
Abstract
Finite saturated regular carbon nanotubes (CNTs) are predicted to exhibit higher capacity as hydrogen storage media compared to unsaturated regular CNTs. In the present study, molecular hydrogen physisorption energies (MHPEs) for finite saturated and unsaturated bumpy defected CNTs were calculated by density functional theory (DFT-D3) methods at the B3LYP/6-31G(d) theory level, with rigorous inclusion of van der Waals interactions. The calculated MHPEs for both regular and bumpy defected armchair, chiral and zigzag CNTs with similar diameters and lengths, with and without nitrogen doping, were compared in terms of Eph/H2, defined as the MHPE per hydrogen molecule adsorbed inside the nanotube. For all studied systems, Eph/H2 increased with the number of physisorbed hydrogen molecules. Nitrogen doping of regular and bumpy CNTs resulted in an increase in the Eph/H2 values, with the exception of bumpy chiral nanotubes. The results of this study demonstrate that bumpy defects are important nanotube structural features whose effects depend on nanotube chirality. For instance, bumpy defects were beneficial for undoped and doped zigzag nanotubes, resulting in a decrease in Eph/H2 values for regular structures from 0.5 and 0.74 to 0.26 and 0.42 eV, respectively. By contrast, for doped armchair regular structures with an Eph/H2 value of 0.38 eV, bumpy defects increased Eph/H2 to 0.45 eV. These Eph/H2 values for bumpy doped armchair and the zigzag nanotubes are all within the range of 0.1–0.5 eV/H2 reported as ideal for reversible hydrogen storage under environmental conditions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Carbon nanotubes (CNTs) are interesting carbon structures because their properties are compatible with many applications in various fields [1–7]. The properties of CNTs depend on their molecular structure. The structure of nanotubes can change depending on the arrangement of carbon atoms, described as chirality. Nanotube diameter and length also affect the properties of CNTs. Structural changes affect the surface area, local charge distribution, radius of curvature of CNTs and, consequently, the interaction of CNTs with other structures. For example, CNTs can incorporate other molecules and have consequently been assessed as drug carriers [8, 9] or hydrogen storage materials [10–22].
Several studies, some of them controversial [19, 23–25], have concluded that the structure of nanotubes allows both hydrogen chemisorption and physisorption, achieving values near those recommended by the U.S. Department of Energy (DOE) [26]. However, the consensus of the analysis of hydrogen uptake by CNTs is that CNTs have a very small storage capacity at room temperature. For economical use, the content of adsorbed hydrogen must be improved while allowing desorption of hydrogen at room temperature and moderate pressure. Thus research is needed on new structural features that facilitate both requirements.
In addition to their regular structure, nanotubes can exhibit defective structures. Regular structures include only six-membered rings in specific arrangements (also called chirality) that give rise to armchair, zigzag and chiral nanotubes. Defects appear in nanotubes that contain other ring sizes, e.g., five-, seven- or eight-membered rings [27]. Depending on the distribution of these rings, bumpy-type, zipper, multizipper and Stone–Wales nanotubes may form, among other defects [28]. For example, nanotubes with bumpy-type defects generated by the presence of five- and seven-membered rings, which produce a change in the curvature of the nanotube and the inner and outer surfaces, have a higher local volume at the defect site. Consequently, the properties of nanotubes with defects are likely different from those of regular nanotubes.
We previously demonstrated that hydrogenation and the presence of N increased the potential of regular nanotubes as hydrogen storage media [29, 30]. We then wished to investigate the properties of bumpy nanotubes and how the presence of defects (five- and seven-membered rings) contributes to the potential of nanotubes as a hydrogen storage material.
Hydrogen physisorption energies were determined for bumpy defected nanotubes with hydrogenation and with different chirality in the presence or absence of N via density functional theory (DFT) calculations at B3LYP/6-31G(d) level. These energies were compared with the hydrogen physisorption energies for the respective regular nanotubes calculated under similar conditions. In both cases, the dispersion forces, which represented a significant component of the total energy, were evaluated.
