saafi and Borthakur (2002) developed an in vitro regeneration protocol for leucaena using hypocotyl and cotyledon segments as the start explant material. Although that study was successful in developing a plant regeneration method for leucaena, we were not able to obtain transgenic leucaena plants using hypocotyl- or cotyledon-derived explants. We used different plant regeneration methods and different explant materials to transform leucaena with A. tumefaciens. Successful production of transgenic leucaena plants was achieved only with the use of explants derived from immature zygotic embryos. Our success in obtaining transgenic leucaena using immature embryo-derived explants is probably due to the fact that by removing most excess plant material from the embryos and splitting them in halves, we increased the exposure of actively dividing plant cells to A. tumefaciens, facilitating infection and subsequent transfer of the T-DNA into the plant cell. It has been previously shown that host-cell division is required for successful Agrobacterium transformation (Binns and Thomashow 1988).
The use of vacuum infiltration during co-cultivation of explants with A. tumefaciens also proved to be essential for the successful development of transformed leucaena. In previous trials in our laboratory, no transformants were obtained without the use of vacuum (data not shown). This observation is consistent with other studies that found the use of vacuum infiltration to be important in increasing transformation efficiency in woody plants (Charity et al. 2002). Transformation efficiency often depends on the strain of A. tumefaciens used (Hood et al. 1993; Wenck et al. 1999). We tested three different strains (LBA4404, EHA105 and C58C1) for transformation of leucaena. Only co-cultivation with the nopaline strain C58C1 resulted in successful generation of transformed leucaena plants. Igasaki et al. (2000) obtained similar results during Agrobacterium-mediated transformation of R. pseudoacacia, showing that the nopaline strain GV3101 gave much higher frequency of transformation compared with the octopine strain LBA4404 and the agropine strain EHA101.
Previously, Rastogi and Dwivedi (2006) reported the development of a transformation protocol for leucaena. In their report, they obtained 13–20% transformation efficiency using mature nodes and cotyledonary nodes as the explant material, and kanamycin as the selective agent. After repeated attempts to reproduce their results, we were not successful in generating transgenic leucaena plants. The main obstacle that we encountered when trying to replicate their protocol was that all the leucaena cultivars tested, including the ones used by them (K-8, K-29, and K-850), were highly resistant to kanamycin under our conditions, which made it impossible to detect transformed cells using this selective agent. High resistance to kanamycin was also observed in other leguminous trees such as A. mangium, a member of the Fabaceae family, which was reportedly resistant to kanamycin up to a concentration of 300 mg l−1 (Xie and Hong 2002) and R. pseudoacacia, which was shown to be resistant to the use of kanamycin as a selective agent during the development of an Agrobacterium-mediated transformation protocol (Igasaki et al. 2000). We also tested the protocol using the binary plasmid pCAMBIA3201, which carries the bar gene as the selectable marker, and again we could not obtain transformed leucaena plants using explants derived from mature nodes and cotyledonary nodes. Using immature zygotic embryos as explants, co-cultivated with the Agrobacterium strain C58C1 and under the selective pressure of the herbicide phosphinothricin, we were able to generate four transgenic leucaena plants, obtaining a transformation efficiency of 2%.
