
Abstract
The N-methyl-D-aspartate (NMDA) is the family of glutamate receptor, which is involved in controlling synaptic plasticity and memory function; but overactivation of this receptor results to excess intracellular calcium formation, triggers neuronal injury and also involves in several pathologies. Both ligand- and structure-based quantitative structure-activity relationship (QSAR), pharmacophore, docking and simulation studies have been performed on a set of structurally diverse inhibitors to explore prime molecular structural features involve for specific binding to NMDA, and vis-à-vis inhibiting enzyme activity. 3D QSAR studies, comparative molecular field analysis (CoMFA) and comparative molecular similarity indices analysis (CoMSIA) models showed the importance of steric, electrostatic and hydrophobic features; while hydrogen bond acceptor and hydrophobic features are depicted as important pharmacophore features of the molecule. Molecular docking and simulation studies corroborated the consequence of the features obtained from ligand-based Bayesian model (AUROCcv = 0.878); 3D QSAR CoMFA (R2 = 0.895, se = 0.513, Q2 = 0.602, R2pred = 0.673); CoMSIA (R2 = 0.877, se = 0.555, Q2 = 0.615, R2pred = 0.727); hologram QSAR (Q2 = 0.812, R2 = 0.941, R2pred = 0.772), and pharmacophore models (Q2 = 0.926, R2 = 0.927, R2pred = 0.621). Presence of aromatic ring, hetero and halogen atoms along with alkyl group of molecular scaffold shows their importance for binding affinity to NMDA receptor. Stability of the complex is adjudged by both docking and simulation studies.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
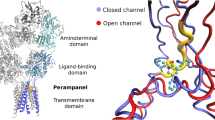
N-methyl D-aspartate (NMDA) [1, 2] participates in different patho-physiological processes, such as neuronal development, learning and memory, motor activity and nociception. The NMDA receptors (NMDARs) have been targeted for various types of neurological disorders, like Alzheimer’s disease (AD), and neuropsychiatric illnesses stimulated by alcohol and psychotropic agents [3]. The NMDA is an analogue of glutamate, an excitatory neurotransmitter that selectively binds with the ion channel receptors and also permeable to Ca2+, K+ and Na+ ions. The receptor consists of four separate domains: an amino-terminal domain, a ligand-binding domain which participates in interactions with agonists, a transmembrane domain that forms an ion channel and an intracellular C-terminal domain [4]. Stimulation of the receptor and activation of the cation current in the membrane are caused by binding of glutamate and glycine simultaneously in the ligand domain. Membrane depolarization and signal transduction have occurred due to influx of positively charged particles. At the same time, increased Ca+2 concentration in neurons triggers various signalling procedures. Pathophysiology of neurodegenerative disorders and excitotoxicity have the strong evidence for glutamate dysregulation [5]. Competitive glutamate and glycine antagonists are efficient to inhibit NMDAR activities [6]. Thus, receptor antagonists and ion channel blockers are effective as neuroprotector agents, specifically for the treatment of AD [7].
In the present work, ligand- and structure-based studies have been performed with NMDA inhibitors for the modelling study. Ligand-based methods, such as 2D and 3D quantitative structure-activity relationships (QSARs), finger printing and pharmacophore mapping studies have been performed for finding associations among chemical structure or structural properties and biological activity or target property of the compounds. The 3D QSAR studies provide information of drug activity on a particular target through the molecular descriptors which involve in structural and binding mode of ligand on the target protein [8]. Pharmacophore mapping is a spatial representation of molecular features in 3D space, which are important for bioactivity. Structure-based docking and dynamics studies provide information about the interaction pattern and stability of the ligand and receptor.
Structurally diverse NMDA receptor inhibitors were used to build 2D and 3D QSAR models [9]. The QSAR analyses depicted the importance of topological descriptors of pyrazine derivatives for predicting antagonistic activity of selective NMDAR. Pharmacophore model also suggested the importance of aromatic ring and hydrogen bond (HB) donor and acceptor features of the molecular scaffold for inhibiting the NMDAR functionality. 3D QSAR study also showed the importance of an electronegative oxygen atom (i.e. acceptor) and phenyl ring (i.e. aromatic ring). QSAR descriptors and pharmacophoric features obtained from the docking studies have been found to support each other [9]. Quinoxaline derivatives were also used to develop pharmacophore and QSAR models. Zinc database was screened against the hypothesis, and the filtered molecules were further docked. The GlideScore and molecular interactions with catalytic amino acids were considered as crucial features to identify final hits and finally, five hit compounds were selected [10]. Additionally, bioactive compounds of Mentha spicata L. were used to generate SAR models and compared molecular features with standard drugs, donepezil, galantamine and memantine. Solvent-accessible area hydrophobicity, energy of frontier molecular orbitals and counts of aromatic ring and rotatable bonds were obtained as most relevant descriptors. The compound 1,8-cineole has physicochemical similarity with memantine but dissimilar with donepezil in respect to hydrophobicity, energy of LUMO and the solvent accessible surface areas [11]. The multiple linear regression (MLR) model of phenyl-amidine derivatives was designed to explain 83.6% of the biological data variance. Molecular models predicted the activity and pharmacokinetics and toxicity profiles of the molecules, and also hypothised the interaction at the catalytic site, led them as a promising class [12]. Adaptive neuro-fuzzy inference system (ANFIS) was also utilized for predicting binding affinity of phencyclidine derivatives. On comparison, it has been observed that the ANFIS model achieved satisfactory accuracy [13] than traditional (MLR and partial least squares (PLS)) methods. Two types of quantitative models were also developed based on docking energies of quinoxaline derivatives. A significant outcome of the structural features for glycine binding site has been observed when steric and electrostatic contours of comparative molecular field analysis (CoMFA) models were correlated with the homology-based model [14]. 3D models of the ion channel were constructed to localize the binding sites for the blockers of the ion channel, although models share low homology with the potassium channels. Based on the determined geometry of the protein-ligand complexes, a quantitative model has been constructed using CoMFA method [15]. The present work explored 2D and 3D QSAR (CoMFA, comparative molecular similarity indices analysis (CoMSIA) and hologram QSAR (HQSAR)) and pharmacophore models, which depict physicochemical and structural requirement for imparting potential inhibition of the enzyme.