The hydrogenated armchair and zigzag nanotubes, with or without N, with bumpy defects exhibited a hydrogen physisorption energy within the range of 0.1–0.4 eV/molecule considered ideal for reversible hydrogen storage [12, 13, 31].
Methods
Four categories of nanotube models were built [28]: (i) regular CNTs; (ii) bumpy defected CNTs (hereafter referred to as bumpy nanotubes); (iii) exo-hydrogenated or saturated regular CNTs; and (iv) exo-hydrogenated or saturated bumpy CNTs. The carbon atoms on nanotubes in categories (i) and (ii) are sp 2 hybridized, whereas the carbon atoms on nanotubes in categories (iii) and (iv) are sp 3 hybridized. The four categories were nitrogen doped or undoped. For each category, the nanotube chirality could be zigzag (n,0), chiral (n,m) or armchair (n,n) [27, 32].
Each individual structure and the systems containing molecular hydrogen inside the nanotubes were optimized via an initial AM1 (Austin Model 1) optimization as implemented in the program Hyperchem [33], followed by a subsequent DFT optimization step over the AM1-optimized geometry by means of the program Jaguar v8.1 [34] at the level B3LYP/6-31G(d). For a better understanding, Fig. 1 shows the optimized structure of a C164N4H210 saturated bumpy nanotube, depicting the distribution of physisorbed H2 molecules. Real harmonic vibrational frequencies at the same level of theory indicated that all of the structures considered were at a minimum. Finally, a DFT-D3 van der Waals correction was performed using the approach of Grimme et al. [35, 36], which has been proven to be a very accurate method compared with other approaches for the determination of dispersion forces [37]. This methodology was validated through DFT-non-local methods employed in the evaluation of hydrogen physisorption energies for some regular, nitrogen-doped CNTs in a previous work [30]. No symmetry constraints were imposed. Different initial geometries gave final results with no significant final differences, probably because the initial AM1 optimization arranges H2 molecules in a similar way regardless of the input distribution: H2 molecules are arranged along the nanotube axis with a higher number of H2 molecules in the bumpy region as shown in Fig. 1. In regular nanotubes, several H2 molecules were often launched outside the nanotube. In that case, a smaller number of H2 molecules were used for those nanostructures. So, results are perceived as representative.

Representation of the B3LYP/6-31G(d) optimized structure of a saturated bumpy nanotube with 15 H2 molecules inside (C164N4H210), showing the final distribution of the physisorbed H2 molecules colored in red for better visualization. Lateral and front views (Color figure online)
The hydrogen physisorption energy for one hydrogen molecule, Eph/H2, was calculated according to the following expression (1):
where \(E_{{({\text{nanotube}}\, + \,h{\text{H}}_{2} )}}\) and E (nanotube) are the total energies of the saturated CNTs with and without physisorbed molecular hydrogen, respectively; h is the number of physisorbed hydrogen molecules inside the nanotube; and EH2 is the total energy of a hydrogen molecule. These parameters were all calculated at the same level of DFT [B3LYP/6-31G(d)]. vdW is the contribution of dispersion energy from the van der Waals interactions among hydrogen molecules and nanotube systems, as calculated by the DFT-D3 method implemented in Jaguar v8.1. A positive value of Eph/H2 indicates that the hydrogen physisorption process is endothermic.
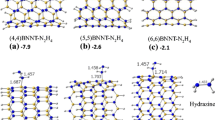
The notation used in this work first specifies the Hamada indices for indicating chirality [(zigzag (n,0); chiral (n,m); armchair (n,n)], followed by the number of nitrogen atoms in the nanotube (pyrimidinic nitrogens, i.e., in a six-membered ring comprising N1–C2–N3–C4–C5–C6, as shown in Fig. 2). The third component of the notation denotes the hydrogenation state. “H” indicates a fully exo-hydrogenated nanotube. The absence of “H” indicates an unsaturated nanotube (sp 2 hybridization). “B” indicates bumpy nanotubes. “R” is used for regular nanotubes. Occasionally, FB indicates a fully bumpy nanotube, whereas PB indicates a partially bumpy nanotube, as shown in Fig. 3. Finally, the number of carbon-atom layers defining the nanotube length is indicated. For example, the notation (5,5)-4N-H-B-15mol-16cl indicates an armchair (5,5) nanotube with four nitrogen atoms (located in the central part of the nanotube) that is saturated and has bumpy defects (fully), 15 physisorbed H2 molecules and 16 carbon-atom layers as represented in Fig. 4.

