
Abstract
A computational study of the isomerization reaction of a series of halodiazirines to halodiazo compounds (cyclic to open-chain RXCN2 species) has been carried out in order to establish the effect of the substituent groups on the isomerization rates and to obtain computational evidence of reaction mechanisms. Fluorine and chlorine were present as the halogen (X) atom, and the groups R=H, CH3, C2H5, n-C3H7, i-C3H7, cyclo-C3H5, phenyl, OCH3 and OH were used. Thermochemical calculations and natural bond orbital analyses were carried out at the B3LYP/6-31+G(d,p) level of theory. The results allowed us to discuss a reaction mechanism that proceeds in two steps: The first is the extrusion of nitrogen and formation of a carbene through a cyclic transition state that promotes the simultaneous breaking of the two C–N bonds, and the second one is described as the rebounding between the carbene and one of the nitrogen atoms of molecular nitrogen, both formed in the first step. The enthalpies of formation of halodiazirines and halodiazoalkanes have been calculated at the G3 level of theory.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Diazirines are compounds with a three-membered cycle containing one carbon atom and two double-bonded nitrogen atoms. They are very unstable on exposure to heat and light. The diazirines containing halogen atoms, such as F, Cl or Br, are named halodiazirines, and they are relatively stable and accessible [1] and have been used as sources of carbenes [2–5].
The isomerization of diazirines to diazoalkanes (Scheme 1) can occur either thermally or photochemically. The physical and chemical properties of both types of compounds, cyclic and open-chain isomers, are very different taking into account their stability on exposure to heat and light, and their interconversion [1].

Isomerization of diazirines to diazoalkanes
It has been experimentally found that diazirines decompose thermally and photolytically by reactions exhibiting first-order kinetics, and the products could be: (1) the respective open-chain isomer (a diazo compound), (2) a carbene intermediate and molecular nitrogen or (3) a mixture of (1) and (2). It has been found that the relative preference for the isomerization process or for the formation of carbene and molecular nitrogen, and the rate at which these reactions can occur are related to the type of substituent in the diazirine [1, 6], but there are experimental problems to directly detect the products forming by decomposition of the diazirines, precluding the determination of the ratio of the formation of the carbene intermediate regarding the diazo compound [4].
From the discovery of diazirines, there have been a large number of studies on the thermal decomposition and photolysis of diazirines carried out in gas phase and in solution [4, 7]; however, there is still no clarity on the process by which it occurs [8, 9]. Stevens et al. [10] studied the thermal decomposition of 3-chloro-3-methoxydiazirine and 3,3-dimethyldiazirine [11] observing first-order kinetics producing molecular nitrogen and the corresponding carbenes and then afterward transforming to more stable products. Stevens et al. [12] also studied the thermal decomposition of 3-(3-methyldiazirin-3-yl)propan-1-ol and 3-(3-methyldiazirin-3-yl)propanoic acid. They found that reactions are unimolecular and the preferential products are alkenes derived from the corresponding carbenes.
The thermal decomposition of phenylchlorodiazirine, phenyl-n-butyldiazirine and 2-adamantane-2,3′-[3H]diazirine in solution and in the presence of C60 (in order to evaluate the quantity of carbene and/or diazo compound formed in the reaction) was studied by Liu et al. [4]. The results showed that phenylchlorodiazirine gave only the carbene, phenyl-n-butyldiazirine gave the diazo intermediate, and 2-adamantane-2,3′-[3H]diazirine gave a mixture of the carbene and the diazo compound. For the aryldiazirine p-methoxy-3-phenyl-3-methyldiazirine, the formation of the corresponding diazo compound and carbene was confirmed [8, 13].
The purpose of this work has been the computational study of the isomerization reaction of halodiazirines, RXCN2, to the respective halodiazo compounds. Two halogen atoms (X=F, Cl) and several substituent groups (R=H, CH3, C2H5, n-C3H7, i-C3H7, cyclo-C3H5, phenyl, OCH3 and OH) have been introduced for determining the variation of the activation energy of the reaction and for the characterization of the transition states proposed for the reaction.
