
Abstract
Assuming aromaticity (cyclic continuous conjugation, planarity, and obeying the Hückel 4n + 2 rule), effects of one and two fused six-membered heterocyclic rings are investigated on the energy lowering (stabilization) of 22 novel singlet (s) and triplet (t) carbenes, at B3LYP/AUG-cc-pVTZ and M06-2X/AUG-cc-pVTZ. Results display that (1) exclusive of triplet pyridine-4-ylidene, and s and t states, other species appear as ground state, so every s Hammick carbene exhibits more stability than its corresponding t state; (2) the highest stability is demonstrated by unsubstituted pyridine-4-ylidene as reference carbene, and the lowest stability is shown by carbene situated between two nitrogen heteroatoms of two fused rings, in a “W” arrangement; (3) regarding the relationship between carbenic center (CC) and substituted heteroatom, the order of stabilization for fused rings is meta > para > ortho; (4) regardless of how organized, fusion of one six-membered ring, in a given arrangement, has more stabilizing effect than two six-membered rings; (5) contrary to our expectation, t Hammick carbenes show higher band gap (ΔΕHOMO-LUMO) than their corresponding s species; (6) based on the NICS (nuclear independent chemical shift) results, the least stable carbene has the most aromaticity in its pyridine ring; and (7) according to proposed homomolecular isodesmotic reactions, all s states are stabilized via π-donor/σ-acceptor substitution more than the t states.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Carbenes as reactivate intermediates are of great current interest, because of their individual structural properties, their catalytic reactions in transition metal complexes or metal-free organocatalysts, their metallopharmaceuticals, and their coordination to p-block elements in many fields of applied chemistry [1, 2]. The size and substitution pattern can have a large effect on the properties of N-heterocyclic carbenes (NHCs) [3]. Initially, Buchner and Curtius discovered carbenes that seemed impossible to isolate [4]. Nevertheless, Bertrand successfully synthesized and isolated five-membered NHCs containing α-nitrogen atoms [4,5,6,7,8]. Kühn et al. synthesized and compared tetrazolylidenes with transition metal complexes [7]. Variation among normal and abnormal substitution pattern brings an effect on σ-donor abilities of the ligands (Scheme 1).

Classes of NHCs including normal A, reduced nitrogen atom stabilized B (CAAC), remote C, and abnormal D
Substituent effects on the five-membered ring is investigated with imidazole-2-ylidene; imidazoline-2-ylidene; 1,2,4-triazole-5-ylidene; and tetrazole-5-ylidene [8]. Clearly, the number and position of substituted atoms are significant. The nucleophilicity (N), and global electrophilicity (ω) of the corresponding CC is decreased, and is increased, respectively, owing to the less inductive electron withdrawal from the neighboring carbon atom, and lack of p-donation, respectively [9,10,11]. Also, Kassaee et al. theoretically have compared steric effects of tetrazol-5-ylidens and diaminocarbenes [12, 13]. They have reported N is a crucial factor for the coordination of NHCs’ excellent σ-donors to transition metal complexes, N increases as the size of the substituent increases, ω trend takes on an exactly opposite direction, and both normal and abnormal carbenes become more stable in the presence of heteroatoms. Evidently, the nitrogen-substituents or other groups situated adjacent to CC have the largest influence on the steric environment at the CC. In this manuscript, we try to respond to the important question of: “How fused rings effect on the stability of Hammick carbene?” Hence, we are probed normal substituted pyridine-4-ylidenes (1–5) and abnormal derivatives (6–11) (Scheme 2).

Hammick carbenes containing pyridine-4-ylidenes (1–5) and abnormal derivatives (6–11)
That is, three classes of pyridine-derived NHCs are identified: normal, abnormal, and normal remote (rNHCs). The difference among these three classes is the relationship between substituted heteroatom and CC, which is ortho, meta, and para, respectively [9,10,11,12,13,14,15,16,17,18].
