
Abstract
Cathepsin K and S are two isoforms of cysteine protease with diverse biological functions in the aspect of osteoporosis and autoimmune diseases. Accordingly, the homologous sequence and similar binding site features among CTSK/S may lead to unselective inhibition and side effects. To address such issue, various computational strategies were applied in the current study to explore the selectivity mechanism of CTSK/S inhibitors, including sequence alignment, molecular docking, MD simulations, MM/GBSA energy calculation, and so on. Our findings highlight the notable effects of CTSK residues Glu59 and Tyr67, as well as CTSS residue Asn67, on inhibition selectivity. Overall, this study provides an informative guideline for the rational design of CTSK/S selective inhibitors.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cathepsins (CTS) are closely related to many human diseases and are currently attracting much attention. More than 20 CTS have been identified in the biological world, 11 of which are present in the human body. It has been proved that CTS overexpression promotes the development of diseases such as neurodegenerative diseases [1], osteoporosis[2], and autoimmune diseases [3], which has caused a large number of scholars to conduct extensive research on CTS inhibitors. Due to the highly conserved active site of CTS [4], research interest in recent years has focused on selective inhibition [5,6,7].
Cathepsin K and S (CTSK and CTSS) are two isoforms of cysteine proteases involved in different human physiological functions. CTSK was found in cells like osteoclasts and macrophages, as well as playing a major role in osteoclastic bone resorption for type I collagen degradation [8]. Variants in the expression of the human CTSK gene lead to dense osteogenesis imperfecta [9, 10], implying that CTSK inhibitors may serve as an effective antiresorptive therapy for osteoporosis [11] and osteoarthritis [12].
While CTSS is mainly in lymph nodes involved in the occurrence and development of diseases, including immune system [13], pulmonary fibrosis [14], cardiovascular [7], and cancer [15]. CTSS is highly expressed in the major histocompatibility complex (MHC) II and plays a key role in antigen presentation [16, 17]. Sophia [3] et al. found that CTSS inhibition suppresses the inflammatory response resulting from autoimmunity. Mice in which the CTSS gene was knocked out exhibit greater resistance than wild-type mice in the development of autoimmune diseases [18]. Thusly, CTSS is considered a potential target for the treatment of autoimmune diseases [19].
Odanacatib is the only CTSK inhibitor so far that has entered phase III clinical trials (NCT01803607, date of registration: 28/02/2013), but its development was discontinued in 2016 as a result of its ability to reduce fracture risk while leading to an increased risk of cardiovascular disease, particularly stroke [20]. Considering the side effects of CTSK/S homology, it is necessary to develop novel CTSK/S inhibitors with high selectivity.
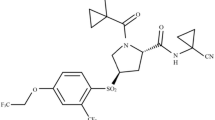
Two target compounds were screened from 17 compounds (Table S1). Among them, compound 1 is a potent small molecule inhibitor binding to CTSK [21]; correspondingly, compound 2 selectively inhibits CTSS [22] (Fig. 1). To reveal the mechanism of selective inhibition, we conducted a systematic study of CTSK/S and their complexes. By comparing the protein structures of CTSK and CTSS (Tanimoto scores: 0.036), analyzing the interaction patterns between CTS and inhibitors, and applying multiple computational methods [23, 24] to verify the structural basis of selective inhibitions of CTS, which will provide a hint for the design and development of effective and selective inhibitors in future.

