
Abstract
Intramolecular halogen bonds have been the subject of several current experimental and theoretical studies. In this work, intramolecular halogen bonds in a series of 1,2-aryldiyne molecules were investigated using density functional theory calculations at the M06-2x level of theory. For comparison, some dimeric complexes between halogenated aryldiynes and quinolinyl compounds were also considered. The calculated interatomic distances and interaction angles of intramolecular halogen bonds compare fairly well with those determined experimentally, and the triangle motifs retain almost perfectly planar in all the studied molecules. Many of the well-known properties of conventional halogen bonds are reproduced in intramolecular halogen bonds: the interaction strength tends to increase with the enlargement of the atomic radius of halogens (I > Br > Cl); the attachment of electron-withdrawing moieties to halogens leads to much stronger intramolecular halogen bonds; the X···N (quinolinyl) interactions are stronger than the X···O (carbonyl) halogen bonds. On the basis of the shorter interatomic distances and the larger values of electron densities at the bond critical points, intramolecular halogen bonds become stronger in strength than corresponding intermolecular halogen bonds. However, these interactions have similar structural, energetic, atoms in molecules (AIM), and noncovalent interaction index (NCI) characteristics to traditional halogen bonds. Therefore, these interactions can be recognized as halogen bonds that are primarily electrostatic in nature. Particularly, the formation of intramolecular halogen bonds gives rise to the essential coplanarity of the molecules, whereas the two subunits in the dimeric complexes deviate from planarity to a large degree. In addition, a small number of crystal structures containing intramolecular halogen bonds were retrieved from the Cambridge Structural Database (CSD), to provide more insights into these interactions in crystals. This work not only will extend the knowledge of noncovalent interactions involving halogens as electrophilic centers but also could be very useful in molecular design and synthetic chemistry.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Noncovalent interactions, such as hydrogen bonding (HB), ion-π, π–π stacking, electrostatic, hydrophobic, and van der Waals (vdW) forces, play a crucial role in the fields of chemical and biological sciences. HB, the most frequently used tool for crystal engineering of solid-state materials and molecular recognition of biomolecules, has been extensively studied in the last few decades [1–4]. Recently, a specific interaction in which halogen atoms (X = Cl, Br, I) act as electrophilic centers has attracted increasing interest, due to its great importance in material science, molecular design, and medicinal chemistry [5–12]. This interaction has been termed as halogen bonding (XB) to emphasize its analogies with HB. It is well known that covalently bonded halogens (even the fluorine atom) show a region of positive electrostatic potential (ESP) [13] at the outmost portion of these atoms along the R–X bonds. Therefore, an electron-rich atom or group prefers to approach the positive region, giving rise to a linear XB. Politzer et al. have previously developed the σ-hole concept to rationalize the anisotropic ESP distribution of halogens, and moreover, they also extended this concept to interpret other specific noncovalent interactions, such as chalcogen and pnicogen bonding [14–20].
Conventional intermolecular XB interactions continue to be an active research area in current years. The group of Metrangolo and Resnati firstly explored the excellence of the N···X–PFC synthon, where PFC denotes a perfluorocarbon moiety and N is either sp2 (typically pyridine derivative) or sp3 (tertiary amine) nitrogen, in solid-state chemistry [5, 8]. Ho et al. [12] then revealed the importance of the interactions between organic halogens and backbone carbonyl oxygens (C–X···O=C) in protein–ligand complexes. The wide application of XB interactions in anion recognition was also reported nowadays [21–23].