Materials and methods
Structurally diverse compounds (n = 87) (Suppl. Fig. S1) [16,17,18,19,20,21,22,23] have been used to derive 2D and 3D QSAR, and pharmacophore models to find out the features of small molecules required for binding specificity to NMDA receptor. The present work has been performed to establish relationship between the chemical structure and corresponding bioactivity (Ki) by the statistical methods and deduce a pharmacophore map through ligand-based modelling technique, and finally, the derived models are extrapolated with structure-based docking and simulation studies. The dataset was divided through k-means clustering (k-MCA) method [24] into training set (n = 66) that contains both most and least active compounds for QSAR model generation, and test set (n = 21) to validate the derived models. The k-MCA method divides the descriptors into clusters based on the biological activity. Clusters are arranged according to their Euclidian distances in multi-dimensional space. Thirty compounds have been selected in the training set for pharmacophore model generation. The splitting technique was validated through analysis of score plot, derived through principle components analysis (PCA) [25], to verify the presence of test set compounds in close vicinity of neighbouring training set compounds (Suppl. Fig. S2). Different statistical parameters have been considered to assess the models that include R2 (correlation coefficient), se (standard error of estimate), Q2 cross-validated correlation (CV), F with degree of freedom (df), R2bs (bootstrapped correlation coefficient) and sb (standard error of bootstrapped correlation) for 3D QSAR study, while different cost factors have been used for pharmacophore mapping study. The R2pred and sp. (standard error of prediction) of the test set have also been estimated to assess the prediction power of the model.
Comparative field and similarity analyses
In drug design, the molecular field analysis is considered the interactions between the receptor and ligand, which mainly emphasizes on understanding non-bonding interactions. Based on the overall orientations and depending upon alignment rules imposed to the aligned compounds, results are obtained that consider factors like lattice shifting step size and type of probe atom [26]. Structural alignment of the molecules determines the prediction accuracy of the model and reliability of the contour model. Therefore, to align molecules, molecular docking has been performed for all structurally diverse compounds in the dataset. Docked pose conformers have been used for model generation. First of all, the molecules were heated at 700 K for 1000 fs and annealing was done at 200 K for 1000 fs. Using the Tripos force field [27], steric (s) and electrostatic (e) interactions were calculated with a distance-dependent dielectric constant at regularly spaced (2 Å) grid, taking an sp3 carbon atom as steric probe and + 1.0 charge as electrostatic probe. The energy cutoff has been set to 30 kcal/mol to avoid excessively high and unrealistic energy values within the molecule. To derive molecular similarity models, different molecular fields, like hydrophobic (p), hydrogen bond (HB) donor (d) and acceptor (a), and s and e interactions were generated. Using cross-validated PLS method (leave one out), regression analysis has been carried out for identifying the relationship between the molecular structure and its properties [28]. The minimum sigma (column filtering) was set to 2.0 kcal/mol to speed up the analytical process and reduce noise. The model has been finalized on the basis of best correlation coefficient with the optimum number of components used in the model generation [28].
Bayesian model
A Bayesian model [29] has the potential to categorize the activity status of the compounds qualitatively. The model was developed by the help of ‘Create Bayesian model’ tool in Discovery Studio (DS) [30]. To generate the model, the dataset was categorized into active and inactive molecules on the basis of the threshold value of Ki. The 2D descriptors such as molecular function class fingerprints of maximum diameter 6 (FCFP_6) and molecular fractional polar surface area (MFPSA); AlogP; molecular weight (MW); and number of rotatable bonds (nRB), rings (nR), aromatic rings (nArR) and hydrogen bond donors (nHBD) and acceptors (nHBA) were selected for model development. Good fingerprints are emphasized from the developed model, which are favourable for binding affinity. Some bad fingerprints are also highlighted which may reduce the affinity towards NMDAR. Validation of the model was performed based on generation of cross-validated receiver operating characteristic value (ROCCV), specificity, sensitivity and concordance of the dataset. The predictive ability of the model was verified on the test set compounds.
Hologram QSAR model
Correlation between structural data of molecular fragments with the response parameter is known as HQSAR. Various feasible molecular fragments comprising branched, linear, cyclic and overlapping features of the molecules were explored as hologram. For model generation, the training set molecules were divided based on different structural fragments, such as linear, branched, cyclic and overlapping. The occurrence of different structural fragments was encoded as hashed fingerprints that were further split into strings at a fixed interval, as determined by a hologram length (HL) parameter. A number of parameters affect hologram generation, such as HL; fragment size and fragment distinction significantly affect model quality [31]. With the help of Sybyl [32], various combination of molecular fragments as well as fragment-generation parameters have been developed for each HL [32]. At optimum component number based on the Q2 and cross-validated standard error (SEcv), the best model was short-listed. By calculating the R2pred of test set compounds, prediction capability of the model has been estimated.