Chemical structures of potent and highly selective CTSK and CTSS antagonists
Experimental section
Protein and ligand preparation
The CTSK and CTSS crystal structures were downloaded from the RCSB PDB databank (http://www.rcsb.org) with the PDB codes 1VSN and 2R9M, while protein sequences were retrieved from the Uniprot (https://www.uniprot.org/) and aligned by applying Discovery Studio and PyMOL. Through the Protein Preparation Wizard module within Schrödinger package 2020, the protein structures were protonated and minimized at pH 7.0 ± 0.2 while removing water molecules and adding hydrogen atoms. Ligand structures were prepared and optimized by using the LigPrep module of the Schrödinger package.
Protein contacts atlas
The crucial residues of CTSK and CTSS were identified via the Protein Contacts Atlas program (http://www.mrc-lmb.cam.ac.uk/pca/).
Molecular docking
Docking of inhibitors and CTSK/S were implemented by Glide module in the Schrödinger package [25]. Grid files were generated within 20 Å of any specified position of the ligands, then extra precision (XP) and flexible docked conformational modes were selected. Docking results were evaluated and obtained using multiple scoring functions, including Glide GScore, Glide Energy, and Glide Emodel.
Molecular dynamics simulation
Molecular dynamics (MD) simulations of CTSK and CTSS complexes were performed by Desmond in the Schrödinger suite 2020. Firstly, the system was set up as a simple point charge (SPC) solvent model and OPLS_3e force field [26], with Na+ and Cl− ions added to keep the system electroneutral. Subsequently, the MD simulation of 100 ns in NPT mode with a maximum iteration for minimization set to 2000 was monitored at 300.0 K and 1.01325 bar, and the time step was chosen to be 2 fs. The root mean square deviation (RMSD) and root mean square fluctuation (RMSF) were calculated to examine the structural stability of the protein–ligand complexes over time.
Dynamic cross correlation matrix analysis
Dynamic cross-correlation matrix (DCCM) analysis is expected to quantify the correlation coefficient of movement between atoms. Relying on displacement from a uniquely determined average coordinate, DCCM analysis gives insight into the correlated motion of the atoms. The DCCM between the i th and j th atoms is defined by the following equation [27]:
where \({r}_{i}\left(t\right)\) represents a vector of the ith atomic coordinates as a function of time t; \({\langle \cdot \rangle }_{t}\) represents the time ensemble average and \({\Delta {r}_{i}\left(t\right)={r}_{i}\left(t\right)-{[r}_{i}\left(t\right)]}_{t}\).
Molecular mechanics/generalized born surface area calculation
The trajectories of the ligand–protein complexes during the molecular dynamics simulation process were extracted, and the binding free energy was calculated by the prime module of Schrödinger. Parameters were set to the variable dielectric generalized-born (VSGB) solvent model and OPLS_3e force field, the energy calculation involves the following formulas [28].
To avoid solvent to solvent interactions, the second formula is usually adopted for practical calculations, among which, \({\Delta G}_{\mathrm{bind},\mathrm{solv}}\) is the binding free energy of the complex in solvent; \({\Delta G}_{\mathrm{bind},\mathrm{vacuum}}\), \({\Delta G}_{\mathrm{complex},\mathrm{solv}}\), \({\Delta G}_{\mathrm{receptor},\mathrm{solv}}\), and \({\Delta G}_{\mathrm{ligand},\mathrm{solv}}\) refer to the binding free energies of the receptor and ligand in the eukaryotic complex, solvent, respectively; \({\Delta G}_{\mathrm{solv},\mathrm{polar}}\) and \({\Delta G}_{\mathrm{solv},\mathrm{nonpolar}}\) refer to the solvation free energies of polar and non-polar, respectively.
Alanine scanning mutation analysis
Alanine scanning mutagenesis (ASM) is a technique to study the extent to which specific residues contribute to the free energy in enzyme–substrate interactions. Taking advantage of the Schrödinger package to mutate specific residues on the non-main chain of the protein to alanine without affecting the protein conformation, by combining the principle of free energy disassembly, the energy changed before, and after mutation (\({\Delta \Delta G}_{\mathrm{bind}}\)) of a single residue was calculated according to the following formula.
Generation of structure-based pharmacophore models analysis
Protein–ligand complexes conformations were extracted after MD simulation to generate structure-based pharmacophore models by applying Ligand Scout 4.3. The model analyzed the chemical characteristics and steric hindrance relationships of the active site, such as hydrogen bonding interactions, hydrophobic groups, and cations, to obtain the best mode of target-ligand binding [29].
Quantum mechanics/molecular mechanics minimization
Quantum mechanics/molecular mechanics (QM/MM) combines the accuracy of QM calculations and the speed advantage of MM to study molecular interactions between protein and inhibitor. Specifically, the ligands and related residues were set up as the QM region with zero charge and other atoms as the MM region. The DFT-B3LYP method was used with the MM force field set to OPLS_3e via the Qsite module of Schrödinger, and default values were chosen for the remaining parameters.
Results and discussion
Structural comparisons between CTSK and CTSS
To compare the structural similarities and differences between CTSK and CTSS, the crystal structures of CTSK (PDB code: 1VSN) and CTSS (PDB code: 2R9M) were aligned using PyMOL and Discovery Studio. As demonstrated in Fig. 2, CTSK exhibited a similar tertiary structure as CTSS, with an amino acid sequence identity of 55.0% and an overall similarity of 71.1%. Meanwhile, several important amino acids in the CTSK active pocket, such as Glu59, Gly64, Gly66, Try67, and Asn158, overlapped notably with the corresponding residues Tyr61, Asn67, Gly69, Phe70, and Asn163 in CTSS (Fig. 3), suggesting that the residues within CTSK/S pockets are highly identical. Remarkably, CTSK residues Cys25, His159, and Asn175, as well as CTSS residues Cys25, His164, and Asn184, all exerted proteolytic effects in the cleft between the N-lobe and C-lobe segments. In a word, CTSK and CTSS show impressive similarities and account for the difficulty of selective inhibition.