When formed from a HB donor and acceptor within the same molecule, the resultant “intramolecular” HB leads to the creation of the so-called quasi-ring and thus is capable of stabilizing conformation. This particular class of HB has been the subject of numerous theoretical and experimental studies [24–30], because of its critical structural and functional roles in biological and synthetic molecular systems. In 2012, Nagy investigated the solvent effect on O–H···X–C intramolecular HBs (X = F, NH2, NO2) using density functional theory (DFT) calculations and estimated the free energy needed for maintaining these interactions in ligand–protein systems [24]. Then, intramolecular C=O···H–O interactions in a family of O-carbonyl hydroquinones displaying anticancer and antioxidant activity were studied via the B3LYP, MP2, and 1H-NMR methods [25]. Very recently, Gerothanassis and coworkers have collated the experimentally and theoretically derived descriptors for intramolecular HBs in the families of phenol-containing natural products and model compounds [26].
As compared to traditional XBs and intramolecular HBs, far less attention has been focused on intramolecular XB interactions to date [31–36]. Bowling et al. have recently applied single-crystal X-ray crystallography to study intramolecular XBs between aryl halide donors and suitable acceptors (carbonyl and quinolinyl groups) that are held in proximity by 1,2-aryldiyne linkers [31]. Subsequently, those authors examined the temperature and solvent effects on intramolecular XB interactions using 15N, 13C, and 19F NMR spectroscopy, and they found that 13C NMR chemical shifts of the alkynyl carbons are good indicators of these bonds [32]. In 2008, Palusiak and Grabowski [33] performed the Cambridge Structural Database (CSD) search and ab initio MP2 calculations to characterize five-membered pseudo-rings closed through Cl···O intramolecular contacts. Some of their findings indicated that Cl···O contacts are attractive and stabilizing ones. However, Jablonski [34] pointed out that these contacts indeed are nonbonding and repulsive, as a result of the positive values of interaction energies obtained by a few estimating methods. More recently, intramolecular halogen···halogen bonding was demonstrated to be of an unusually strong vdW type [35]. Despite these attempts, several issues of intramolecular XB interactions remain largely elusive. What is the nature of these bonds? Do intramolecular XBs have similar properties to intermolecular XB interactions? Can they be classified as conventional XBs? What is the difference between intramolecular XB and HB interactions?
In this work, a systematic theoretical study at the DFT (M06-2x) level of theory was undertaken to examine intramolecular XB interactions in a series of aryldiyne molecules 1–12, as displayed in Fig. 1. The X-ray crystal structures of some of these compounds have been determined recently [31]. For comparison, four halogen-bonded dimeric complexes 13–16 (see Fig. 2) of halogenated aryldiynes with quinolinyl compounds were also taken into account. To gain a deeper understanding of these peculiar interactions, the atoms in molecules (AIM) [37] and noncovalent interaction index (NCI) calculations [38] were performed. In addition, a survey of the CSD was also undertaken to provide more insights into intramolecular XBs in crystals.

Chemical structures of the studied aryldiyne molecules 1–12

Chemical structures of the halogen-bonded complexes under study 13–16
Computational methods
The geometries of all the molecules under study were fully optimized by means of the hybrid M06-2x functional developed by Zhao and Truhlar [39], which has proved to be reliable in the description of various types of noncovalent interactions [40]. The basis set of Peterson et al. [41], cc-pVTZ-PP, obtained from the EMSL Basis Set Exchange, was used for the I atom. The (–PP) notation indicates that relativistic effective core potentials were employed for the core electrons, that is, 1s22s22p63s23p63d10 for I. For the remaining atoms, the Dunning’s triple-zeta correlation-consistent basis set, cc-pVTZ [42], was applied. No symmetry or geometry constraint was imposed during the optimizations. The ultrafine Gaussian integration grid (99,590) was used, and the self-consistent-field convergence was set at 10−8. Frequency calculations were performed at the same theoretical level to ensure that all the structures are genuine minima on the potential energy surface. All of these calculations were carried out with the Gaussian 09 suite of programs [43].
The AIM calculations were performed by the AIM 2000 software [44], using the wave functions generated with M06-2x/cc-pVTZ(–PP). The NCI analysis was undertaken with the Multiwfn program [45] and visualized using the VMD package [46].