Pharmacophore space modelling
Pharmacophore hypothesis explores probable interactive features between ligand and receptor. A pharmacophore model portrays the features through which structurally diverse ligands can bind to a common receptor site. The model can also be used to screen novel ligands through de novo design or virtual screening. The ‘Pharmacophore generation’ tool of DS [30] was used for pharmacophore mapping of NMDA inhibitors. The chemical features, such as HB a and d, p, ring aromatic (r) and positive ionization (i) were used for pharmacophore generation, setting the feature selection criteria with a minima and maxima of 1 and 5, respectively. The different control parameters, like spacing, uncertainty and weight variation have been implemented for hypothesis generation (hypogen process). The extent of magnitude of the compound’s activity is demonstrated by each feature. Spacing is varied from 1.000 to 3.000 pm, weight variation parameter is varied from 0.300 to 0.500 and values of 1.5 to 3.0 have been considered in uncertainty parameter. While generating hypothesis, a total cost function is minimized comprising with three terms: weight, error and configuration cost. Proximity of total to fixed cost determines the acceptability of the hypothesis, as it is nearer to the ideal hypothesis. Difference between the total and null costs as 60 bits has been considered for hypothesis optimization. The configuration cost is equal to the entropy of hypothesis space and should have a value < 17 for a good pharmacophore model. The greater the difference (Δcost) between the total and the null costs, it is more likely that the hypothesis does not reflect a chance correlation. The developed hypothesis was further validated to nullify over prediction of bioactivity for inactive compounds through a process, known as Hyporefine. In this process, the steric interactions of the compound have been considered in the hypothesis generation, and if steric properties are crucial for bioactivity, then these are portrayed in the validated (refined) hypothesis. A cross-validation procedure, CatScramble based on Fischer’s randomization test has been used to judge the quality of hypothesis [33]. Initiation of hypogen process and randomization of biological activity data within a fixed chemical dataset were carried out. The randomization technique proves that pharmacophore model generated before scrambling is better than hypothesis generated after scrambling.
Molecular docking
Molecular interaction between the ligand and receptor at the active site of protein has been determined by the molecular docking study using Grid-Based Ligand Docking with ‘Glide’ in Schrodinger [34]. Based on structural resolution (2.6 Å) of the crystalline NMDAR, (PDB id: 3QEL [35]), the docking analysis was carried out. Self-docking was performed for validation of the method, where the bound ligand was re-docked at the active site of the protein. For calculation of root mean square deviation (RMSD), conformer of the original bound ligand was superimposed to the docked poses. Reports suggest that low RMSD (< 2 Ǻ) of original bound ligand validates the docking procedure [36]. ‘Protein preparation wizard’ module was used for preparation of protein molecule [37]. Addition of hydrogen atoms and minimization and optimization of energy by OPLS2005 force field have been executed. With the help of ‘PROPKA’, the protonation states were assessed [38, 39] at physiological pH 7.4. During protein preparation, the water molecules were retained up to 5 Ǻ around the active site and the rest were removed. LigPrep module [40] was used for the ligand structure preparation. Based on the active site of protein, the grid was generated subsequent to ligand and protein preparation. In order to enclose the receptors within 3 Å from the central domain of the residues, the grid-enclosing box was centred to the catalytic sites of the respective 3D conformation of the receptor. For the allocated binding site, docking calculation has been performed with Glide XP mode [40] that establishes all possible orientation for each low-energy conformer.
Molecular dynamics
Protein residues responsible for binding to the ligand and the interactions pattern between them have been analysed by molecular dynamics (MD) simulation method. The MD study provides information regarding thermodynamically stable conformation of the specific protein. It provides an idea about the stability of protein-ligand complexes, using ‘Desmond Molecular Dynamics’ module [41] of Schrodinger Maestro [40] utilizing OPLS2005 molecular mechanics force field. For simulation of MD, the ligand-protein complex was solvated with TIP3P water model [42]. For avoiding the direct association of protein complex with its own periodic image, the orthorhombic boundary box at 10 Å was designed. Calibration of the system was done by setting other parameters kept as default. The MD simulation was executed for 30 ns at Nose-Hoover chain thermostat at 300 K, Martyna-Tobias-Klein barostat at 1.013 bar pressure and at time step of 10 ps. In order to cross-check the stability of model system, the RMSD, root mean square fluctuation (RMSF), radius of gyration (Rg) and H bonds were verified [43]. For analysis of MD trajectory equilibration, RMSD happens to be an essential parameter, while RMSF characterizes the local changes throughout the protein chain. In order to gain an idea regarding the structural compactness of the protein with respect to active and inactive ligands, the Rg was analysed.
Results and discussion
A set of structurally diverse non-selective NMDA inhibitors (n = 87) (Suppl. Fig. S1) [16,17,18,19,20,21,22,23] have been considered for QSAR, pharmacophore mapping, docking and dynamics analyses. All the molecules of the dataset were docked in NMDA protein structure (PDB id: 3QEL [35]), and the docked confirmers were considered for alignment to generate 3D QSAR models. Bayesian model and HQSAR have been developed to explore the important fragments for imparting bioactivity.