Comparison of structures and sequences of CTSK and CTSS. A 3D superimposed diagram and key residues of CTSK and CTSS. B Sequence alignment of CTSK (1VSN) and CTSS (2R9M)

Visualization and the asteroid plots analysis of protein–ligand contacts. A CTSK crystal structure. B CTSS crystal structure. The inner shell residues are immediate residues that formed intermolecular contacts with ligands, and the outer shell residues indirectly contact with the ligand
Binding affinity and modes analysis of CTSK and CTSS complexes
Re-docking of the CTSK/S co-crystalline complexes generated RMSD values of 0.916 and 0.541 Å for CTSK (PDB code 1VSN) and CTSS (PDB code 2R9M), indicating the docking process is credible. As disclosed from the results in Table 1, the docking scores of the complexes were consistent with bioactivity results, revealing that compound 1 shows a better binding affinity toward CTSK whereas compound 2 prefers to interact with CTSS.
As shown in two-dimensional receptor-ligand interaction diagrams (Fig. 4A), the phenyl of compound 1 formed π–π stacking with CTSK residue Tyr67, while the carbonyl oxygen and the positively charged tertiary amine on piperazinyl of compound 1 formed hydrogen bonds with Gly66 and Glu59, respectively. Comparatively, compound 2 only formed one hydrogen bond with CTSK residue Gln19 (Fig. 4B), suggesting compound 1 exhibited prior binding toward CTSK other than compound 2. While for the CTSS system, only one hydrophobic contact was formed in CTSS/compound 1 complex (Fig. 4C), but various interactions were established for CTSS/compound 2 complex, such as π–π stacking of the phenyl with Phe70, hydrogen bonds of the sulfone group with Gly69, and acylamino with Asn67 (Fig. 4D), showing compound 2 fits CTSS cavity better than compound 1. In summary, residues Glu59, Gly66, and Tyr67 are crucial for CSTK ligand binding, while residues Asn67, Gly69, and Phe70 contribute significantly to CTSS ligand interaction.

Predicted binding patterns of CTSK/S and inhibitors. A CTSK/compound 1. B CTSK/compound 2. C CTSS/compound 1. D CTSS/compound 2
Analysis of molecular dynamics trajectories
To evaluate the conformational stability of CTSK/S complexes, all systems were evaluated using RMSD values of protein α-carbon (Cα) atoms (Fig. S1). As shown in Fig. 5, the RMSD values of all the CTSK/S complexes eventually leveled off after slight fluctuations, indicating their conformations were stable during the MD simulation.

RMSD line charts and bar charts of MD simulations. A CTSK apo and complexes. B CTSS apo and complexes. The mean values are labeled on the top of each bar and the error bars indicate the highest value of RMSD during the simulations
To further investigate the structural flexibility of the local protein, RMSF values were calculated by comparing the transient position of residues to the average one. As displayed in Fig. 6, similar RMSF curves were characterized for CTSK/S complexes, suggesting that different ligands exhibit similar binding modes within CTS sites. Besides, lower peaks of conserved residues in CTSK, such as Cys25, Gly66, Tyr67, His159, and Asn175, as well as CTSS residues like Cys25, Asn67, Gly69, His164, and Asn184, illustrated constrained protein conformation upon ligand binding.