Results and discussion
Geometries
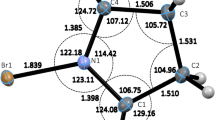
The key geometric parameters of the aryldiyne molecules 1–12 and halogen-bonded dimers 13–16 are summarized in Table 1. The optimized structures of some representative aryldiyne compounds and the dimeric complexes under study are displayed in Fig. 3 and Fig. S1, respectively. As can be seen, the interatomic X···Y distances in the aryldiyne molecules are predicted within the range from 2.895 to 3.074 Å, a reduction of 7.6–16.3 % of the vdW radius sum of the atoms involved [47]. All the intramolecular X···Y interactions in 1–12 are rather linear (<(C–X···Y) > 165°), quite similar to the geometric features of conventional intermolecular XBs [8]. However, the computed interatomic distances in the aryldiyne species are shorter than those in the corresponding halogen-bonded complexes. For example, the I···N separation in 9 is calculated about 0.07 Å less than that in 14 (2.983 vs 3.052 Å), and the reduction in the vdW radius sum amounts to 15.5 and 13.5 %, respectively. Furthermore, as compared to the XB dimers, the interactions in 1–12 to some extent deviate from the linearity. This can be attributed to the rigid 1,2-aryldiyne linkers in these molecules, which restrains the formation of ideal interactions. In addition, the deviation becomes greater as the atomic radius increases; that is, the larger size of halogen σ-hole leads to nonideal XB angles to reduce angle strain in the aryldiyne backbone, opposite to the known characteristics of traditional XBs (cf. Table 1). Notably, the C=O···X angles in 1–6 are estimated to be about 120°, thus indicating the O lone pair electrons as electron donor. This structural feature of XB has been frequently observed in biological molecules [12]. From Table 1, it is also seen that the calculated geometric data of intramolecular X···Y interactions compare fairly well with the X-ray crystallographic data [31], especially the linearity of these bonds.

Optimized structures of four representative aryldiyne compounds 2, 5, 8, and 11. Distances are in angstroms and angles in degrees
As expected, the interatomic X···Y distances in the aryldiyne compounds tend to elongate with the increase in the size and polarizability of halogen atoms. The reduction in the sum of the vdW radii, however, appears to be gradually greater when going down the periodic table, which implies the strength order (I > Br > Cl) of intramolecular X···Y interactions. Furthermore, the attachment of strong electron-withdrawing groups to the donor X atoms results in shorter interatomic separations. For instance, the X···Y distances in 7–9 with the pentafluoro-phenyl moiety bound to halogens are predicted about 0.09 Å less than those in 10–12 where halogen atoms are bonded to the phenyl ring. This trend of the bonding distances was also found in the crystal structures of 2 and 5 (2.751 vs 2.909 Å), in which the donor Br atom is attached to the pentafluoro-phenyl group and the CF3-substituted benzene, respectively. Here, it is worth mentioning that these structural properties of intramolecular X···Y interactions have been well established in conventional intermolecular XBs [8].
As shown in Fig. 3, the formation of intramolecular X···Y interactions in 1–12 yields triangular structures, the so-called quasi-ring that occurs commonly in intramolecular HB cases. The C(X)···Y distances are predicted to be within the range of 4.593–5.138 Å, which indicates the flexibility of the 1,2-aryldiyne bridge that can accommodate chloro, bromo, and iodo substituents. In addition, this nonalkyne side of the triangle also has similar dimension to the other two aryldiyne sides (about 4.1 Å). In light of these findings, flexible aryldiyne linkers provide an ideal template for a variety of intramolecular XB interactions. Notably, the triangular structures maintain almost perfectly planar in all the aryldiyne molecules; the dihedral angles of the two linkers are very close to zero (see Table 1). However, the two subunits in the dimeric complexes are not in the same plane; the dihedral angles of the two alkyne groups amount to about 60°. As a consequence, the rigidity of the aryldiyne bridge between the XB donor and acceptor in 1–12 retains the triangles almost coplanar.