Computational methods
The reliability of various, the most popular density functional theory (DFT) [19,20,21,22], B3LYP [23,24,25,26,30], and M06-2X [31] is already evaluated for the study of bond dissociation energies, heats of formation, molecular properties, geometrical parameters, polarizability, and hyperpolarizability not only for atoms and small molecules but also for large systems [36–42]. Conversely, the advantages of using DFT for determining single-bond torsional potentials in π-conjugated systems are less obvious. In accordance with the previous calculations, the B3LYP [23,24,25,26,27] method has been well-used to the theoretical valuations on divalent species so that it could provide rather responsible results contrasted to those achieved through various basis sets [41,42,43,44,45,46]. In this work, full geometry optimization of Hammick s and t carbenes is carried out without any symmetry constraints using the GAMESS program package [47, 48]. The restricted hybrid functional B3LYP and M06-2X methods are employed for s states, and unrestricted broken spin-symmetry UB3LYP and UM06-2X are used for t states due to its excellent performance-to-cost ratio as compared with correlated wave function theory [19,20,21,22,23,24,25,26,27]. The applied 6-311+G* basis set is confined of Pople’s famous basis set and an extra plus owing to the significance of diffuse functions [49,50,51,52]. To reach more accurate energetic data, single-point calculations are accomplished at (U)B3LYP/AUG-cc-pVTZ//(U)B3LYP/6-311+G*. In order to confirm the nature of the stationary species, the harmonic vibration frequency (υmin) calculations are carried out, at (U)M06-2X/AUG-cc-pVTZ//(U)M06-2X/6-311+G* [53, 54]. The natural bond orbital (NBO) calculations involving the charge distributions are done at the (U)M06-2X/AUG-cc-pVTZ [55,56,57,58]. To obtain the magnetic data, NICS calculations are applied at GIAO/B3LYP/AUG-cc-pVTZ [59,60,61] including NICS (0, 0.5, 1, 1.5, 2) values, at rings centers, 0.5, 1.0, 1.5, and 2.0 Å above the plane of rings showing NICS1 for singlet pyridine-4-ylidene ring, 1, along with NICS2 (values in italic) for its fused rings, respectively. The nucleophilicity index, N, is calculated as N = ΔEHOMO(Nu)−ΔEHOMO(TCNE), where tetracyanoethylene (TCNE) is preferred as the reference [62]. The global electrophilicity (ω), chemical potential (μ), and chemical hardness (η) are obtained via the expression of ω = (μ2/2η), μ = (EHOMO + ELUMO)/2, and η = EHOMO−ELUMO [63,64,65]. DFT calculations are implemented to identify the stability of the scrutinized carbenes through appropriate isodesmic reactions.
Results and discussion
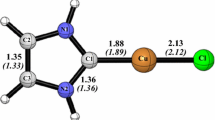
Succeeding our search for stable NHCs, we have probed s and t states of pyridine-4-ylidene, and its derivatives, (1–11, Scheme 2) and contrasted their stability based on singlet-triplet energy difference (ΔEs−t), structural parameters such as bond length (R), divalent angle (A), dihedral angle (D), dipole moment (DM), NBO charge distribution, and MEP maps at B3LYP/AUG-cc-pVTZ, and M06-2X/AUG-cc-pVDZ which approve the higher stability of singlet carbenes. Beneficial results are attained from these species including υmin, N, ω, μ, and ΔΕHOMO-LUMO. Additionally, we have estimated relative stability through aromaticity (NICS), and isodesmic reactions. Except for 1t with an imaginary frequency of − 222.1 cm−1, all full-optimized geometries turn out as minima on their potential energy surfaces for displaying no negative force constant (Table 1).
Fortunately, both s and t states of 2, 4, 6, 7, and 8 carbenes have υmin values more than 100 cm−1 [66]. All s Hammick carbenes appear as ground state, showing more stability than their corresponding t congeners. In this work, because of the reliability between the thermodynamic characters (ΔEs−t, ΔHs−t, and ΔGs−t) also for the sake of time saving, we confine our results and discussion to ΔEs−t. The total stability trend recommended by these parameters is 1 (22.5) > 2 (14.9) > 7 (14.0) ≥ 6 (13.8) > 8 (12.5) > 10 (12.0) > 3 (10.5) > 4 (9.1) > 9 (8.6) > 11 (6.9) > 5 (0.3 kcal/mol). There is an inconsistent relationship among ΔΕHOMO-LUMO trend of s species; 1s (70.7) > 7s (62.3) ≥ 2s (62.1) > 10s (60.1) > 6s (59.3) ≥ 8s (59.2) > 3s (58.6) > 9s (55.8) > 4s (54.2) > 11s (53.6) > 5s (46.1 kcal/mol) and ΔΕHOMO-LUMO trend of t species; 1t (115.4) > 4t (102.1) > 6t (98.9) > 7t (94.3) > 5t (91.2) > 2t (89.8) = 8t (89.8) > 10t (87.6) > 9t (87.0) > 3t (78.3) > 11t (77.0 kcal/mol) (Table 2).