The reaction was modeled following a mechanism in two steps shown in Scheme 2 (proposed by Liu et al. [4]): the first one being the simultaneous breaking of the two C–N bonds of the diazirine, forming a carbene with the extrusion of molecular nitrogen, and the second one being the formation of a bond between the carbene and one of the N atoms of molecular nitrogen. Liu et al. [4] also proposed another mechanism for the isomerization of diazirines to diazomethanes in one step via a non-cyclic transition state with the breaking of only one of the C–N bonds, but this transition state is not well described by density functional theory because of its multiconfigurational character [4].

Reaction mechanism in two steps proposed for the isomerization of halodiazirines to halodiazo compounds
A computational study of the thermochemistry of halodiazirines and their isomeric halodiazomethanes has also been carried out.
Computational details
Calculations were made using the Gaussian 09 computational package [14]. The geometric parameters of reactants, transition states and products of the reactions studied were fully optimized at DFT level with the B3LYP functional and the 6-31+G(d,p) [15] basis set. Additional calculations at the M06-2X/6-311G(d,p) and MP2/cc-pVTZ levels for species with selected substituents were also carried out in order to validate the results obtained at the B3LYP/6-31+G(d,p) level. The calculation of the vibrational frequencies allows an evaluation of zero-point vibrational energies (ZPE) and the kinetic and thermochemical parameters of the reaction. ZPE values were scaled by the factor 0.9857 [16]. Thermal corrections to enthalpy and entropies were evaluated at 298.15 K.
Intrinsic reaction coordinate (IRC) calculations [17] were performed in order to verify that localized transition states connect with the corresponding minima stationary points associated with reactants and products.
The bonding characteristics of reactants, transition states and products have been investigated using a population partition technique, the natural bond orbital (NBO) analysis of Reed and Weinhold [18, 19]. The NBO formalism provides the values for the atomic natural total charges and also provides the Wiberg bond indexes [20] used to follow the progress of the reaction. The NBO analysis has been performed using the NBO program [21] implemented in Gaussian 09 computational package [14].
The rate constants have been calculated according to the classical transition state theory [22, 23] through the Eyring–Polanyi equation:
where K B , h, R and ΔG ≠(T) are Boltzmann’s constant, Planck’s constant, the universal gas constant and the standard-state Gibbs energy of activation, at the absolute temperature T, respectively.
Results and discussion
Isomerization of halodiazirines to halodiazoalkanes
The isomerization reactions of a selected set of halodiazirines to halodiazo compounds were studied at the B3LYP/6-31+G(d,p) level of theory, following the mechanism shown in Scheme 2. Additional calculations at the M06-2X/6-311G(d,p) and MP2/cc-pVTZ levels were carried out for species involving groups R=H, CH3 and OCH3 and X=Cl and F, in order to validate the results obtained at the B3LYP/6-31+G(d,p) level.
Table 1 shows a comparison of the Gibbs free activation energies (ΔG ≠) obtained for both steps of the reactions using the different methods of calculation. As it can be seen, the values calculated with the MP2 method are close to those calculated with B3LYP, except the value calculated for the reaction of fluoromethoxydiazirine. The values calculated with M06-2X are slightly higher than those obtained with the other methods, but the trend is similar at the three levels. The reaction profiles are represented in Fig. 1, in order to appreciate the similar trends.

Gibbs free energy profiles for the isomerization reactions from halodiazirines to halodiazoalkanes, with the substituents H (a, b), CH3 (c, d) and OCH3 (e, f), calculated at the B3LYP/6-31+G(d,p), M06-2X/6-311G(d,p) and MP2/cc-pVTZ levels of theory
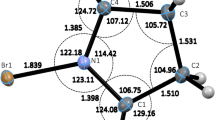
The optimized geometries of the species involved in the isomerization reaction of chlorodiazirine to chlorodiazomethane are represented in Fig. 2, and the IRC plots corresponding to the two transition states of the same reaction are drawn in Fig. 3.