Overall, the combination of flexibility and rigidity allows the 1,2-aryldiyne linkers to template a large variety of intramolecular X···Y interactions that deviate from the linearity to some degree, leading to the creation of coplanar, triangular structures.
ESP analysis
The ESP surfaces of the isolated donor and acceptor molecules in the intermolecular complexes 13–16, together with the most positive surface ESP (V s,max) for the Br and I atoms and the most negative surface ESP (V s,max) for the N atom, are shown in Fig. 4. At the M06-2x/cc-pVTZ(–PP) level of theory, the interaction energies including counterpoise correction for basis set superposition error (BSSE) [48] corrections are computed to be −4.77, −6.84, −3.49, and −5.02 kcal/mol, respectively, for 13–16. Cleary, attractive linear interactions occur between the positive σ-holes of the X atoms and the negative ESP region of the N atom in 13–16. As anticipated, the I atom forms much stronger X···N interactions with respect to the Br atom, due to the larger value of V s,max; the attachment of the stronger electron-withdrawing C6F5 group to the donor X atom leads to greater V s,max and thus stronger X···N interactions. Notably, these strength trends of intermolecular XBs are reproduced in the X···N interactions in 1–12, although these intramolecular bonds exhibit shorter X···N distances and also to some extent deviate from the linearity. These provide strong evidence for intramolecular XB interactions in 1–12.

ESP surfaces of the donor and acceptor molecules, together with V s,max for the X atoms and V s,min for the N atoms. The ESP ranges from negative (red) to neutral (green) and to positive (blue) (Color figure online)
AIM analysis
The AIM theory has been widely employed for charactering and quantifying various kinds of noncovalent interactions [49–52]. The most immediate evidence of weak interactions is the existence of a bond critical point (BCP) accompanied by a bond path linking the donor atom and the acceptor. In particular, the topological parameters at the BCPs, e.g., the electron density (ρ b) and its Laplacian (∇2 ρ b), correlate with the strength of the interactions.
For each of the X···Y interactions in 1–16, a BCP between the donor X and the acceptor O/N atoms has been identified via the AIM method, as shown in Fig. 5 and Fig. S2. The triangular structures in 1–12 are also indicated by a ring CP (yellow point in Fig. 5). The topological parameters (ρ b and ∇2 ρ b) at the BCPs for the studied systems are given in Table 2. It can be seen that 102 ρ b is estimated to vary from 1.135 to 1.879 au for intramolecular X···Y interactions, within the range of HB suggested by Koch and Popelier (0.2–3.5 au) [49, 50]. In addition, all the values of ∇2 ρ b are calculated to be positive, thereby suggesting the typical closed-shell kind of interactions. Table 2 also includes the energetic properties at the BCPs, i.e., local one-electron kinetic energy density G(r), local potential energy density V(r), and the electronic energy density H b [kinetic G(r) plus potential V(r) energy density]. The sign of H determines whether the interaction is electrostatic dominant (H > 0) or covalent dominant (H < 0) [52]. As evident from this table, H b is computed to be positive for all intramolecular X···Y interactions in 1–12, which implies the electrostatic character of these bonds. Note that the AIM properties of intramolecular X···Y interactions reported here behave very much like those of well-studied traditional XBs [53].