Completely planar geometries are demonstrated by all species, while their symmetries are Cs and/or C1 (Table 3).
Located non-bonding electrons in the σ-orbital of CC, which is orthogonal to π-system and the ring current, leads to higher DM in s structures (1s–11s) than their corresponding triplets (about 1.5–3 times). For example, 5s (9.56 Debye) displays a higher DM than 5t (6.53 Debye). Also, in polar environment, 9s (1.93 Debye) and 9t (0.97 Debye) are expected to be stabilized to a smaller extent than the other species. The average polarizability (α), as a criteria of interaction of one molecule with its surrounding polar species, increases from 64.31 a.u. for 1t to 179.66 a.u. for 3t compared with 1s, and 3s (68.34, and 174.23 a.u., respectively). This result reveals that substituting the mentioned groups leads to increasing α and activity of fused carbenes (Table 3). Triplet carbenes have less N than their corresponding s states, showing the most and the least value for 1s (4.87 eV) and 1t (2.34 eV), respectively (Table 4).
More participation of the unpaired non-bonding electron on CC in delocalization causes the s structures more prone to N than their related t states. Here, the substituent effect plays a different role in ω of the scrutinized s and t Hammick carbenes. The N data show no noticeable correlations with the corresponding ω values for s and t species. For instance, 11s shows higher ω (4.16 eV) than 11t (3.47 eV), while 1s shows lower ω (1.52 eV) than 1t (2.13 eV). Also, every s carbene shows lower absolute value of chemical potential (|μ|), and global hardness (η) as global reactivity descriptor than its corresponding t structure. Succeeding our attention to stable synthesized carbenes, here, we compare the substituent effect on the N and ω of some of common NHCs with five-membered rings (12s–19s, Scheme 3).

The common NHCs 12–19
Because of the smaller carbenic angle, all five-membered rings exhibited less N and less ω than the six-membered rings (Table 5).
The highest N and the highest ω of the synthesized NHCs are considered for 12s and 17s with the values of 3.78 and 1.56 eV, respectively. We hope the higher N of six-membered carbenes along with their thermodynamic stability will make them worthy of synthetic attentions. The overall aromaticity, according to NICS1 values for pyridine-4-ylidene rings and NICS2 values for fused rings, is considered for substituted species specially 2s and 5s more than those of unsubstituted species, 1 (Table 6).
The trend of aromaticity for the parent unsubstituted molecules is 1 < 2 < 3, and for their corresponding substituted species is ortho > meta > para. However, substitution increases differences in aromaticity between the parent molecules (1, 2, and 3) and their corresponding derivatives. In addition, by going down from 1s to 3s and from 1t to 3t, the electron density distribution on CC decreases (− 0.727, − 0.681, − 0.156, − 0.276, − 0.074, and 0.456 e, respectively, Figs. 1 and 2 and S1–S2).

The NBO charge distribution on C, N, and H atoms of selected s species (1s, 2s, 3s, and 5s) at M06-2X/AUG-cc-pVTZ

The NBO charge distribution on C, N, and H atoms of selected t species (1t, 2t, 3t, and 5t) at M06-2X/AUG-cc-pVTZ
Contrary to benzene molecule, the pyridine group increases the electron density distribution on CC, revealing the most negative charge (−2.774 and − 1.526 e) for 5s, and 5t, respectively. Hence, pyridine ring has the weaker resonance stabilization than benzene (resonance energy in pyridine and benzene is 28.0 and 35.8 kcal/mol, respectively) [67]. Indeed, NLP in the plane of the ring as the π-electron donoring group especially in a “W” arrangement has a more effect on charge distribution of s species than those of t species by more strongly interacting with the ring π-system.
Here, the electrostatic potential values of MEP maps are specified with different intensities for s carbenes compared with their corresponding t states in the range of − 0.0318 a.u. (deepest red) to + 0.0318 a.u. (deepest blue) (Figs. 3 and 4, and S3–S4) [37,38,39,40, 68, 69].