RMSF and ΔRMSF plots of CTSK (green) and CTSS (blue) structures. (A–C) Apo and complexes of CTSK. (D–F) Apo and complexes of CTSS. Background in blue of RMSF plots indicates residues in the β-sheet and the red indicates residues in the α-helix secondary structure. Compound 1 is shown in purple and compound 2 is displayed as yellow
In addition, ΔRMSF values of Cα atoms were obtained by RMSFComplex − RMSFAPO. For the favorable complexes of CTSK/compound 1 and CTSS/compound 2, lower negative ΔRMSF values were detected (Fig. 6), indicating compound 1 preferably stabilizes CTSK conformation and compound 2 selectively inhibits CTSS.
Intermolecular interaction analysis by MD simulation
As disclosed from the protein–ligand interaction snapshots obtained from MD simulations, compound 1 was found to form an array of interactions with CTSK (Fig. 7A, C, E), including hydrogen bonds with Glu59 (63%), Gly66 (80 and 94%), and Asn158 (55%), water bridges with Gly64 (60%), and hydrophobic interaction with Tyr67 (94%) (Fig. S2). Comparatively, only one stable H-bond was formed between compound 2 and CTSK residue Gln19 (Fig. 7B, D, E), suggesting compound 1 selectively inhibits CTSK. These obviously showed that CTSK formed more stable H-bonds and hydrophobic interactions to compound 1 than compound 2.

Protein–ligand interactions obtained from MD simulations of CTSK. A CTSK/compound 1 pocket. B CTSK/compound 2 pocket. C Interaction fraction of CTSK/compound 1. D Interaction fraction of CTSK/compound 2. E Line charts and bar charts of hydrogen bond distance. The mean values are labeled on the top of each bar and the error bars indicate the max H-bond distance during the simulations. F Detailed compound 1 atom interactions with the CTSK residues. G Detailed compound 2 atom interactions with the CTSK residues
For the CTSS complexes (Fig. 8), compound 2 formed more stable hydrogen bond interactions with CTSS residues Gly69 and Asn67 compared to compound 1, as also verified by hydrogen bond distance detection (Fig. 8E), indicating that compound 2 showed better binding affinity toward CTSS than compound 1. Briefly, CTSK residues Glu59 and Tyr67, as well as CTSS residue Asn67, have an effect on selective inhibition.

Protein–ligand interactions obtained from MD simulations of CTSS. A CTSS/compound 1 pocket. B CTSS/compound 2 pocket. C Interaction fraction of CTSS/compound 1. D Interaction fraction of CTSS/compound 2. E Line charts and bar charts of hydrogen bond distance. The mean values are labeled on the top of each bar and the error bars indicate the max H-bond distance during the simulations. F Detailed compound 1 atom interactions with the CTSS residues. G Detailed compound 2 atom interactions with the CTSS residues
Dynamic cross correlation matrix analysis
The dynamic correlation matrix (DCCM) plot described the fluctuating correlations of the Cα atoms of the complexes during the MD simulation. As disclosed in Fig. 9, the CTS-inhibitor complexes showed a significantly correlated motion compared to the apo CTS structure. In addition, the CTSK residues C25-E59, T139-N158, and H170-N175, as well as CTSS residues C25-F70, S135-N163, and N184-Y195, exhibited positively correlated motions in the DCCM plots, confirming that bound inhibitors contribute significantly to the molecular interactions with CTS proteins.

Dynamics cross-correlation map for the Cα atom pairs within CTSK/S complexes. Correlation coefficient (Cij) was shown as different colors. Cij with values from 0 to 1 (blue) represents positive correlations, whereas Cij with values from − 1 (pink) to 0 represents negative correlations
Binding free energies calculation for CTSK/S and inhibitors
To investigate the binding affinities of the inhibitors toward CTS proteins, the binding free energies during the 100 ns MD simulations were decomposed and calculated. As presented in Table 2 and Fig. 10, CTSK bound tighter to compound 1 (− 62.17 kcal/mol) rather than compound 2 (− 52.38 kcal/mol), while CTSS displayed a greater binding affinity for compound 2 (− 48.74 kcal/mol) rather than compound 1 (− 43.83 kcal/mol). In addition, the better Coulomb energy of CTSS/compound 1 was offset by the solvation-free energy, further supporting the preference of CTSS for compound 2. Besides, the MM/GBSA values of the CTS-inhibitor systems fluctuated smoothly during the simulation, suggesting that the binding of CTS toward inhibitors is stable. For the favorable complexes of CTSK/compound 1 and CTSS/compound 2, the Coulomb and Solv_GB energy terms are the main contributors that distinguish the selective inhibition of CTSK/S.