B3LYP/6-31+G(d,p)-optimized geometries of the species involved in the isomerization reaction of chlorodiazirine to chlorodiazomethane

B3LYP/6-31+G(d,p)-calculated intrinsic reaction coordinate (IRC) plots for the isomerization reaction of chlorodiazirine to chlorodiazomethane. a step I, b step II
The calculated Gibbs free energies of activation and rate constants for all the reactions studied in this work are collected in Table 2. It can be observed, in all cases, the Gibbs free energies of activation of the first step are higher than those of the second one and so the first step is the rate-determining one, except in the cases of compounds with the substituents hydroxy and methoxy in which the barrier of the second step is higher than that of the first one. Some examples of the reaction profiles for the isomerization of fluoro- and chlorodiazirines are shown in Fig. 4.

Gibbs free energy profiles, calculated at the B3LYP/6-31+G(d,p) level, for the isomerization reactions from halodiazirines to halodiazoalkanes, with the substituents H, CH3, phenyl and OCH3
As it can be observed, in the case of fluoro- and chlorodiazirine with substituent R=OCH3, the carbenes formed in the first step are very stable compared to the initial halodiazirines, and the barriers of the second step are very high, so the formation of the diazo compounds from the corresponding carbenes is very unlikely. The same occurs in the case of R=OH. The opposite situation occurs for chlorodiazirine with R=H. In this case, the barrier of the second step is lower than that of the first one, the carbene formed is less stable, and the halodiazomethane is very stable, being favoring its formation.
Calculated kinetic constants for the first step of the reactions range from 10−6 to 10−13 s−1. They are several orders of magnitude lower than those reported for the thermolysis of some diazirines; however, it has to be taken into account that the experimental conditions were different, especially the temperatures. There are values reported for the kinetic constant of the thermolysis of 3-chloro-3-aryldiazirines [24, 25] in cyclohexane of ca. 10−4 s−1, at temperatures higher than 60 °C.
Other values reported in the literature are those for the thermal decomposition in the gas phase of diazirines with X=Cl and R=CH3, C2H5, n-C3H7 and i-C3H7 [24, 26, 27]. The authors reported values for the entropies of activation ranged from 3.2 to 3.9 cal mol−1 K−1, in good agreement with the values calculated by us, around 2.0 cal mol−1 K−1 (see Table S1 of the Supporting information).
As it can be seen in Table 2, the groups with high electron-donating capacity such as OH and OCH3 decrease the Gibbs free energy of activation of the first step of the reaction, which could be related to the stabilization of the cyclic carbon of diazirine in the transition state because this atom loses electronic charge for the simultaneous breaking of the two C–N bonds. The calculated kinetic parameters show that both steps of the isomerization reactions of chlorodiazirines are in almost all the cases more rapid than the reactions of the corresponding fluorodiazirines. This fact could be explained by the weak electron-withdrawing capacity of halogens that destabilize the transition state, more in the case of fluorine because of its higher electronegativity.
There are values in the literature [4] for the activation energy of the rebound reaction between the nitrogen and the carbene in the thermal decomposition of some diazirines in the gas phase, the values ranging from 9 to 60 kJ mol−1. The authors conclude that these values depend solely on the substituent on the ring. Our results suggest that the rate of the reactions depends on the capacity of stabilization of carbene by the substituents groups which can interact with the empty orbital of carbene through electron delocalization. Strongly activating groups such as OH and OCH3 have significant effects on the activation energies, whereas weakly activating groups, such as alkyl groups, have a lower effect.