The resulted MEP maps of selected s species (5s, 9s, 10s, and 11s) at M06-2X/AUG-cc-pVTZ

The resulted MEP maps of selected t species (5t, 9t, 10t, and 11t) at M06-2X/AUG-cc-pVTZ
Also, MEP plots of both s and t states indicate blue color for hydrogen atoms via their positive charges, red color for carbon atoms via their negative charges, and the electron cloud in middle of ring(s). Also, MEP maps of s and t states display symmetrically and differentially electron current in the centers of the rings. As abovementioned, conjugation of the nitrogenʼs lone pairs with the vacant p orbitals of the CC in the most delocalized species, i.e., 5s and 5t increase their stability individually and decrease the corresponding ΔΕs−t. The electron-donating NLP in the plane of the ring affects inversely charge distribution and electrostatic potential on the surfaces of 9s, 10s, 11s, 9t, 10t, and 11t, that is when nitrogen atoms of pyridine are placed far from reach in meta and para positions of CC via decreasing the effective π-overlap and the resulted aromaticity.
Based on suggested homomolecular isodesmotic reactions (Scheme 4) [70, 71], ΔEs and ΔEt are the energy released by s and t carbenes when the corresponding CC is converted to a saturated carbon via addition of two hydrogen atoms from the corresponding homomolecular saturated carbon; also, ΔEtotal and ΔErelative are defined as ΔEt−ΔEs and ΔEs/ΔEt, respectively (Table 7).

The suggested isodesmic reactions for Hammick s and t carbenes 1–11
We considered stabilization of both s and t carbenes, although with different degrees. The more heat of hydrogenation, the lower stability it has. The s species are stabilized more than the t states. The trends of absolute value of ΔEtotal as well as ΔErelative are somewhat consistent with the ΔEs−t results; |ΔEtotal|: 1 (25.6) > 2 (17.9) = 7 (17.9) > 6 (16.1) > 4 (13.9) > 10 (10.9) = 8 (10.9) > 3 (10.5) > 9 (9.4) = 11 (9.4) ≥ 5 (9.3 kcal/mol). Evidently, compared with the completely conjugated carbenes 1–11, the designed non-planar carbenes 1′−11′ only benefit from substitutient effects of fused benzene or pyridine rings with contribution of non-planar cyclohexa-2,5-dienic moiety. The absolute values of ΔEtotal along with ΔErelative seem much more (approximately 1.4–2.7 times) in unsubstituted carbene, 1, than those of other substituted carbene, 2–11, perhaps owing to the higher electronic energy barrier to planarity of N—atom in two rings compared with one ring.
Conclusion
Using B3LYP/AUG-cc-pVTZ and M06-2X/AUG-cc-pVTZ computations, 22 s and t Hammick carbenes (1–11) were inspected. Some of thermodynamic and kinetic factors including ΔΕs−t, ΔΕ, ΔΕHOMO-LUMO, DM, α, N, ω, μ, η, NICS, NBO charge, MEP plots, and relative energies of isodesmic reactions (ΔEs, ΔEt, and ΔEtotal) support differently substitution effect on s and t states. Apart from t pyridine-4-ylidene, all studied Hammick carbenes emerge as minima, and s states display more thermodynamic stability than the corresponding t states. The most stable carbene is unsubstituted s pyridine-4-ylidene, and the least stable species is considered with CC situated by two nitrogen heteroatoms of two fused rings, in a “W” arrangement. According to the situation of CC and heteroatoms, stabilization for fused rings is meta more than para, and para more than ortho; also, the substitution effect of one six-membered ring is significant than that of two six-membered rings. Every t state exhibits higher kinetic stability than its corresponding s state. Regarding to proposed isodesmic reactions, all s states are stabilized through π-donor/σ-acceptor substitution, more than the corresponding t states.