Abbreviations
- 2,4-D:
-
2,4-Dichlorophenoxyacetic acid
- BA:
-
Benzyladenine
- C/S/RIM:
-
Callus/shoot/root induction medium
- GUS:
-
β-Glucuronidase
- IBA:
-
Indole butyric acid
- NAA:
-
Naphthalene acetic acid
References
Archilletti T, Lauri P, Damiano C (1995) Agrobacterium-mediated transformation of almond leaf pieces. Plant Cell Rep 14:267–272. doi:10.1007/BF00232026
Ashby AM, Watson MD, Loake GJ, Shaw CH (1988) Ti plasmid-specified chemotaxis of Agrobacterium tumefaciens C58C1 toward vir-inducing phenolic compounds and soluble factors from monocotyledonous and dicotyledonous plants. J Bacteriol 170:4181–4187
Binns AN, Thomashow MF (1988) Cell biology of Agrobacterium infection and transformation of plants. Annu Rev Microbiol 42:575–606. doi:10.1146/annurev.mi.42.100188.003043
Bishop-Hurley SL, Zabkiewicz RK, Grace L, Gardner RC, Wagner A, Walter C (2001) Genetic transformation and hybridization: conifer genetic engineering: transgenic Pinus radiata (D. Don) and Picea abies (Karst) plants are resistant to the herbicide Buster. Plant Cell Rep 20:235–243. doi:10.1007/s002990100317
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein dye binding. Anal Biochem 72:248–254. doi:10.1016/0003-2697(76)90527-3
Charity JA, Holland L, Donaldson SS, Grace L, Walter C (2002) Agrobacterium-mediated transformation of Pinus radiata organogenic tissue using vacuum-infiltration. Plant Cell Tissue Organ Cult 70:51–60. doi:10.1023/A:1016009309176
Corredoira E, Montenegro D, San-Jose MC, Vieitez AM, Ballester A (2004) Agrobacterium-mediated transformation of European chestnut embryogenic cultures. Plant Cell Rep 23:311–318. doi:10.1007/s00299-004-0804-0
Cote C, Rutledge RG (2002) An improved MUG fluorescent assay for the determination of GUS activity within transgenic tissue of woody plants. Plant Cell Rep 21:619–624
Doty SL, Shang TQ, Wilson AM, Moore AL, Newman LA, Strand SE, Gordon MP (2003) Metabolism of the soil and groundwater contaminants, ethylene dibromide and trichloroethylene, by the tropical leguminous tree, Leucaena leucocephala. Water Res 37:441–449. doi:10.1016/S0043-1354(02)00291-9
Franche C, Diouf D, Le QV, Bogusz D, N’Diaye A, Gherbi H, Gobe C, Duhoux E (1997) Genetic transformation of the actinorhizal tree Allocasuarina verticillata by Agrobacterium tumefaciens. Plant J 11:897–904. doi:10.1046/j.1365-313X.1997.11040897.x
Gamborg OL, Mill R, Ogima K (1968) Nutrient requirements of suspension cultures of soybean root cells. Exp Cell Res 50:151–156. doi:10.1016/0014-4827(68)90403-5
Gartland JS, McHugh AT, Brasier CM, Irvine RJ, Fenning TM, Gartland KMA (2000) Regeneration of phenotypically normal English elm (Ulmus procera) plantlets following transformation with an Agrobacterium tumefaciens binary vector. Tree Physiol 20:901–907
Hoekema A, Hirsch PR, Hooykaas PJJ, Schilperoort RA (1983) A binary plant vector strategy based on separation of vir and T-region of Agrobacterium tumefaciens Ti plasmid. Nature 303:179–180. doi:10.1038/303179a0
Hood EE, Gelvin SB, Melchers LS, Hoekema A (1993) New Agrobacterium helper plasmids for gene transfer to plants. Transgenic Res 2:208–218. doi:10.1007/BF01977351
Igasaki T, Mohri T, Ichikawa H, Shinohara K (2000) Agrobacterium tumefaciens-mediated transformation of Robinia pseudoacacia. Plant Cell Rep 19:448–453. doi:10.1007/s002990050754
Jefferson RA, Kavanagh TA, Bevan MW (1987) GUS fusion: β-glucuronidase as a sensitive and versatile gene fusion marker in higher plants. EMBO J 6:3901–3907
Jones RJ (1979) The value of Leucaena leucocephala as a feed for ruminants in the tropics. World Anim Rev 31:13–23