Molecular graphs of four representative aryldiyne molecules 2, 5, 8, and 11. The electron densities at the BCPs are in au
It has been well documented that ρ b at the BCPs can serve as a convenient measure for different types of HB or XB interactions with a wide range of strength [49, 54]. Not surprisingly, larger values of ρ b are predicted for the aryldiyne molecules with respect to the corresponding halogen-bonded dimers, consistent with the shorter X···Y distances in the former cases (vide supra). Therefore, intramolecular X···Y interactions become stronger in strength than corresponding intermolecular XBs. As expected, ρ b increases with the enlargement of the atomic size of halogens, thus indicating the strength order (I > Br > Cl) of these bonds. Furthermore, the presence of strong electron-withdrawing groups into the donor X atoms gives rise to greater values of ρ b. For example, 102 ρ b is calculated to be 1.879 au for the compound 9 in which the I atom is attached to the pentafluoro-phenyl moiety, much larger than that for 12 (1.607 au) with the I atom bound to the phenyl ring. In addition, the computed ρ b for X···N interactions is higher than that for corresponding X···O bonds, which implies stronger X···N interactions in the studied molecules. These trends of the AIM results are in line with those observed in the interatomic X···Y distances.
As expected, the interaction energies follow a good linear relationship with the values of ρ b at the BCPs for the four XB dimers under study, as shown in Fig. S3. In light of the correlation equation established in this figure, the bonding energies of the intramolecular interactions in 1–12 can be roughly evaluated, approximately in a magnitude of −3.0 to −9.0 kcal/mol. This indicates medium-strong intramolecular bonds, in accordance with the positive ∇2 ρ b and H b at the BCPs.
NCI analysis
The NCI index is based on the relationship between the electron density ρ(r) and the reduced density gradient s, which is expressed as:
It allows isosurfaces of the reduced density gradient at low densities to visualize the position and nature of noncovalent interactions in 3D space [55]. Regions with low electron density ρ(r) and reduced density gradient s correspond to the occurrence of noncovalent interactions. This method has been recently used to study intramolecular HBs, because it can overcome some limitations of the AIM theory and thus provides a more global description of noncovalent bonding [56].
The NCI isosurfaces of the intramolecular system 11 and the intermolecular complex 15 are illustrated in Fig. 6, where strong attractive interactions are represented in blue, weak interactions in green, and repulsive interactions in red. Both images obviously indicate a typical Br···N interaction (green in color). However, in the molecule 11 a second weak interaction is evident from the green isosurface between the donor Br and the H atom in the quinolinyl group, consistent with the existence of a BCP between these two atoms within the AIM analysis (cf. Fig. 6). In addition, a hollow disk-shaped shadow is also presented in this molecule, which is related to the ring CP for the triangle.

NCI isosurfaces and plots of the reduced density gradient versus the electron density multiplied by the sign of the second Hessian eigenvalue for 11 and 15
To provide a better understanding of chemical bonding, the Laplacian is often decomposed into the contributions along the three principal axes of maximal variation, \(\nabla_{\rho }^{2}\) = λ 1 + λ 2 + λ 3. A negative value of λ 2 suggests attractive interaction (with an accumulation of density perpendicular to the bond), while positive λ2 indicates steric repulsion (density depletion). The plots of s and the sign of second eigenvalue λ 2 for 11 and 15 are also given in Fig. 6. Clearly, the first low-gradient spike lying at negative value indicates the Br···N interaction, while the second spike at negative sign of λ 2 corresponds to the secondary weak Br···H interaction. The peaks located at positive values indicate the repulsive steric interactions due to ring formation. In general, the plot of 11 is not qualitatively different from that of 15, except that the first peak in 11 is deeper still and resides at more negative value of λ 2. This suggests stronger intramolecular X···Y interactions with respect to corresponding intermolecular ones coincide with the geometric and AIM results as mentioned above.
CSD survey
The CSD is a convenient and reliable storehouse for structural information. The small molecule crystallography and the CSD have been widely used for analyzing geometric features of noncovalent interactions. To gain more insights into intramolecular XB interactions in crystals, a survey of the CSD (version 5.35, updates May 2014) was undertaken herein. We only considered crystal structures with no disorder and no errors as well as R-factor less than 0.1. The following search criteria were utilized: (1) Both the XB donors and acceptors are within the same molecule; (2) the interatomic X···Y (X = Cl, Br, I and Y = O, N, S) distances are shorter than the sum of the vdW radii of the X and Y atoms; (3) the interaction angles <(C−X···Y) are larger than 150°. According to our survey of the CSD, 34 X-ray crystal structures containing intramolecular X···O interactions and one crystal structure with intramolecular Cl···N bond were exacted, as summarized in Supplementary material.