References
Danopoulos AA, Simler T, Braunstein P (2019) N-Heterocyclic carbene complexes of copper, nickel, and cobalt. Chem Rev 119(6):3730
Smith CA, Narouz MR, Lummis PA, Singh I, Nazemi A, Li C-H, Crudden CM (2019) N-Heterocyclic carbenes in materials chemistry. Chem Rev 119(88):4986
Melaimi M, Soleilhavoup M, Bertrand G (2010) Stable cyclic carbenes and related species beyond diaminocarbenes. Angew Chem Int Ed 49:8810
Buchner E, Curtius T (1885) Ueber die Einwirkung von Diazoessigäther auf aromatische Kohlenwasserstoffe. Ber Dtsch Chem Ges 8:2377
Igau A, Grützmacher H, Baceiredo A, Bertrand G (1988) Analogous .alpha.,.alpha.′-bis-carbenoid, triply bonded species: synthesis of a stable .lambda.3-phosphino carbene-.lambda.5-phosphaacetylene. J Am Chem Soc 110:6463
Bourissou D, Guerret O, Gabbaï FP, Bertrand G (2000) Stable Carbenes. Chem Rev 100:39
Schaper L-A, Wei X, Altmann PJ, Öfele K, Pöthig A, Drees M, Mink J, Herdtweck E, Bechlars B, Herrmann WA, Kühn FE (2013) Synthesis and comparison of transition metal complexes of abnormal and normal tetrazolylidenes: a neglected ligand species. Inorg Chem 52:7031
Schumacher M, Goldfuss B (2015) Quantifying N-heterocyclic carbenes as umpolung catalysts in the benzoin reaction: balance between nucleophilicity and electrophilicity. New J Chem 39:4508
Nelson DJ, Nolan SP (2013) Quantifying and understanding the electronic properties of N-heterocyclic carbenes. Chem Soc Rev 42:6723
Huynh HV, Frison G (2013) Electronic structural trends in divalent carbon compounds. J Org Chem 78:328
Frison G, Huynh HV, Bernhammer JC (2013) Electronic structure trends in N-heterocyclic carbenes (NHCs) with varying number of nitrogen atoms and NHC—transition-metal bond properties. Chem Eur J 19:12892
Rezaee N, Ahmadi A, Kassaee MZ (2016) Nucleophilicity of normal and abnormal N-heterocyclic carbenes at DFT: steric effects on tetrazole-5-ylidenes. RSC Adv 6:13224
Khorshidvand N, Kassaee MZ, Ahmadi AA, Cummings PT (2018) Steric effects on normal and abnormal acyclic, cyclic-saturated, and cyclic-unsaturated diaminocarbenes using DFT method. J Phys Org Chem 32:e3898
Schuster O, Yang L, Raubenheimer HG, Albrecht M (2009) Beyond conventional N-heterocyclic carbenes: abnormal, remote, and other classes of NHC ligands with reduced heteroatom stabilization. Chem Rev 109:3445
Han Y, Huynh HV (2007) Preparation and characterization of the first pyrazole-based remote N-heterocyclic carbene complexes of palladium(II). Chem Commun 10:1089
Schneider SK, Rentzsch CF, Krüger A, Raubenheimer HG, Herrmann WA (2007) Pyridin- and quinolinylidene nickel carbene complexes as effective catalysts for the Grignard cross-coupling reaction. J Mol Catal A Chem 265:50
Schneider SK, Roembke P, Julius GR, Loschen C, Raubenheimer HG, Frenking G, Herrmann WA (2005) Extending the NHC concept: C–C coupling catalysis by a PdII carbene (r NHC) complex with remote heteroatoms. Eur J Inorg Chem 15:2973
Schneider SK, Julius GR, Loschen C, Raubenheimer HG, Frenking G, Herrmann WA (2006) A first structural and theoretical comparison of pyridinylidene-type rNHC (remote N-heterocyclic carbene) and NHC complexes of Ni(II) obtained by oxidative substitution. Dalton Trans 9:1226
Chong DP (1997) Recent advances in density functional methods, parts I and II. World Scientific, Singapore.