Bar charts and curves of protein–ligand complexes binding energy contributions during the whole MD simulation determined by MM/GBSA calculations. A Mean binding energy of complexes. B CTSK/compound 1. C CTSK/compound 2. D CTSS/compound 1. E CTSS/compound 2
Alanine scanning mutagenesis analysis
Alanine scanning mutagenesis analysis provided further insight into key residues participating in molecular interactions, such as CTSK residues Cys25, Glu59, and Tyr67, as well as CTSS residues Ser21, Phe70, Thr72, and Val162 (Fig. 11). In addition, residues Asp61CTSK, Asn158CTSK, Asn67CTSS, and Phe211CTSS showed negative ΔΔG values, indicating their side chains could lead to spatial resistance during interaction with the inhibitors. The mentioned results further illustrate that CTSK residues Glu59 and Tyr67, as well as CTSS residue Asn67, affect the selectivity of inhibition of CTSK/S.

Alanine scanning mutagenesis analysis plots
Pharmacophore features for CTSK/S selective inhibitors
To elucidate the chemical characteristics necessary for the binding of the inhibitors to CTSK/S, the complexes obtained from MD simulations were generated as three-dimensional pharmacophores. As shown in Fig. 12, the phenyl group of compound 1 exhibited a hydrophobic interaction, and the cyano group of compound 1 acted as a hydrogen bond acceptor. While for compound 2, both the trifluorotoluene and the trifluoromethyl groups were hydrophobic spheres, as well as the sulfone and the amino group acted as hydrogen bond acceptors and donors, respectively. Moreover, the MD trajectories of the favorable complexes of CTSK/compound 1 and CTSS/compound 2 were clustered, and the obtained conformations were analyzed pharmacologically, proving the reliability and validity of the above (Fig. S3). In conclusion, hydrophobic compounds with a sulfone and amino group as hydrogen bond acceptors and donors can selectively bind to CTSK/S receptors.

Structure-based pharmacophore models generated by the last frame of MD simulation. Hydrophobic features are depicted as yellow spheres, hydrogen bond donors are displayed as green arrows, and hydrogen bond acceptors are depicted as red arrows
Quantum mechanics calculation
Natural bond orbitals (NBO) and Mulliken calculations were performed to explore the effect of inhibitors on hydrogen bond formation. As disclosed from the local charges of the inhibitors shown in Fig. 13, the H38, H51, and H52 atoms of compound 1 displayed positive charges as electron donors, while the O23 and O24 atoms were negatively charged as electron receptors. Likewise, the positively charged N22 in compound 2 served as a hydrogen bond donor, and the negatively charged O29 and O30 served as hydrogen bond acceptors.

Atomic charges calculated with NBO and Mulliken methods
QM/MM calculations were carried out to obtain frontier molecular orbitals HOMO–LUMO. As displayed in Fig. 14, the HOMO orbital of CTSK/compound 1 was located in the phenyl region, and the LUMO orbital focused on the phenylamide group, indicating that the phenyl group is prone to electron transfer. Correspondingly, the HOMO of CTSS/compound 2 was distributed in the amide group and the LUMO was mainly in the aryl group, revealing that the amide group was the key factor to generate selectivity for CTSK/S.