Three representative crystal structures, SURXUZ [57], YOBXIX [58], and ENIGEN [59], are selected and depicted in Fig. 7. In the crystal structure of SURXUZ, the Cl atom in the dinitrobenzoate molecule established a intramolecular XB with the O atom in the nitro moiety (d(Cl···O) = 3.270 Å, <(C–Cl···O) = 171.4°). In the YOBXIX structure, a intramolecular Br···O interaction (d(Cl···O) = 3.033 Å, <(C–Cl···O) = 161.2°) is formed in the bromo-nitrobenzoate molecule. In the crystal structure of ENIGEN, the hypervalent I atom in the o-nitrodiphenyliodonium compound is involved in a intramolecular I···O bond (d(I···O) = 2.735 Å, <(C–Cl···O) = 157.5°) with the O atom in the nitro group. These intramolecular bonds, although completely unrealized by the original authors, play a central role in the conformations of halogenated molecules in crystals.

Selected fragments of the three selected crystal structures. Distances are in angstroms and angles in degrees
Present database includes 29 % Cl···O, 21 % Br···O, and 50 % I···O interactions with the average interatomic distances of 3.15, 3.18, and 2.91 Å, respectively. The much shorter average I···O distance can be ascribed to the fact that the donor I atoms in most of the iodinated systems are of hypervalent iodine, i.e., C–I+–R (see Fig. 7), which is capable of forming much stronger interactions with electronegative atoms. Notably, the average interaction angle for all the X···O bonds retrieved from the CSD amounts to about 164°, thus indicating the rather good linearity of these interactions in solid state. This geometric feature is quite different from intramolecular HBs with a wide range of interaction angle, which is reminiscent of the obvious distinction between conventional XB and HB interactions [8].
In a word, despite a small number of crystal structures, intramolecular XBs exhibit a good linearity in crystals and contribute significantly to the stability of relevant halogenated compounds.
Implications for molecular design
According to the above analyses, intramolecular X···Y interactions show the approximately same kind of geometric, energetic, AIM, and NCI behaviors as conventional intermolecular XBs. These interactions, consequently, can be classified as XBs. However, as compared to corresponding intermolecular XBs, intramolecular XB interactions to some extent deviate from the linearity and become somewhat stronger in strength. The rigidity of the 1,2-aryldiyne bridge keeps the molecule almost in the same plane, thereby reducing the steric hindrance between the aryldiyne moieties. On the other hand, the formation of intramolecular XB interactions gives rise to the triangular structures in the molecules. The resulting coplanar triangles held the donor X and the acceptor O/N atoms in more close proximity, leading to shorter interatomic distances and hence relatively stronger interactions.
Similar to traditional XBs, intramolecular XB interactions are mainly electrostatic in nature. Particularly, several well-known trends of conventional XBs are reproduced in intramolecular XB systems. For instance, the interaction strength increases with the enlargement of atomic radius (I > Br > Cl), and the attachment of electron-withdrawing groups to the donor X atoms results in much stronger interactions. As a consequence, the well-established properties of traditional XBs could be used to tune intramolecular XB interactions. In addition, as compared to intramolecular HBs with a wide range of interaction angle, intramolecular XB interactions are rather linear in both gas phase and solid state.
As noted above, intramolecular XBs have a pronounced influence on the conformations of the molecules which are closely related to their function. These interactions, accordingly, can be employed as structure-promoting elements in designed molecular systems that show biological activity or synthetic function. Furthermore, the properties of these interactions can be readily tuned by the common tools applied in conventional XBs, which would be very useful in the conformational switching of functional molecules. Considering the importance of intramolecular XBs in molecular design and crystal engineering, much more future efforts should be devoted to exploring these interactions from physical characterization to synthetic application.