NBO calculations have been carried out in order to obtain the Wiberg bond indexes, \(B_{i}\), and the natural atomic charges. Moyano et al. [28] defined the parameters \({\updelta }B_{i} = \left( {B_{i}^{TS} - B_{i}^{R} } \right)/(B_{i}^{P} - B_{i}^{R} )\) that gives us the relative variation of the bond index at the transition state for every bond involved in the chemical reaction, where R, TS and P refer to reactant, transition state and product, respectively, and the percentage of evolution, %EV = 100 \({\updelta }B_{i}\), which gives us an idea of the progress of transformation of any bond involved in the reaction. Calculated values of the Wiberg bond indexes corresponding to the bonds that are forming or breaking in the isomerization reaction from halodiazirines to halodiazomethanes are collected in Table 3. The results corresponding to all of the reactions studied are shown in Table S3 of the Supporting information. Numbering of the atoms involved in the isomerization reaction of halodiazirines to halodiazo compounds is shown in Scheme 3.

Numbering of the atoms involved in the isomerization reaction of halodiazirines to halodiazo compounds
The results show that the Wiberg bond indexes calculated for chloro compounds are slightly higher than those calculated for fluoro compounds. It is worth noting that in the transition states, the calculated Wiberg bond indexes are different for the two C–N bonds, which indicates that their breaking is not synchronic. This is also observed in the calculated NBO charges (Table 5), where the values for both nitrogen atoms in chlorodiazirine are the same, −0.068, whereas in the transition state, the charges of the nitrogen atoms are different, −0.030 and 0.001. In the case of fluorodiazirine, the charge of the nitrogen atoms is −0.094, and in the transition state, −0.338 and −0.061.
The percentages of evolution of the bonds are more advanced for chlorodiazirines in the first step of the isomerization reactions, but the behavior is opposite in the second step, where the percentages of evolution of the bonds are more advanced for fluorodiazirines. The non-homogeneous breaking of the two C–N bonds is also observed in the values of the percentages of evolution of the bonds, being 33.1 and 47.1 % for the reaction of chlorodiazirine, and 30.5 and 42.3 % for the reaction of fluorodiazirine.
Another useful measure [28] of the degree of evolution of bonding at the transition state along the reaction path is obtained from the average value, δB av = 1/n \(\sum {\updelta }B_{i}\), and a convenient concept to characterize the reaction or reaction steps is the synchronicity, Sy, which is calculated from the Wiberg bond indexes. Sy = 1 − A, where A is the asynchronicity of a chemical reaction, calculated as [28]:
The values for the degree of evolution of bonding at the transition state, δB av, and the absolute synchronicities, Sy, calculated for all the reactions studied are collected in Table 4. δB av values are generally lower than 0.5, indicating that the transition states are early, nearer to the reactants than to the products, this behavior being very different for halodiazirines with OH and OCH3 substituents. In this case, δB av values are ca. 0.3, indicating very early character of the transition states of the first step, but the values are very high for the second step, 0.6 for chlorodiazirines and ca. 0.9 for fluorodiazirines, indicating transition states nearer to the products than to the reactants.
Synchronicity values are close to 0.90 for the first step of the isomerization reactions of chloro- and fluorodiazirines with hydrogen or alkyl substituents, whereas values lower than 0.90 have been obtained for halodiazirines with phenyl, OH and OCH3 substituents, indicating that they are asynchronous processes. The second step of the reactions has a very different behavior, and the Sy values are clearly lower than 0.90 for halodiazirines with alkyl substituents, especially with hydrogen, indicating very asynchronous processes, but they are higher than 0.90 for phenyl, OH and OCH3, indicating very synchronous processes, especially for fluorohydroxydiazirine (Sy = 0.98) and fluoromethoxydiazirine (Sy = 0.97).
In Table 5 are collected the NBO natural atomic charges for the species involved in the isomerization reactions of fluoro- and chlorodiazirine. Results for the species involved in the isomerization reactions of all the halodiazirines studied in this work are collected in Table S4 in the Supporting information. A polarization of the C–X bond in the transition states with migration of the negative charge toward the C atom can be observed.