CoMFA model
In the field analysis study, s and e factors individually develop the models with correlation of 0.890 and 0.855, respectively. The s and e features together develop the most suitable model (model 1: nTr = 66, R2 = 0.895, se = 0.513, Q2 = 0.602). The steric favourable green contour shows conformational rigidity, and unfavourable region has been demonstrated by yellow contour. Blue and red contours depict the electrostatic favourable and unfavourable regions, respectively. The model exhibits steric and electrostatic domains with contributions of 46 and 54% (Fig. 1a). The predictive ability of the model (nTs = 21, R2pred = 0.673) signifies the potency of the model (Table 1). This model correctly estimated 57.58% active molecules, and 56.32% molecules are accurately estimated among the total dataset molecules. The estimated binding affinity of the compounds as per model 1 is represented in Fig. 2, and also listed in Suppl. Table S1. The active molecule (cpd C62) mapped in the contour map suitably (Fig. 1a (I)), whereas the less active compound (cpd C42) (Fig. 1a (II)) failed to bind in the model. Presence of electron donating oxygen of keto group and favourable inductive effect of alkyl chain make the region favourable for electrostatic activity. Nitrogen atoms in triazole ring donate electrons to each other and develop an electron cloud which makes the region favourable for electrostatic activity. The alkyl and amino groups show the steric favourable region for their conformational rigidity. The importance of steric and electrostatic features is also adjudged by the models developed with quinolone and quinoxaline derivatives [15].

Contour maps of a CoMFA and b CoMSIA models fitted in I active cpd C62 and II inactive cpd C42. Steric: green = favourable, yellow = unfavourable; electrostatic: blue = favourable, red = unfavourable; hydrophobic: cyan = favourable, white = unfavourable

Observed vs predicted binding affinity as per QSAR and pharmacophore models of NMDA inhibitors
CoMSIA model
In a similar study, a, d, p, s and e features individually develop models with correlation of 0.791, 0.749, 0.835, 0.890 and 0.855, respectively. But combination of steric, electrostatic and hydrophobic features develop the best model. The CoMSIA model (model 2 nTr = 66, R2 = 0.877, se = 0.555, Q2 = 0.615) shows steric, electrostatic and hydrophobic regions with 12.40, 46.30 and 41.40% contributions, respectively (Table 1). Figure 1b represents the contour map of model 2. Cyan contour shows the hydrophobic favourable region, while white contour shows the hydrophobic unfavourable region. Steric and electrostatic contours are depicted in same colours as in CoMFA model. Efficiency of estimation (nTs = 21, R2pred = 0.727) depicts the robustness of the model. The model accurately estimated 52% active molecules, 9% molecules are over estimated and 39% molecules are under estimated. The predicted activity of the compounds as per model 2, is represented in Fig. 2, and also tabulated in Suppl. Table S1. It is observed that triazole ring along with alkyl chain show steric favourable region for their conformational rigidity. Presence of electron donating oxygen of keto group in pyrazine ring and positive inductive effect of alkyl chain make the regions favourable for electrostatic activity. Nitrogen atoms of triazole ring contain lone pair of electron and contribute electrons to each other, which make an electron dense region that is favourable for electrostatic activity. Phenyl ring, chlorine atoms and alkyl chain act as hydrophobic for their non-polar property.
Bayesian model
For development of Bayesian model, seven explanatory descriptors (AlogP, MW, nHBA, nHBD, nR, nArR and MFPSA) and one fingerprint feature FCFP_6 (molecular function class fingerprints of maximum diameter 6) were implemented. Cross-validated ROC plot (AUROCcv = 0.878) and the parameters, like specificity (0.929), sensitivity (0.714), concordance (0.857) and enrichment data (Table 2) of the developed Bayesian model (model 3) specify its capability to separate active and inactive compounds efficiently. The fivefold cross-validation result, AUCROCTest (0.942) authenticates the developed Bayesian model and also indicates the acceptability of the model (Fig. 3). Depending on Bayesian score and FCFP_6, the best five favourable and unfavourable molecular fragments are specified for NMDA inhibitory activity, which are listed in Fig. 4. Among the favourable features, piperidine ring along with alkyl chain with hydroxyl substitution, imidazolidine scaffold and suitable alkyl and hydroxy alkyl chains are depicted their importance. Pyrazolo-pyrimidine and cyclobutene rings, and isopropenylamine, amide or amine groups are depicted as bad contributing molecular fragments for the activity.

ROC plot for test set

Good and bad fingerprints depicted in Bayesian model
HQSAR model
Based on the optimization of different fragment features (A/B/C/H/Ch/D-A) and the hologram length, HQSAR analysis has been performed with the training set (nTr = 66) molecules. Top six hypotheses are presented in Table 3. Owing to the highest values of Q2 and smallest cross-validated standard error (SEcv), the most suitable fragment features are selected. The statistical results (Q2 = 0.812, R2 = 0.941 with eight components) of the best model (model 4) is depicted in Table 1. The model contains bonds (B), connections (C) and hydrogen (H) as fragment distinction and having a size of 6–8. A better estimation of test compounds (nTs = 21, R2pred = 0.772) substantiates the strength of the model (Table 1). The model predicts 64.37% molecules accurately, 19.54% over estimated and 16.09% molecules are under estimated in the dataset. Predicted value of the compounds according to model 4 is represented in Fig. 2, and also listed in the Suppl. Table S1. Contribution of various colours of atom or fragment to the molecular activity has been depicted by contour maps of the HQSAR model (Fig. 5). The significance of the different colours is (i) red-orange colour denotes an inferior contribution of − 0.107 to − 0.064, (ii) white colour suggests an average contribution of − 0.043 to 0.102, (iii) yellow colour indicates a superior contribution of 0.102 to 0.153 and (iv) green colour signifies the highest contribution of 0.255 or above. In the dataset, most active compound (cpd C62) shows the good fragments in its molecular scaffold, while the least active compound (cpd C42) contains bad fragments in its molecular structure, depicted in the contour map. From the contour map (Fig. 5), it can be concluded that pyrazine ring, associated cyclic alkyl ring and alkyl chain act as important fragments for binding to the receptor. But backbone chain along with keto and amino groups depict as bad fragments for receptor binding.