HOMO–LUMO contours and molecular electrostatic potential (MEP) surface of the inhibitors calculated through DFT method. A CTSK/compound 1. B CTSK/compound 2. C CTSS/compound 1. D CTSS/compound 2. MEP surface color ranges from violet to red indicating from electropositive to electronegative region
In addition, as revealed from molecular electrostatic potential (MEP) plots (Fig. 14), the amide nitrogen of compound 1 was surrounded by positive charges, thereby serving as a hydrogen bond donor to bind with CTSK, while the secondary amino group of compound 2 was a region of high electron density and formed hydrogen bond with CTSS. In a nutshell, these data further support the conclusion that hydrogen bonds between inhibitors and targets are required.
Conclusion
The current study provides valuable information for understanding the selective inhibition mechanism of CTSK/S, revealing CTSK residues Glu59 and Tyr67, as same as CTSS residue Asn67, are key factors contributing to the selectivity of the isoforms, which is of great and profound significance for further development of highly potent and selective CTS inhibitors.
Data availability
Not applicable.
Materials availability
Not applicable.
References
Hook V, Yoon M, Mosier C, Ito G, Podvin S, Head BP et al (2020) Cathepsin B in neurodegeneration of Alzheimer's disease, traumatic brain injury, and related brain disorders. Biochim Biophys Acta Proteins Proteom 1868(8):140428. https://doi.org/10.1016/j.bbapap.2020.140428
Drake MT, Clarke BL, Oursler MJ, Khosla S (2017) Cathepsin K Inhibitors for osteoporosis: biology, potential clinical utility, and lessons learned. Endocr Rev 38(4):325–50. https://doi.org/10.1210/er.2015-1114
Sophia Thanei, Michel Theron, Ana Patricia Silva, Bernhard Reis, Branco L, Lucia Schirmbeck et al (2017) Cathepsin S inhibition suppresses autoimmune-triggered inflammatory responses in macrophages. Biochem Pharmacol 146:151–64. https://doi.org/10.1016/j.bcp.2017.10.001
Turk V, Stoka V, Vasiljeva O, Renko M, Sun T, Turk B et al (2012) Cysteine cathepsins: from structure, function and regulation to new frontiers. Biochim Biophys Acta 1824(1):68–88. https://doi.org/10.1016/j.bbapap.2011.10.002
Xiao-Yu Yuan, Ding-Yi Fu, Xing-Feng Ren, Xuexun Fang, Lincong Wang, Zouc S et al (2013) Highly selective aza-nitrile inhibitors for cathepsin K, structural optimization and molecular modeling. Org Biomol Chem 11(35):5847–52. https://doi.org/10.1039/c3ob41165f
Robichaud J, Bayly C, Oballa R, Prasit P, Mellon C, Falgueyret JP et al (2014) Rational design of potent and selective NH-linked aryl/heteroaryl cathepsin K inhibitors. Bioorg Med Chem Lett 14(16):4291–5 https://doi.org/10.1016/j.bmcl.2004.05.087
Ahmad S, Siddiqi MI (2017) Insights from molecular modeling into the selective inhibition of cathepsin S by its inhibitor. J Mol Model 23(3):92. https://doi.org/10.1007/s00894-017-3255-6
Lu J, Wang M, Wang Z, Fu Z, Lu A, Zhang G (2018) Advances in the discovery of cathepsin K inhibitors on bone resorption. J Enzyme Inhib Med Chem 33(1):890–904. https://doi.org/10.1080/14756366.2018.1465417
Mukherjee K, Chattopadhyay N (2016) Pharmacological inhibition of cathepsin K: a promising novel approach for postmenopausal osteoporosis therapy. Biochem Pharmacol 117:10–9. https://doi.org/10.1016/j.bcp.2016.04.010
Novinec M, Lenarcic B (2013) Cathepsin K: a unique collagenolytic cysteine peptidase. Biol Chem 394(9):1163–79. https://doi.org/10.1515/hsz-2013-0134
Yamashitaa DS, Dodds RA (2000) Cathepsin K and the design of inhibitors of cathepsin K. Curr Pharm Des 6(1):1–24. https://doi.org/10.2174/1381612003401569
Yasuda Y, Kaleta J, Bromme D (2005) The role of cathepsins in osteoporosis and arthritis: rationale for the design of new therapeutics. Adv Drug Deliv Rev 57(7):973–93. https://doi.org/10.1016/j.addr.2004.12.013
Saegusa K, Ishimaru N, Yanagi K, Arakaki R, Ogawa K, Saito I, et al. Cathepsin S inhibitor prevents autoantigen presentation and autoimmunity. J Clin Investig 110(3):361–9. https://doi.org/10.1172/jci200214682
Small DM, Brown RR, Doherty DF, Abladey A, Zhou-Suckow Z, Delaney RJ et al (2019) Targeting of cathepsin S reduces cystic fibrosis-like lung disease. Eur Respir J 53(3):1801523. https://doi.org/10.1183/13993003.01523-2018