Barone V, Bencini A (1999) Recent advances in density functional methods, part III. World Scientific, Singapore
Adamo C, Matteo A d, Barone V (2000) From classical density functionals to adiabatic connection methods. The state of the art. Adv Quantum Chem 36:45
Ess DH, Houk KN (2005) Activation Energies of pericyclic reactions: performance of DFT, MP2, and CBS-QB3 methods for the prediction of activation barriers and reaction energetics of 1,3-dipolar cycloadditions, and revised activation enthalpies for a standard set of hydrocarbon pericyclic reactions. J Phys Chem A 109:9542
Becke AD (1988) Density-functional exchange-energy approximation with correct asymptotic behavior. Phys Rev A 38:3098
Becke AD (1993) Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys 98:5648
Becke AD (1996) Density-functional thermochemistry. IV. A new dynamical correlation functional and implications for exact-exchange mixing. J Chem Phys 104:1040
Lee C, Yang W, Parr RG (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B 37:785
Krishna R, Frisch MJ, Pople JA (1980) Contribution of triple substitutions to the electron correlation energy in fourth order perturbation theory. J Chem Phys 72:4244
Torkpoor I, Janjanpour MHN, Salehi N, Gharibzadeh F, Edjlali E, (2018) Insight into Y@X2B8 (Y= Li, CO2 and Li-CO2, X = Be, B and C) nanostructures: A computational study. Chem Rev Lett, 1:2-8
Sarvestani MRJ, Majedi S (2020) A DFT study on the interaction of alprazolam with fullerene (C20) J Chem Lett, 1:32-38
Gharibzadeh F, Gohari S, Nejati K, B. Hashemzadeh B, Mohammadiyan S (2018) The Be atom doping: An effective way to improve the Li-atom adsorptionin boron rich nanoflake of B24. Chem Rev Lett 1:16-22
Zhao Y, Truhlar DG (2008) The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor Chem Accounts 120:215
Rostamoghli R, Vakili M, Banaei A, Pourbashir E, Jalalierad K (2018) Applying the B12N12 nanoparticle as a sensor for CO, CO2, H2O and NH3 gasses. Chem Rev Lett 1:31-36
Majedi S, Behmagham F, Vakili M (2020) Theoretical view on interaction between boron nitride nanostructures and some drugs. J Chem Lett, 1:19-24
Janjanpour MHN, VakiliM, Daneshmehr S, Jalalierad K, Alipour F (2018) Study of the ionization potential, electron affinity and HOMO-LUMO gaps in the small fullerene nanostructures. Chem Rev Lett 1:45-49
Moladoust R. (2019) Sensing performance of boron nitride nanosheets to a toxic gas cyanogen chloride: Computational exploring. Chem Rev Lett 2:151-156
Koohi M, Bastami H (2020) Structure, stability, MEP, NICS, reactivity, and NBO of Si–Ge nanocages evolved from C20 fullerene at DFT. Monatsh Chem 151:693
Koohi M, Bastami H (2020) Substituent effects on stability, MEP, NBO analysis, and reactivity of 2,2,9,9-tetrahalosilacyclonona-3,5,7-trienylidenes, at density functional theory. Monatsh Chem 151:11
Koohi M, Bastami H (2020) A density functional theory perspective on 2,2,9,9-tetrahalostannacyclonona-3,5,7-trienylidenes. J Phys Org Chem 33:e4031
Koohi M, Bastami H (2020) A quest for stable 2,2,9,9-tetrahaloplumbacyclonona-3,5,7-trienylidenes at density functional theory. Struct Chem 31:877
Koohi M (2020) Estimating the stability and reactivity of cyclic tetrahalo substituted germylenes: a density functional theory investigation. J Phys Org Chem 33:e4032
Vessally E, Nikoorazm M, Esmaili F, Fereyduni E (2011) Substitution effects at α-position of divalent five-membered ring XC4H3M (M = C, Si and Ge). J Organomet Chem 696:932
Vessally E, Edjlali L, Shabrendi H, Rezaei M (2012) Electronic states of XC3H3Si five-membered rings (X = CH, N, P, and As). Russ J Phys Chem 86:595
Kassaee MZ, Koohi M (2013) Breathing viability into cyclonona-3,5,7-trienylidenes via α-dimethyl and ά-moieties at DFT. J Phys Org Chem 26:540
Kassaee MZ, Koohi M, Mohammadi R, Ghavami M (2013) 2,2,9,9-Tetramethylcyclonona-3,5,7-trienylidene vs. its heterocyclic analogues: a quest for stable carbenes at DFT. J Phys Org Chem 26:908
Koohi M, Kassaee MZ, Haerizade BN, Ghavami M, Ashenagar S (2015) Substituent effects on cyclonona-3,5,7-trienylidenes: a quest for stable carbenes at density functional theory level. J Phys Org Chem 28:514