HQSAR contribution maps: a most active cpd C62 and b inactive cpd C42. White = average contribution (− 0.097 to 0.102), yellow = good contribution (0.102 to 0.153), green = maximum contribution (0.254 or above), cyan = maximum common substructure
Pharmacophore mapping
On the basis of R2, RMSD and cost values, best combinations of the features have been standardized for HypoGen model development. Final HypoGen model is further used for HypoRefine model generation with addition of excluded volume. Results of the top six hypotheses are summarized in Table 4. Based on cost difference (Δcost = 164.826), low RMSD (0.992), best cross-validated correlation (Q2 = 0.926) and test set prediction (R2pred = 0.621) (Table 1), the most suitable hypothesis (H6) is characterised. Figure 6 highlights the mapped pharmacophore features. The hypothesis H6 (model 5) demonstrates more than 93% correlation to the bioactivity. The hypothesis is verified with Fischer’s randomization test at 98% confidence level, demonstrated no hypothesis other than H6 (model 5) generates better correlation. Predicted value of the compounds as per model 5 is represented in Fig. 2 and Suppl. Table S1. Approximately 46% of the active molecules are accurately estimated by the model, 42% under estimated and 12% molecules are over estimated. The model depicts that HB acceptor (a) and hydrophobic (p) features along with three excluded volumes (ev) might have a role as chief biophores for activity. Inter-feature distances of the mapped features are represented in Fig. 6a, which are critical for imparting selective inhibitory activity of NMDA. Most active compound (cpd C62) maps the features very well (Fig. 6b) but the least active compound (cpd C42) do not map all the features properly (Fig. 6c). Feature map shows that pyrazine, and associated oxygen and nitrogen atoms act as HB acceptors for their electronegetivity. Halogen atom and alkyl chain portray as hydrophobic for non-polarity. Excluded volumes are important around the features for steric hindrance. The observations can be corroborated with the work of Zambre et al., where selective NMDA receptor antagonists were used to generate pharmacophore model that suggests the importance of two aromatic rings, one HB donor and HB acceptor features [9]. Ugale et al. also reported the contribution of HB acceptor, donor and ring aromatic features for the models developed with quinoxaline derivatives [10]. The model developed with quinolones derivative also revealed the importance of HB acceptor, electropositive HB region and hydrophobic features for imparting activity [15]. Therefore, HB acceptor and hydrophobic features have significant influence for binding affinity to NMDAR, as also suggested in present study.

a Pharmacophoric features of NMDA inhibitors with inter-feature distances, mapped with b most active (cpd C62) and c least active (cpd C42) compounds. Pharmacophore features are green = HB donor (d), cyan = hydrophobic (p) and grey = excluded volume (ev)
Docking study
Docking study of active (cpd C62) and inactive compounds (cpd C42) has been carried out at the catalytic site of the crystal structure, 3QEL [35] (Fig. 7). Amino acids present (within 4 Ǻ) at the catalytic site include Glu70, Arg79, Glu106, Tyr109, Gln110, His120, Asn121, Asp139, Tyr175, Glu236 (polar amino acids) and Val15, Ala72, Ala107, Ile111, Phe114, Leu123, Leu135, Phe176 (non-polar amino acids). It has been observed that catalytic residues, Glu106 interacts with the amino group of pyrazine ring and Phe176 forms pi-pi stacking interaction with chloro-benzene, are involved in the hydrophobic interaction. The Glu236 forms an HB interaction with the amino group, while Tyr175 and Met207 both interact with the carboxyl group to form HB interactions. Therefore, amino and carboxyl groups and pyrazine ring may play an important role as per the interactions occurred. Further dynamics study has been performed to investigate the interaction and binding stability of the complex.