Numbering of both the six-membered ring with the pyrimidine disposition N1–C2–N3–C4–C5–C6 present in the N-CNTs and the five-membered ring as used in Table 1

Structures of a a FB nanotube with four bumpy defects and any six-membered rings around the defect zone comprising coupled five-membered rings only and b a PB nanotube with two bumpy defects exhibiting some six-membered rings in the nanotube defect zone

Schematic representation of the specific nanotube notation employed in this work
Results and discussion
In this section, the structural characteristics of the CNTs, such as C–C and C–N distances, angle values and net charges on each atom of the pyrimidine rings contained in the N-CNTs, and the hydrogen physisorption energies for saturated and non-saturated, regular and bumpy, doped and undoped CNTs are presented. For clarity, Fig. 5 presents the types of nanostructures studied and their structural relationships.

Structural relationships of the different molecular nanostructure types studied in this work
Structural features
Saturated regular CNTs are stable compounds, particularly exo-hydrogenated structures [10], and most of the regular and bumpy nanotubes studied in this work are stable as well. Table 1 presents the C–N and C–C distance values for both pyrimidine rings in non-saturated and saturated (partial and full) bumpy doped nanotubes. Chiral and zigzag saturated FB structures that contain nitrogen atoms in two imidazole rings (instead of pyrimidine rings) are also considered (the last two rows of Table 1).
The optimized, non-saturated PB N-CNTs exhibited 1.42 Å average C–C distances, in agreement with both the C–C bond lengths in graphene (1.42 Å) and the average value of the C–C distance in optimized [B3LYP/6-31G(d)] non-saturated regular nanotubes [38, 39]. The average C–N bond lengths of 1.40–1.43 Å are in agreement with the 1.42 Å average C–N bond length reported for regular armchair N-CNTs [40]. The average C2–N bond lengths (C–N Av1 in Table 1) were shorter than the other average C–N bond lengths (C–N Av2 in Table 1) for non-saturated N-CNTs, indicating that the C2–N bond is stronger than the rest of the C–N bonds in these structures. The armchair and chiral non-saturated PB nanotubes exhibited C2–N bond length values very close to the X-ray crystal structure values of 1.341 and 1.335 Å for C2–N bond lengths in a pyrimidine derivative [41]. Bond lengths between 1.375–1.383 and 1.377–1.379 Å for C–C bonds and between 1.322–1.353 and 1.329–1.344 Å for C–N bonds have been reported for pyrimidine rings in crystallized derivative structures [42]. The saturated bumpy N-CNTs, including PB and FB structures with pyrimidine or imidazole rings, exhibited average C–C bond lengths of 1.57 Å and average C–N bond lengths of 1.47–1.49 Å. These values are expected for sp 3 hybridization [a typical (sp 3)C–C(sp 3) bond length is 1.54 Å; the C–N bond length for amines is 1.479 Å] and are similar to those of saturated regular N-CNTs [39]. The H–H bond length for physisorbed H2 molecules of 0.733 Å, on average, was almost 0.010 Å shorter than the H–H bond length for a free H2 molecule.
Table 2 presents the corresponding angles for all atoms of both pyrimidine rings for non-saturated and saturated bumpy nanotubes and imidazole rings for saturated chiral and zigzag bumpy N-CNTs. The non-saturated structures exhibited N1–C2–N3, C–N–C, N–C–C and C–C–C angles of approximately 116°–122°. These values are consistent with those reported for pyrimidine rings of crystallized derivative structures [42] and with the expected values for sp 2 hybridization. The saturated structures exhibited N1–C2–N3, C–N–C, N–C–C and C–C–C angles of 100°–120°. The C2–N3–C4 angles in both six-membered rings exhibited lower values of approximately 100°–103°, smaller than the expected values for sp 3 hybridization. The saturated FB nanotubes containing imidazole rings instead of pyrimidine rings exhibited much smaller angles compared to the corresponding six-membered rings.