Conclusions
In the present work, DFT calculations using the M06-2x method have been performed to investigate intramolecular halogen bonds in a series of aryldiyne compounds, which were recently characterized via single-crystal X-ray crystallography. For comparison, some halogen-bonded dimers of halogenated aryldiynes with quinolinyl compounds were also calculated. The combination of flexibility and rigidity allows the 1,2-aryldiyne linkers to template a large variety of intramolecular XB interactions, leading to the creation of coplanar, triangular structures. These interactions, although somewhat stronger in strength, have similar geometric, energetic, AIM, and NCI characteristics to conventional intermolecular XBs. Therefore, these bonds can be recognized as XBs that are mainly electrostatic in nature. Particularly, these interactions can be readily tuned by the common tools used in traditional XBs, which would be very useful in the conformational switching of functional molecules.
According to the survey of the CSD, a small number of crystal structures involving intramolecular XBs were extracted. These interactions exhibit a rather good linearity in crystals and contribute significantly to the stability of relevant halogenated compounds. Intramolecular XBs thus can be employed as structure-promoting elements in designed molecular systems that show biological activity or synthetic function. We hope that the results reported in this work will assist in the characterization of intramolecular XB interactions in catalyst design and function as well as in modulation of photophysical and electronic structure of functional materials.
References
Jeffrey G (1997) An introduction to hydrogen bonding. Oxford University Press, New York
Scheiner S (1997) Hydrogen bonding: a theoretical perspective. Oxford University Press, New York
Desiraju G, Steiner T (1999) The weak hydrogen bond, in structural chemistry and biology. Oxford University Press, Oxford
Steiner T (2002) Angew Chem Int Ed Engl 41:49
Priimagi A, Cavallo G, Metrangolo P, Resnati G (2013) Acc Chem Res 46:2686
Meyer F, Dubois P (2013) CrystEngComm 15:3058
Aakeroy CB, Baldrighi M, Desper J, Metrangolo P, Resnati G (2013) Chem Eur J 19:16240
Metrangolo P, Neukirch H, Pilati T, Resnati G (2005) Acc Chem Res 38:386
Sun A, Lauher JW, Goroff NS (2006) Science 312:1030
Rissanen K (2008) CrystEngComm 10:1107
Metrangolo P, Meyer F, Pilati T, Resnati G, Terraneo G (2008) Angew Chem Int Ed 47:6114
Auffinger P, Hays FA, Westhof E, Ho PS (2004) Proc Natl Acad Sci USA 101:16789
Bauzaa A, Alkorta I, Frontera A, Elguero J (2013) J Chem Theory Comput 9:5201
Murray JS, Politzer P (2011) WRIES Comput Mol Sci 1:153
Politzer P, Murray JS, Clark T (2010) Phys Chem Chem Phys 12:7748
Murray JS, Riley KE, Politzer P, Clark T (2010) Aust J Chem 63:1598
Politzer P, Riley KE, Bulat FA, Murray JS (2012) Comput Theor Chem 998:2
Politzer P, Murray JS (2013) ChemPhysChem 14:278
Politzer P, Murray JS, Clark T (2013) Phys Chem Chem Phys 15:11178
Politzer P, Murray JS, Clark T (2015) J Mol Model 21:52
Beale TM, Chudzinski MG, Sarwar MG, Taylor MS (2013) Chem Soc Rev 42:1667