Docking interaction of a most active cpd C62 and b least active cpd C42 compounds
Dynamics study
The MD simulation study has been undertaken for 30 ns in an aqueous solution in order to get an idea of the interactions of ligand in a dynamic environment. The docked poses of active (cpd C62) and inactive (cpd C42) compounds are short-listed for MD simulation. The RMSDs of the complexes are analysed (Fig. 8) through dynamic stability of both ligand and proteins. The RMSD plot of active compound (Fig. 8a) shows that the complex is stable. Maximum stability has been observed at 17 to 30 ns. In case of inactive compound (Fig. 8b), RMSD plot suggests that complex is not stable throughout time length. Although the variations between the side chain and backbone for both the complexes are alike, RMSF plot is also beneficial for characterization of local alterations along with the protein chain. Analyses of RMSF graph for both complexes (Fig. 8) exhibit that the active inhibitor has better RMSF distribution and dynamic features (Fig. 8a) in contrast to the less active compound (Fig. 8b). Amino acids, Ala72, His120 and Leu123 maintained stability of the protein ligand contact during fluctuation. Therefore, the binding pocket showed adequate stability, as RMSF is limited to 4.5 Å in active compound (cpd C62); on the contrary, cpd C42 showed less stability. Residue Ala72 has HB interaction with active compound, which further changed to water-bridge bond with the ligand that confirms the stability of the complex. It maintained 40% stability of the full time length (Fig. 8a). The residue His120 also formed HB interaction with active compound, maintained its stable interaction contact up to 33% of the time length and further changed into water-bridge bond. The Leu123 maintained its hydrophobic interaction through simulation. Additionally, Arg79 formed an HB interaction, then changed into ionic bond interaction, and further changed into water-bridge interaction. But the interactions are not stable throughout the time length. The Glu70 formed an ionic bond and Val15 formed a hydrophobic bond with the compound, but are not stable throughout the time length. The least active compound does not maintain stable HB interaction (Suppl. Fig. S3). Only Asp139 maintained a stable interaction of 38% on total time length (Fig. 8b). Residues His120, Asn121 and Ala72 also tried to interact with the ligand, but interactions are converted to water-bridge bond and did not maintain their stability. Residues Leu123 and Val15 showed hydrophobic interactions but these are unstable too. Stability of the protein-ligand complex with more stable HB interactions are the key fragrances of the study.

a RMSD plot. b RMSF plot. c Interaction during simulation study of I most active (cpd C62) and II least active (cpd C42) compounds
In the present study, developed QSAR and pharmacophore models explored the essential features which are further substantiated by docking and dynamics studies. The features of QSARs and pharmacophore models are adjudged through the interaction sites (Fig. 9). Importance of steric functionality of heteroatom in models 1 and 2 is substantiated by HB interactions with Glu106 and Glu236. Simulating stable interaction of the heteroatom of triazole ring with His120 is also substantiated the steric property in model 2. Electrostatic functionality of keto group in models 1 and 2 is substantiated with the interactions of residues Tyr175 and Met207. Importance of oxo group in model 5 as HB acceptor is substantiated with stable interaction of Ala72 during simulation study. Significance of aromatic ring, halogen atom along with alkyl group as hydrophobic fragments is substantiated by the interaction with Phe176 in models 2–5. Heterocyclic rings, such as pyrazine, imidazolidine, piperidine and alkyl chains can improve binding property as depicted in both models 3 and 4, but pyrazole, pyrimidine and amide linkage can decrease the activity property. Oxygen atom in pyrazine ring portrayed its importance in models 1, 2 and 5. Halogen atom associated with aromatic ring can improve the activity, suggested in models 1 and 5. Long alkyl chain also improves the activity, depicted in models 2, 3 and 5. In model 1, heteroatom present in triazole ring shows unfavourable for activity, but small alkyl chain in triazole ring is favourable for activity, observed in model 2. Finally, all such models demonstrate that the chemical features and groups happen to be the prime structural features can be effectively utilized for the formulation of potential hit compounds for NMDA inhibition.

Schematic representation of pharmacophore features of potent NMDA inhibitors
Conclusion
In the present work, structurally diverse compounds having wide range of activity (pKi = 3.721 to 9.125) were utilized to explore important chemical features and determine binding modes of ligands towards NMDA receptor for therapeutic invasion. Both ligand- and structure-based multi-chemometric methods have been adopted to corroborate the facts from the observations of the previously developed 2D and 3D QSAR models [9, 11,12,13,14,15] for explaining binding affinity of NMDAR. It was also observed that a particular QSAR model may not be effective for prediction of compounds. There should not be any biasness on prediction; multiple models can be reliable than single model prediction [44]. Thus, multi-chemometric models explored in the present study may be successively used for more reliable and precise prediction. Generated models prove the importance of the features essential for binding affinity. The HB acceptor, hydrophobicity, steric and electrostatic features of the molecular scaffold might have a crucial role in NMDA inhibition, as observed in developed models generated through both structure- and ligand-based studies. Docking and MD studies infer that Ala72, Glu106, His120, Tyr175, Phe176, Met207 and Glu236 are involved as catalytic residues in receptor-ligand interactions. Thus, the key structural requirements (as depicted in the models) are essential to formulate hit compounds for inhibition of NMDA. Physicochemical significance and biological implications of the models are justified with the features of models, and vis-à-vis the binding interactions at the active site of the enzyme. A 2D schematic representation (Fig. 9) provides specific information (importance of HB acceptor, hydrophobic, steric and electrostatic features that may be associated for interaction with amino acids at the enzyme’s active site) of the structural and characteristic features of a molecule for effective binding at the catalytic site.