The net charges of the N1, C2 and N3 atoms in each pyrimidine and imidazole ring in the nanotube structures as well as their dipole moments are presented in Table 3. The net charges did not differ significantly for nitrogen atoms located in six-membered or five-membered rings. Chirality also did not appear to affect net charge atomic values. The net charge of the C2 atom decreased by half when non-saturated structures were compared with saturated structures, regardless of whether the saturated nanotubes were PB, FB or had imidazole rings. The charges of the N1, C2 and N3 atoms in both non-saturated bumpy pyrimidine rings did not differ significantly from those in the corresponding regular nanotubes [39]. That is, neither the presence of a bumpy defect itself in a nanotube nor the presence of five-membered rings necessarily altered the net atomic charges or the dipole moments, which remained less than 3.7 D for the different structures, as shown in Table 3.
A bumpy defect itself did not significantly change the atom–atom distances, angles or net atomic charges of the atoms present in the defect region of the nanostructure or the molecular dipole moment. The only exception was saturated structures, which exhibited angles smaller than the expected value for sp 3 hybridization, with no significant differences in charge.
Hydrogen physisorption energies
The term physisorption energy refers to the interaction energy between molecular hydrogen and the nanotube without the formation of a covalent bond or any associated carbon hybridization change. In this work, the only molecular hydrogen positions considered were inside the nanotube, and no molecular hydrogen was located on the external nanotube surface. The saturated nanotubes considered here were full exo-hydrogenated finite open full bumpy hydrogen-terminated nanotubes.
To analyze the ability of bumpy defects to enhance the hydrogen storage capability of the nanotubes, the hydrogen physisorption energy associated with a single hydrogen molecule Eph/H2, was calculated for different bumpy nanotubes containing different numbers of hydrogen molecules according to Eq. (1), taking into account the dispersion forces due to van der Waals interactions [35, 36]. The DFT-D3 method for calculating van der Waals interactions used in this work has been reported to be very precise compared with other methods used to perform the same calculations [37].
Figure 6 presents the Eph/H2 values for both regular and bumpy defected saturated armchair (5,5), chiral (6,3) and zigzag (8,0) CNTs with seven physisorbed H2 molecules. We have used seven physisorbed H2 molecules for practical reasons because for that condition, it was possible to obtain the data necessary to compare the hydrogen physisorption energies for regular and bumpy nanotubes with different chirality. Some regular nanotubes are not able to physisorb more than seven H2 molecules. The nanotubes become “saturated” with a much lower number of H2 molecules than bumpy analog structures. For instance, regular (5,5)-0N-H-R-8cl nanotube is not able to physisorb more than 7 hydrogen molecules compared to the more than 18 hydrogen molecules physisorbed by their bumpy counterpart, (5,5)-0N-H-B-8cl nanotube. The regular nanotubes considered in Fig. 6 had nearly identical diameters (7.46–7.50 Å) and lengths (eight carbon-atom layers in each nanotube: 8.8, 7.2 and 6.9 Å for armchair, chiral and zigzag nanotubes, respectively). The armchair and zigzag bumpy nanotubes exhibited lengths of 8.4 and 8.1 Å, respectively, whereas the chiral bumpy structure exhibited a length of 11.7 Å. The columns in white represent data for undoped nanotubes, and the columns in red represent data for nanotubes doped with four nitrogen atoms in each nanotube.

Representation of the dispersion-corrected B3LYP/6-31G(d)-calculated Eph/H2 for both a regular armchair (5,5), chiral (6,3) and zigzag (8,0) nanotubes; and b bumpy armchair (5,5), chiral (6,3) and zigzag (8,0) nanotubes, all featuring eight carbon-atom layers and seven physisorbed hydrogen molecules inside. White and red columns represent undoped and nitrogen-doped structures, respectively (Color figure online)
Doped and undoped regular nanotubes with different chirality (Fig. 6a) exhibited similar relative behaviors: All of the nitrogen-doped regular CNTs exhibited higher physisorption energies than the undoped regular nanotubes, and the energies of both doped and undoped CNTs increased in the order armchair < chiral < zigzag.