Koohi M (2019) Cyclonona-3,5,7-trienylidene and its Si, Ge, Sn, and Pb analogs versus their α-halogenated derivatives at B3LYP and MP2 methods. J Phys Org Chem 32:e4013
Schmidt MW, Baldridge KK, Boatz JA, Elbert ST, Gordon MS, Jensen JH, Koseki S, Matsunaga N, Nguyen KA, Su SJ, Windus TL, Dupuis M, Montgomery JA (1993) General atomic and molecular electronic structure system. J Comput Chem 14(11):1347
Sobolewski AL, Domcke W (2002) Ab initio investigation of the structure and spectroscopy of hydronium−water clusters. J Phys Chem A 106:4158
Hariharan PC, Pople JA (1974) Accuracy of AH, equilibrium geometries by single determinant molecular orbital theory. J Mod Phys 27:209
Francl MM, Pietro WJ, Hehre WJ, Binkley JS, Gordon MS, DeFrees DJ, Pople JA (1982) Self-Consistent Molecular Orbital Methods. XXIII. A polarization-type basis set for second row elements. J Chem Phys 77:3654
Frisch MJ, Pople JA, Binkley JS (1984) Self-consistent molecular orbital methods 25: supplementary functions for Gaussian basis sets. J Chem Phys 80:3265
Clark T, Chandrasekhar J, Spitznagel GW, Schleyer PR (1983) Efficient diffuse function-augmented basis sets for anion calculations. III. The 3-21+G set for first-row elements, Li-F. J Comput Chem 4:294
Kendall RA, Dunning Jr TH, Harrison RJ (1992) Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J Chem Phys 96:6796
Hehre WJ, Radom L, Schleyer PR, Pople JA (1986) Ab initio molecular orbital theory. Wiley, New York
Weinhold F, Glendening ED, NBO 7.0 Program manual natural bond orbital analysis programs
Weinhold F (2012) Natural bond orbital analysis: a critical overview of relationships to alternative bonding perspectives. J Comput Chem 33:2363
Glendening ED, Landis CR, Weinhold F (2012) Natural bond orbital methods. Wiley Interdiscip Rev Comput Mol Sci 2:1
Zhang G, Musgrave CB (2007) Comparison of DFT methods for molecular orbital eigenvalue calculations. J Phys Chem A 111:1554
Schleyer PVR, Maerker C, Dransfeld A, Jiao H, van Eikema Hommes NJR (1996) Nucleus-independent chemical shifts (NICS): a simple and efficient aromaticity probe. J Am Chem Soc 118:6317
Schleyer PR, Jiao H, van Eikema Hommes NJR, Malkin VG, Malkina OL (1997) An evolution of the aromaticity of inorganic rings: refined evidence from magnetic properties. J Am Chem Soc 119:12669
Schleyer PVR, Manoharan M, Wang Z, Kiran B, Jiao H, Puchta R, van Eikema Hommes NJR (2001) Dissected nucleus-independent chemical shift analysis of p-aromaticity and antiaromaticity. Org Lett 3(16):2465
Domingo LR, Chamorro E, Pérez P (2008) Understanding the reactivity of captodative ethylenes in polar cycloaddition reactions. A theoretical study. J Org Chem 73:4615
Parr RG, Szentpaly L, Liu S (1999) Electrophilicity index. J Am Chem Soc 121:1922
Parr RG, Pearson RG (1983) Absolute hardness: companion parameter to absolute electronegativity. J Am Chem Soc 105:7512
Parr RG, Yang W (1989) Density functional theory of atoms and molecules. Oxford University Press, New York
Hoffmann R, Schleyer PR, Schaefer HF (2008) Predicting moleculesdmore realism, Please! Angew Chem Int Ed Eng 47:7164
Joule JA, Mills K (2010) Heterocyclic Chemistry5th edn. Blackwell Publishing, Chichester
Alam MJ, Ahmad S (2014) Molecular structure, anharmonic vibrational analysis and electronic spectra of o-, m-, p-iodonitrobenzene using DFT calculations. J Mol Struct 1059:239
Koohi M, Bastami H (2020) Substituted Hammick carbenes: the effects of fused rings and hetero atoms through DFT calculations. J Phys Org Chem 33:e4023
Boehme C, Frenking G (1996) Electronic structure of stable carbenes, silylenes, and germylenes. J Am Chem Soc 118:2039
Jursic BS (1999) Hybrid density functional theory study of low reactivity of imidazol-2-ylidine toward insertion and addition reactions. J Chem Soc, Perkin Trans 2 8:1805
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 14331 kb).
Rights and permissions
About this article

Cite this article
Nezhad, P.D.K., Youseftabar-Miri, L., Ahmadi, S. et al. A DFT quest for effects of fused rings on the stability of remote N-heterocyclic carbenes. Struct Chem 32, 787–798 (2021). https://doi.org/10.1007/s11224-020-01650-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-020-01650-5