References
Gitto R, De Luca L, Ferro S, Occhiuto F, Samperi S, De Sarro G, Russo E, Ciranna L, Costa L, Chimirri A (2008) Computational studies to discover a new NR2B/NMDA receptor antagonist and evaluation of pharmacological profile. ChemMedChem 3:1539–1548
Gitto R, De Luca L, Ferro S, Citraro R, De Sarro G, Costa L, Ciranna L, Chimirri A (2009) Development of 3-substituted-1H-indole derivatives as NR2B/NMDA receptor antagonists. Bioorganic Med Chem 17:1640–1647
Yosa J, Blanco M, Acevedo O, Lareo LR (2009) Molecular orbital differentiation of agonist and antagonist activity in the GlycineB-iGluR-NMDA receptor. Eur J Med Chem 44:2960–2966
Sobolevsky AI, Rosconi MP, Gouaux E (2009) X-ray structure, symmetry and mechanism of an AMPA-subtype glutamate receptor. Nature 462:745–756
Hynd MR, Scott HL, Dodd PR (2004) Glutamate-mediated excitotoxicity and neurodegeneration in Alzheimer’s disease. Neurochem Int 45:583–595
Chen HS, Lipton SA (2006) The chemical biology of clinically tolerated NMDA receptor antagonists. J Neurochem 97:1611–1626
Farlow MR (2004) NMDA receptor antagonists. A new therapeutic approach for Alzheimer’s disease. Geriatrics 59:22–27
Kubinyi H (1997) QSAR and 3D QSAR in drug design. Drug Discov Today 2:457–467
Zambre VP, Hambarde VA, Petkar NN, Patel CN, Sawant SD (2015) Structural investigations by in silico modeling for designing NR2B subunit selective NMDA receptor antagonists. RSC Adv 5:23922–23940
Ugale VG, Bari SB (2016) Identification of potential Gly/NMDA receptor antagonists by cheminformatics approach: a combination of pharmacophore modelling, virtual screening and molecular docking studies. SAR QSAR Environ Res 27:125–145
Avram S, Maria M, Bagci E, Hritcu L, Borcan LC, Mihailescu D (2017) Advanced structure-activity relationships applied to Mentha spicata L. subsp. spicata essential oil compounds as AChE and NMDA ligands, in comparison with donepezil, galantamine and memantine—new approach in brain disorders pharmacology. CNS Neurol Disord Drug Targets 16:800–811
Abreu PA, Castro HC, Paes-de-Carvalho R, Rodrigues CR, Giongo V, Paixao IC, Santana MV, Ferreira JM, Caversan OM, Leao RA, Marins LM, Henriques AM, Farias FM, Albuquerque MG, Pinheiro S (2013) Molecular modeling of a phenyl-amidine class of NMDA receptor antagonists and the rational design of new triazolyl-amidine derivatives. Chem Biol Drug Des 81:185–197
Buyukbingol E, Sisman A, Akyildiz M, Alparslan FN, Adejare A (2007) Adaptive neuro-fuzzy inference system (ANFIS): a new approach to predictive modeling in QSAR applications: a study of neuro-fuzzy modeling of PCP-based NMDA receptor antagonists. Bioorganic Med Chem 15:4265–4282
Tikhonova IG, Baskin II, Palyulin VA, Zefirov NS (2003) CoMFA and homology-based models of the glycine binding site of N-methyl-d-aspartate receptor. J Med Chem 46:1609–1616
Tikhonova IG, Baskin II, Palyulin VA, Zefirov NS (2004) 3D-model of the ion channel of NMDA receptor: qualitative and quantitative modeling of the blocker binding. Doklady 396:181–186
Chenard BL, Bordner J, Butler TW, Chambers LK, Collins MA, De Costa DL, Ducat MF, Dumont ML, Fox CB, Mena EE et al (1995) (1S,2S)-1-(4-hydroxyphenyl)-2-(4-hydroxy-4-phenylpiperidino)-1-propanol: a potent new neuroprotectant which blocks N-methyl-D-aspartate responses. J Med Chem 38:3138–3145
Ornstein PL, Schoepp DD, Arnold MB, Leander JD, Lodge D, Paschal JW, Elzey T (1991) 4-(Tetrazolylalkyl)piperidine-2-carboxylic acids. Potent and selective N-methyl-D-aspartic acid receptor antagonists with a short duration of action. J Med Chem 34:90–97
Varano F, Catarzi D, Colotta V, Filacchioni G, Galli A, Costagli C, Carla V (2002) Synthesis and biological evaluation of a new set of pyrazolo[1,5-c]quinazoline-2-carboxylates as novel excitatory amino acid antagonists. J Med Chem 45:1035–1044
Fray MJ, Bull DJ, Carr CL, Gautier EC, Mowbray CE, Stobie A (2001) Structure-activity relationships of 1,4-dihydro-(1H,4H)-quinoxaline-2,3-diones as N-methyl-D-aspartate (glycine site) receptor antagonists. 1. Heterocyclic substituted 5-alkyl derivatives. J Med Chem 44:1951–1962
Kinney WA, Abou-Gharbia M, Garrison DT, Schmid J, Kowal DM, Bramlett DR, Miller TL, Tasse RP, Zaleska MM, Moyer JA (1998) Design and synthesis of [2-(8,9-dioxo-2,6-diazabicyclo[5.2.0]non-1(7)-en-2-yl)-ethyl]phosphonic acid (EAA-090), a potent N-methyl-D-aspartate antagonist, via the use of 3-cyclobutene-1,2-dione as an achiral alpha-amino acid bioisostere. J Med Chem 41:236–246
Torres E, Duque MD, Lopez-Querol M, Taylor MC, Naesens L, Ma C, Pinto LH, Sureda FX, Kelly JM, Vazquez S (2012) Synthesis of benzopolycyclic cage amines: NMDA receptor antagonist, trypanocidal and antiviral activities. Bioorganic Med Chem 20:942–948