Nearly identical behavior was observed for the armchair (5,5) and zigzag (8,0) bumpy CNTs (Fig. 6b), i.e., they exhibited higher physisorption energies when nitrogen doped. In these nanostructures, nitrogen probably enhances molecular local polarizability, in turn enabling higher hydrogen physisorption. This effect was observed in most nitrogen-doped CNTs. By contrast, the undoped chiral (6,3) bumpy CNTs exhibited very high hydrogen physisorption energies compared with the corresponding nitrogen-doped CNTs. The nanotube curvature and geometrical distribution of both nitrogen and carbon atoms in the doped chiral structures, together with the bumpy defect, probably allow these CNTs to behave as a flexible or weak hydrogen host: Hydrogen can easily enter and exit the nanotubes. By contrast, the undoped chiral bumpy nanotubes behaved like a harder host because once hydrogen is adsorbed, higher energy is required for desorption. These results indicate that the bumpy defect, and not the presence of nitrogen, seems to be the main structural feature affecting the hydrogen host behavior of chiral nanotubes. One manifestation of the bumpy defect in chiral nanotubes is exhibited in the length of the nanotube. Chiral nanotubes are significantly longer than armchair and zigzag nanotubes with all of the same number of carbon layers, as was shown previously.
The data in Fig. 6 demonstrate that a bumpy defect is an important nanotube structural feature whose effects differ depending on nanotube chirality. For instance, bumpy defects were beneficial for undoped and doped zigzag (8,0) nanotubes and resulted in decreases in the Eph/H2 values for regular nanotubes from 0.5 and 0.74 to 0.26 and 0.42 eV, respectively. By contrast, for the armchair (5,5) doped regular nanotubes, which had an Eph/H2 value of 0.38 eV, the bumpy defect increased that value to 0.45 eV; although both values and those for the zigzag bumpy nanotubes are all within the ideal range of 0.1–0.5 eV/H2 for reversible hydrogen storage under environmental conditions [31, 43] and in agreement with the range of 0.16–0.4 eV/molecule reported from thermodynamic estimations of hydrogen nanotube binding energy [12, 13, 44].
To further characterize the effect of a full round of bumpy defects (called FB) around the central part of the nanotube versus a pair of bumpy defects (one defect in front of the other, called PB) in that region, the frontier orbitals HOMO (highest occupied molecular orbital) and LUMO (lowest unoccupied molecular orbital) for doped FB and PB zigzag nanotubes are presented in Fig. 7. This analysis again clearly highlights the importance of the bumpy defect: Most of the atomic orbitals contributing to the frontier orbitals belong to atoms in the bumpy defect zone. The contribution of nitrogen orbitals is less important.

Frontier orbitals HOMO (a, c) and LUMO (b, d) for optimized FB (a, b) and PB (c, d) zigzag (8,0) N-CNTs with seven physisorbed hydrogen molecules inside. For each orbital, the lateral and front views are shown in the upper and lower parts, respectively. Nitrogen is contained in a five-membered ring
In the bumpy chiral (6,3) and zigzag (8,0) nanostructures, N is located in five-membered rings (as described in Fig. 2). The Eph/H2 values of doped and undoped regular zigzag nanotubes (Fig. 6a) with nitrogen located in six-membered rings and those of doped and undoped bumpy zigzag nanotubes (Fig. 6b) with nitrogen in five-membered rings followed the same trend: Doped zigzag nanostructures had higher Eph/H2 values than undoped zigzag nanotubes. Bumpy chiral nanotubes, however, behaved differently, as discussed previously. The frontier orbitals of the FB and PB chiral N-CNTs (see Fig. 8) also exhibited a different pattern compared with the zigzag N-CNTs, explaining in part the observed chiral nanotube behavior. Only a few of the atoms of FB chiral N-CNT contribute to the HOMO (Fig. 8a), whereas for the PB chiral N-CNT (Fig. 8c), nearly all of the atoms associated with the bumpy defect contribute to the HOMO. A similar trend was observed for LUMO orbitals (Figs. 6d, 8b). The (n,m) chiral CNTs are special structures. As a function of their relative n, m values, the structures can behave as semiconductors or as conductors if n minus m results in a number divisible by 3.