Le QV, Bogusz D, Gherbi H, Lappartient A, Duhoux E, Franche C (1996) Agrobacterium tumefaciens gene transfer to Casuarina glauca, a tropical nitrogen-fixing tree. Plant Sci 118:57–69. doi:10.1016/0168-9452(96)04386-5
Li Z, Fang F, Liu G, Bao M (2007) Stable Agrobacterium-mediated genetic transformation of London plane tree (Platanus acerifolia Willd.). Plant Cell Rep 26:641–650. doi:10.1007/s00299-006-0271-x
Lin J-J (1995) Electrotransformation of Agrobacterium. In: Nickoloff JA (ed) Methods in molecular biology. Humana Press, Totowa, pp 171–178
Lin RC, Ding ZS, Li LB, Kuang TY (2001) A rapid and efficient DNA minipreparation suitable for screening transgenic plants. Plant Mol Biol Rep 19:379a–379e
Murashige T, Skoog F (1962) A revised medium for rapid growth and bioassay with tobacco tissue cultures. Physiol Plant 15:437–497
Norton BW (1995) The nutritive value of tree legumes. In: Gutteridge RC, Shelton HM (eds) Forage tree legumes in tropical agriculture. CABI, Wallingford, pp 128–132
Polin LD, Liang H, Rothrock RE, Nishii M, Diehl DL, Newhouse AE, Nairn CJ, Powell WA, Maynard CA (2006) Agrobacterium-mediated transformation of American chestnut [Castanea dentata (Marsh.) Borkh.] somatic embryos. Plant Cell Tissue Organ Cult 84:69–78. doi:10.1007/s11240-005-9002-1
Rastogi S, Dwivedi UN (2006) Down-regulation of lignin biosynthesis in transgenic Leucaena leucocephala harboring O-methyltransferase gene. Biotechnol Prog 22:609–616. doi:10.1021/bp050206+
Saafi H, Borthakur D (2002) In vitro plantlet regeneration from cotyledon of the tree legume Leucaena leucocephala. Plant Growth Regul 38:279–285. doi:10.1023/A:1021591212710
Sambrook J, Fritsch EE, Mamiatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor, New York
Sarria R, Calderon A, Thro AM, Torres E, Mayer JE, Roca WM (1994) Agrobacterium-mediated transformation of Stylosanthes guianensis and production of transgenic plants. Plant Sci 96:119–127. doi:10.1016/0168-9452(94)90228-3
Shelton HM, Brewbaker JL (1994) Leucaena leucocephala—the most widely used forage legume. In: Gutteridge RC, Shelton HM (eds) Forage tree legumes in tropical agriculture. CABI, Wallingford, pp 15–29
Soedarjo M, Borthakur D (1996) Simple procedures to remove mimosine from young leaves, seeds and pods of Leucaena leucocephala used as food. Int J Food Sci Technol 31:97–103. doi:10.1111/j.1365-2621.1996.24-321.x
Wenck AR, Quinn M, Whetten RW, Pullman G, Sederoff R (1999) High-efficiency Agrobacterium-mediate transformation of Norway spruce (Picea abies) and loblolly pine (Pinus taeda). Plant Mol Biol 39:407–416. doi:10.1023/A:1006126609534
Xie DY, Hong Y (2002) Agrobacterium-mediated genetic transformation of Acacia mangium. Plant Cell Rep 20:917–922. doi:10.1007/s00299-001-0397-9
Yang M, Xie X, Zheng C, Zhang F, He X, Li Z (2008) Agrobacterium tumefaciens-mediated genetic transformation of Acacia crassicarpa via organogenesis. Plant Cell Tissue Organ Cult 95:141–147. doi:10.1007/s11240-008-9424-7
Zaragoza C, Munoz-Bertomeu J, Arrillaga I (2004) Regeneration of herbicide-tolerant black locust transgenic plants by SAAT. Plant Cell Rep 22:832–838. doi:10.1007/s00299-004-0766-2
Acknowledgments
This work was supported by the National Science Foundation award CBET08-27057. The authors would like to thank Dr. James Brewbaker for providing the leucaena seeds.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Jube, S., Borthakur, D. Development of an Agrobacterium-mediated transformation protocol for the tree-legume Leucaena leucocephala using immature zygotic embryos. Plant Cell Tiss Organ Cult 96, 325–333 (2009). https://doi.org/10.1007/s11240-008-9490-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11240-008-9490-x