Dheilly E, Battistello E, Katanayeva N, Sungalee S, Michaux J, Duns G et al (2020) Cathepsin S regulates antigen processing and T cell activity in non-Hodgkin lymphoma. Cancer Cell 37(5):674–89 e12. https://doi.org/10.1016/j.ccell.2020.03.016
Costantino CM, Ploegh HL, Hafler DA (2009) Processing in human CD4 + HLA-DR+ T cathepsin S regulates class II MHC cells. J Immunol 183(2):945–52. https://doi.org/10.4049/jimmunol.0900921
Beers C, Burich A, Kleijmeer MJ, Griffith JM, Wong P, Rudensky AY (2005) Cathepsin S controls MHC class II-mediated antigen presentation by epithelial cells in vivo. J Immunol 174(3):1205–12. https://doi.org/10.4049/jimmunol.174.3.1205
Chatterjee AK, Liu H, Tully DC, Guo J, Epple R, Russo R et al (2007) Synthesis and SAR of succinamide peptidomimetic inhibitors of cathepsin S. Bioorg Med Chem Lett 17(10):2899–903. https://doi.org/10.1016/j.bmcl.2007.02.049
Kaori Kubo, Yuka Kawato, Koji Nakamura, Yutaka Nakajima, Terry Y Nakagawa, Kaori Hanaoka et al (2018) Effective suppression of donor specific antibody production by cathepsin S inhibitors in a mouse transplantation model. European J Pharmacol 838:145–52. https://doi.org/10.1016/j.ejphar.2018.09.007
McClung MR, O'Donoghue ML, Papapoulos SE, Bone H, Langdahl B, Saag KG et al (2019) Odanacatib for the treatment of postmenopausal osteoporosis: results of the LOFT multicentre, randomised, double-blind, placebo-controlled trial and LOFT Extension study. Lancet Diabetes Endocrinol 7(12):899–911. https://doi.org/10.1016/s2213-8587(19)30346-8
Palmer JT, Bryant C, Wang D-X, Davis DE, Setti EL, Rydzewski RM et al (2005) Design and synthesis of Tri-Ring P3 benzamide-containing aminonitriles as potent, selective, orally effective inhibitors of cathepsin K. J Med Chem 48(24):7520–34. https://doi.org/10.1021/jm058198r
Yoo Y, Choi E, Kim Y, Cha Y, Um E, Kim Y et al (2022) Therapeutic potential of targeting cathepsin S in pulmonary fibrosis. Biomed Pharmacother 145:112245. https://doi.org/10.1016/j.biopha.2021.112245
Stumpfe D, Sisay MT, Frizler M, Vogt I, Gutschow M, Bajorath J (2010) Inhibitors of cathepsins K and S identified using the DynaMAD virtual screening algorithm. Chem Med Chem 5(1):61–4. https://doi.org/10.1002/cmdc.200900457
Wang H, Wang Y, Li C, Wang H, Geng X, Hu B et al (2021)Structural basis for tailor-made selective PI3K α/β inhibitors: a computational perspective. New J Chem 45(1):373–82. https://doi.org/10.1039/d0nj04216a
Bhachoo J, Beuming T (2017) Investigating protein-peptide interactions using the Schrodinger computational suite. Methods Mol Biol 1561:235–54. https://doi.org/10.1007/978-1-4939-6798-8_14
Roos K, Wu C, Damm W, Reboul M, Stevenson JM, Lu C et al (2019) OPLS3e: extending force field coverage for drug-like small molecules. J Chem Theory Comput 15(3):1863–74. https://doi.org/10.1021/acs.jctc.8b01026
Kasahara K, Fukuda I, Nakamura H(2014) A novel approach of dynamic cross correlation analysis on molecular dynamics simulations and its application to Ets1 dimer-DNA complex. PLoS One 9(11):e112419. https://doi.org/10.1371/journal.pone.0112419
Genheden S, Ryde U (2015) The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin Drug Discov 10(5):449–61. https://doi.org/10.1517/17460441.2015.1032936
Luo L, Zhong A, Wang Q, Zheng T (2021) Structure-based pharmacophore modeling, virtual screening, molecular docking, ADMET, and molecular dynamics (MD) simulation of potential inhibitors of PD-L1 from the Library of Marine Natural Products. Mar Drugs 20(1):29. https://doi.org/10.3390/md20010029
Funding
This work was supported by an award from the Taishan Industry Leading Talents Project (2018TSCYCX-03).
Author information
Authors and Affiliations
Contributions
Qinyi Zhong and Jiasi Luan wrote the main manuscript text, and Baichun Hu and Yan Ma prepared the data. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Ethical approval
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhong, Q., Luan, J., Hu, B. et al. Binding mechanism of selective cathepsin K/S inhibition revealed from molecular simulations. Struct Chem 34, 1911–1925 (2023). https://doi.org/10.1007/s11224-023-02136-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-023-02136-w