Varano F, Catarzi D, Colotta V, Calabri FR, Lenzi O, Filacchioni G, Galli A, Costagli C, Deflorian F, Moro S (2005) 1-substituted pyrazolo[1,5-c]quinazolines as novel Gly/NMDA receptor antagonists: synthesis, biological evaluation, and molecular modeling study. Bioorganic Med Chem 13:5536–5549
Kinney WA, Lee NE, Garrison DT, Podlesny Jr EJ, Simmonds JT, Bramlett D, Notvest RR, Kowal DM, Tasse RP (1992) Bioisosteric replacement of the alpha-amino carboxylic acid functionality in 2-amino-5-phosphonopentanoic acid yields unique 3,4-diamino-3-cyclobutene-1,2-dione containing NMDA antagonists. J Med Chem 35:4720–4726
Kanungo TM, Mount DM, Netanyahu NS, Piatko CD, Silverman R, Wu AY (2002) An efficient k-means clustering algorithm: analysis and implementation. IEEE Trans Pattern Anal Mach Intell 24:881–892
Everitt BS, Dunn G (2013) Applied multivariate data analysis2nd edn. Wiley Online Library Arnold, London
Balaji S, Prasanna DS, Rangappa KS (2013) Docking, QSAR and CoMFA studies on arecoline analogues as muscarinic acetylcholine receptor (mAChR) M1 agonists. Proc Indian Natl Sci Acad 79:41–50
Lu X, Lv M, Huang K, Ding K, You Q (2012) Pharmacophore and molecular docking guided 3D-QSAR study of bacterial enoyl-ACP reductase (FabI) inhibitors. Int J Mol Sci 13:6620–6638
Srivastava V, Kumar A, Mishra BN, Siddiqi MI (2008) CoMFA and CoMSIA 3D-QSAR analysis of DMDP derivatives as anti-cancer agents. Bioinformation 2:384–391
Halder AK, Saha A, Jha T (2013) Exploration of structural and physicochemical requirements and search of virtual hits for aminopeptidase N inhibitors. Mol Div 17:123–137
Discovery studio 2.5 (2009) a) LigandFit, (b) Pharmacophore (c) Bayesian model; Accelrys Software Inc., San Diego
Yang XL, Zhou Y, Liu XL (2014) Hologram quantative structure–activity relationship studies on 1-(5-carboxyindol-1-yl) propan-2-one inhibitors of human cytosolic phospholipase A2α. Med Chem Res 23:1512–1518
Sybyl (2006) Tripos Inc.
Jacquez JA, Jacquez GM (2002) Fisher’s randomization test and Darwin’s data—a footnote to the history of statistics. Math Biosci 180:23–28
Ramar V, Pappu S (2016) Exploring the inhibitory potential of bioactive compound from Luffa acutangula against NF-kappaB-A molecular docking and dynamics approach. Comput Biol Chem 62:29–35
Karakas E, Simorowski N, Furukawa H (2011) Subunit arrangement and phenylethanolamine binding in GluN1/GluN2B NMDA receptors. Nature 475:249–253
Taha MO, Habash M, Al-Hadidi Z, Al-Bakri A, Younis K, Sisan S (2011) Docking-based comparative intermolecular contacts analysis as new 3-D QSAR concept for validating docking studies and in silico screening: NMT and GP inhibitors as case studies. J Chem Inf Model 51:647–669
Sastry GM, Adzhigirey M, Day T, Annabhimoju R, Sherman W (2013) Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des 27:221–234
Bas DC, Rogers DM, Jensen JH (2008) Very fast prediction and rationalization of pKa values for protein-ligand complexes. Proteins 73:765–783
Li H, Robertson AD, Jensen JH (2005) Very fast empirical prediction and rationalization of protein pKa values. Proteins 61:704–721
Maestro v9.7. a) Glide, (b) LigPrep; Protien prep wizard. Schrodinger
Tyagi C, Gupta A, Goyal S, Dhanjal J, Grover A (2014) Fragment based group QSAR and molecular dynamics mechanistic studies on arylthioindole derivatives targeting the alpha-beta interfacial site of human tubulin. BMC Genomics 15(Suppl 9):S3
Pradeepkiran JA, Kumar KK, Kumar YN, Bhaskar M (2015) Modeling, molecular dynamics, and docking assessment of transcription factor rho: a potential drug target in Brucella melitensis 16M. Drug Des Dev Ther 9:1897–1912
Kallubai M, Amineni U, Mallavarapu M, Kadiyala V (2015) In silico approach to support that p-Nitrophenol monooxygenase from Arthrobacter sp. strain JS443 catalyzes the initial two sequential monooxygenations. Interdiscip Sci Comput Life Sci 7:157–167
Roy K, Ambure P, Kar S, Ojha PK (2018) Is it possible to improve the quality of predictions from an “intelligent” use of multiple QSAR/QSPR/QSTR models? J Chemom:1–18
Funding
Financial assistance for major research project (MRP) from University Grants Commission (UGC) is thankfully acknowledged. One of the authors, Tabassum Hossain wishes to thank UGC-MANF for awarding senior research fellowship.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Electronic supplementary material
ESM 1
(DOC 814 kb)
Rights and permissions
About this article

Cite this article
Hossain, T., Mukherjee, A. & Saha, A. Exploration of structural and physicochemical properties of small molecules to inhibit NMDA functionality. Struct Chem 29, 1175–1187 (2018). https://doi.org/10.1007/s11224-018-1103-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-018-1103-7