Thermochemistry of halodiazirines and halodiazoalkanes
The enthalpies of formation in the gas phase of the studied halodiazirines and halodiazoalkanes have been calculated at the G3 level of theory using atomization reactions [29], that is the standard procedure to obtain enthalpies of formation in Gaussian-n theories. The calculated values are collected in Tables 6 and 7 for fluoro and chloro compounds, respectively.
We have also calculated the enthalpies of formation of parent compounds, diazirine and diazomethane, the values being 318.9 and 269.8 kJ mol−1, respectively. For these molecules, there are experimental values available in the literature that differ greatly from each other in the case of diazirine. Laufer and Okabe [30] reported values of 267.1 and 215.0 kJ mol−1, based on a photodissociation method, and Paulett and Ettinger [31], 332.0 ± 9.6 and 206.0 ± 9.6 kJ mol−1, from early appearance potential studies based on electron impact experiments, for the enthalpies of formation of diazirine and diazomethane, respectively. Excepting the value for diazomethane measured by Paulett and Ettinger [31], the other experimental values are very different from those calculated by us, especially in the case of diazirine, but it is interesting to note that the difference in stability between diazirine and diazomethane calculated by us, 49.1 kJ mol−1, is very similar to that obtained by Laufer and Okabe, 52.1 kJ mol−1. Several authors have calculated the heats of formation of diazirine and diazomethane through different methods: Catoire and Swihart [32] have reported values of 318.4 and 310.0 kJ mol−1, for diazirine, and 264.4 and 268.6 kJ mol−1, for diazomethane, obtained at the CBS-Q and G2 levels, respectively; Gordon and Kass [33, 34] reported values at the G2 level of 310.9 and 269.0 kJ mol−1, for diazirine and diazomethane, respectively; and Dixon et al. [35] reported a value of 273.2 kJ mol−1 for the enthalpy of formation of diazomethane, computed using the CCSD(T) method and CBS extrapolation. All theoretical values are in agreement with our calculations.
There is another compound with a known enthalpy of formation, chloromethyldiazirine. In this case, the value calculated by us, 245.6 kJ mol−1, is in very good agreement with the values available in the literature, 243.0 [36] and 233 ± 40 [37] kJ mol−1.
As it can be observed (see Tables 6, 7), the behavior of chloro and fluoro compounds is very different. Chlorodiazoalkanes are more stable than chlorodiazirines in all cases, except when the substituent is OH or OCH3, strongly activating groups, whereas fluorodiazirines are ever more stable than the corresponding fluorodiazoalkanes.
Conclusions
Calculations at the B3LYP/6-31+G(d,p) level have been carried out for all the species involved in the thermal isomerization reaction of halodiazirines to halodiazo compounds, with substituent groups R=H, CH3, C2H5, n-C3H7, i-C3H7, cyclo-C3H5, phenyl, OH and OCH3, and halogen X=F and Cl. The studied mechanism occurs in two steps: the first one being the simultaneous but not synchronic breaking of the two C–N bonds in halodiazirines forming a carbene and molecular nitrogen, and the second one being the rebounding between the carbene and one of the N atoms of molecular nitrogen forming the corresponding halodiazoalkane. The transition states have an early character, nearer to the reactants than to the products of the reactions. Substituent groups with high electron-donating capacity, such as OH and OCH3, stabilize the transition state of the first step and so increase the rate of the reactions.
The G3-calculated enthalpies of formation show that chlorodiazirines are less stable than chlorodiazoalkanes in all cases, except when the substituent is OH or OCH3, strongly activating groups, whereas fluorodiazirines are ever more stable than the corresponding fluorodiazoalkanes.