Lim JY, Beer PD (2015) Chem Commun 51:3686
Langton MJ, Robinson SW, Mar ques I, Felix V, Beer PD (2014) Nat Chem 6:1039
Nagy PI (2012) J Phys Chem A 116:7726
Martinez-Cifuentes M, Weiss-Lopez BE, Sanos LS, Araya-Maturana R (2014) Molecules 19:9354
Charisiadis P, Kontoqianni VG, Tsiafoulis CG, Tzakos AG, Siskos M, Gerothanassis IP (2014) Molecules 19:13643
Solha DC, Barbosa TM, Viesser RV, Rittner R, Tormena CF (2014) J Phys Chem A 118:2794
Nagy PI (2013) J Phys Chem A 117:2812
Sanchez-Sanz G, Trujillo C, Alkorta I, Elguero J (2012) Comput Theor Chem 991:124
Trujillo C, Sanchez-Sanz G, Alkorta I, Elguero J, Mo O, Yanez M (2013) J Mol Struct 1048:138
Widner DL, Knauf QR, Merucci MT, Fritz TR, Sauer JS, Speetzen ED, Bosch E, Bowling NP (2014) J Org Chem 79:6269
Thorson RA, Woller GR, Driscoll ZL, Geiger BE, Moss CA, Schlapper AL, Speetzen ED, Bosch E, Erdelyi M, Bowling NP (2015) Eur J Org Chem 8:1685
Palusiak M, Grabowski SJ (2008) Struct Chem 19:5
Jablonski M (2012) J Phys Chem A 116:3753
Johansson MP, Swart M (2013) Phys Chem Chem Phys 15:11543
Murray JS, Concha MC, Politzer P (2011) J Mol Model 17:2151
Bader RFW (1990) Atoms in molecules: a quantum theory. Oxford University Press, New York
Johnson ER, Keinan S, Mori-Sanchez P, Contreras-Garcia J, Cohena AJ, Yang W (2010) J Am Chem Soc 132:6498
Zhao Y, Truhlar DG (2008) Theor Chem Acc 120:215
Zhao Y, Truhlar DG (2008) Acc Chem Res 41:157
Peterson KA, Shepler BC, Figgen D, Stoll H (2006) J Phys Chem A 110:13877
Dunning TH Jr (1989) J Chem Phys 90:1007
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Peralta Jr JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2009) Gaussian: Wallingford
Biegler-Konig F, Schonbohm J, Bayles D (2001) J Comput Chem 22:545
Lu T, Chen F (2012) J Comput Chem 33:580
Humphrey W, Dalke A, Schulten K (1996) J Mol Graphics 14:33
Bondi A (1964) J Phys Chem 68:441
Boys SF, Bernardi F (1970) Mol Phys 19:553
Koch U, Popelier PLA (1995) J Phys Chem 99:9747
Popelier PLA (1998) J Phys Chem A 102:1873
Grabowski SJ (2000) J Phys Chem A 104:5551
Alkorta I, Rozas J, Elguero J (1998) J Phys Chem A 102:9278
Lu YX, Zou JW, Wang YH, Jiang YJ, Yu QS (2007) J Phys Chem A 111:10781
Lu YX, Zou JW, Wang YH, Yu QS (2006) J Mol Struct Theochem 776:83
Contreras-Garcia J, Johnson ER, Keinan S, Chaudret R, Piquemal J-P, Beratan DN, Yang W (2011) J Chem Theory Comput 7:625
Lane JR, Contreras-Garcia J, Piquemal J-P, Miller BJ, Kjaergaard HG (2013) J Chem Theory Comput 9:3263
Hashimoto T, Sakata K, Maruoka K (2009) Angew Chem Int Ed 48:5014
Bee C, Han SB, Hassan A, Lida H, Krische MJ (2008) J Am Chem Soc 130:2746
Nikiforov VA, Karavan VS, Miltsov SA, Selivanov SI, Kolehmainen E, Wegelius E, Nissinen M (2003) Arkivoc 4:191
Acknowledgments
This work was supported by the National Natural Science Foundation of China (21473054).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zhang, Y., Lu, Y., Xu, Z. et al. Intramolecular halogen bonds in 1,2-aryldiyne molecules: a theoretical study. Struct Chem 27, 907–917 (2016). https://doi.org/10.1007/s11224-015-0671-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-015-0671-z