References
Korneev SM (2011) Eur J Org Chem 6153-6175
Pfeiffer F, Rauhut G (2011) J Phys Chem A 115:11050–11056
Moss RA, Tian J, Chu G, Sauers RR, Krogh-Jespersen K (2007) Pure Appl Chem 79:993–1001
Liu MTH, Choe Y-K, Kimura M, Kobayashi K, Nagase S, Wakahara T, Niino Y, Ishitsuka MO, Maeda Y, Akasaka T (2003) J Org Chem 68:7471–7478
Moss RA (2006) Acc Chem Res 39:267–271
Muller-Remmers PL, Jug K (1985) J Am Chem Soc 107:7275–7284
Liu MTH (1987) Chemistry of Diazirines. CRC Press, Boca Raton
Zhang Y, Vyas S, Hadad CM, Platz MS (2010) J Phys Chem A 114:5902–5912
Fedorov I, Koziol L, Mollner AK, Krylov AI, Reisler H (2009) J Phys Chem A 113:7412–7421
Smith NP, Stevens IDR (1979) J Chem Soc Perkin Trans 2:213–216
Frey HM, Stevens IDR (1962) J Chem Soc 3865–3867
Stevens IDR, Liu MTH, Soundararajan N, Paike N (1990) J Chem Soc Perkin Trans 2:661–667
Zhang Y, Burdzinski G, Kubicki J, Vyas S, Hadad CM, Sliwa M, Poizat O, Buntinx G, Platz MS (2009) J Am Chem Soc 131:13784–13790
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery Jr JA, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2010) Gaussian 09, Revision B.01, Gaussian Inc., Wallingford CT
Ditchfield R, Hehre WJ, Pople JA (1971) J Chem Phys 54:724–728
Merrick JP, Moran D, Radom L (2007) J Phys Chem A 111:11683–11700
Kenichi F (1970) J Phys Chem 74:4161–4163
Reed AE, Weinhold FJ (1983) Chem Phys 78:4066–4073
Reed AE, Curtiss LA, Weinhold F (1988) Chem Rev 88:899–926
Wiberg KB (1968) Tetrahedron 24:1083–1096
Glendening ED, Reed AE, Carpenter JE, Weinhold F (1988) NBO version 3.1 Madison, WI
Glasstone S, Laidler K, Eyring H (1941) The theory of rate processes, 1st edn. McGraw Hill, New York
Benson SW (1969) The foundations of chemical kinetics. McGraw Hill, New York
Liu MTH, Toriyama K (1972) Can J Chem 50:3009–3016
Liu MTH, Chien DHT (1974) J Chem Soc Perkin Trans 2:937–941
Bridge MR, Frey HM, Liu MTH (1969) J Chem Soc A 91-94
Frey HM, Liu MTH (1970) J Chem Soc A 1916–1919
Moyano A, Pericas MA, Valentí E (1989) J Org Chem 54:573–582
Notario R, Castaño O, Gomperts R, Frutos LM, Palmeiro R (2000) J Org Chem 65:4298–4302
Laufer QAH, Okabe H (1971) J Am Chem Soc 93:4137–4140
Paulett GS, Ettinger R (1963) J Chem Phys 39:825–827
Catoire L, Swihart MT (2002) J Propul Power 18:1242–1253
Gordon MS, Kass SR (1995) J Phys Chem 99:6548–6550
Gordon MS, Kass SR (1997) J Phys Chem A 101:7922
Dixon DA, de Jong WA, Peterson KA, McMahon TB (2005) J Phys Chem A 109:4073–4080
Archer WH, Tyler BJ (1976) J Chem Soc, Faraday Trans 1(72):1448–1455
Cadman P, Engelbrecht WJ, Lotz S, Van der Merwe SWJ (1974) J S Af Chem Inst 27:149–161
Acknowledgments
We thank the financial support of the Universidad Nacional de Colombia-Medellín through the convocatory “Apoyo a grupos de investigación Facultad de Ciencias” under the project number 201010015547.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zapata, L.A., López, S., Ruiz, P. et al. Halodiazirines and halodiazo compounds: a computational study of their thermochemistry and isomerization reaction. Struct Chem 28, 597–605 (2017). https://doi.org/10.1007/s11224-016-0824-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-016-0824-8