Frontier orbitals HOMO (a, c) and LUMO (b, d) for optimized FB (a, b) and PB (c, d) chiral (6,3) N-CNTs with seven physisorbed hydrogen molecules inside. For each orbital, the lateral and front views are shown in the upper and lower parts, respectively. Nitrogen is contained in a five-membered ring
Data in Fig. 9 depict the hydrogen physisorption energies for doped and undoped armchair (4,4) nanotubes with 8 and 12 carbon-atom layers with 2–24 molecules of physisorbed hydrogen molecules.

Representation of the molecular hydrogen physisorption energy per adsorbed H2 molecule for hydrogenated armchair (4,4) nanotubes as a function of the number of physisorbed hydrogen molecules calculated at the B3LYP/6-31G(d) level considering dispersion forces correction. The filled and empty forms are nitrogen-doped and undoped nanotubes, respectively. Square and triangle forms are 12 carbon-atom layer regular and bumpy nanotubes, respectively. Circle forms are the eight carbon-atom layer bumpy structures
As shown in Fig. 9, the Eph/H2 value increases with the number of physisorbed hydrogen molecules. This effect was more relevant for shorter bumpy defected structures (8 carbon-atom layer) and for regular structures. Nanotube nitrogen doping also clearly increased the value of Eph/H2, particularly for regular structures. By contrast, the bumpy defect in nanotube structures caused a significant decrease in the Eph/H2 values in a more significant manner than for the doped nanotubes. This effect was more relevant for nanotubes with a higher number of physisorbed H2 molecules. For instance, the Eph/H2 values for doped regular and doped bumpy defected nanotubes containing 15 physisorbed H2 in each were 0.82 and 0.35 eV, respectively, thus differing by 0.47 eV. The corresponding Eph/H2 values for undoped structures were 0.62 and 0.29 eV, respectively, resulting in a difference of 0.33 eV. As shown in Fig. 9, a higher number of carbon-atom layers in the nanostructures also decreased Eph/H2 significantly. These data suggest that bumpy defected armchair (4,4) nanotubes (with or without nitrogen doping) with a 12 carbon-atom layer and 10–15 physisorbed H2 molecules will behave as an ideal system for reversible hydrogen storage as a clean source of energy due to their Eph/H2 values in the range of 0.1–0.4 eV [12, 13, 44]. Figure 10 shows a comparison between the two 12 carbon-atom layer nanotubes and a longer one of a 20 carbon-atom layer. All of the nanotubes have Eph/H2 values in the ideal range. As expected, the 20 carbon-atom layer has a lower value of Eph/H2. The trend observed in Fig. 8 related to the increasing values of Eph/H2 for a higher number of physisorbed hydrogen molecules, suggests that longer saturated bumpy nanotubes have a higher and wider range of hydrogen molecules to be physisorbed in the 20 carbon-atom layer case, starting, for instance, with 15 H2 molecules.

Representation of the dispersion-corrected B3LYP/6-31G(d)-calculated Eph/H2 for armchair (4,4) saturated bumpy nanotubes with 12 and 20 carbon-atom layers and 15 physisorbed hydrogen molecules inside for undoped (first column from the left) and nitrogen-doped structures (second and third columns)
Table 4 presents the hydrogen physisorption energies, the vdW term and the dispersion-force-corrected corresponding energies for hydrogenated full bumpy defected armchair (4,4) nanotubes with an 8–20 carbon-atom layer and 2–24 physisorbed H2 molecules (run no. 1–26) and some bumpy nanotubes with different chirality and similar diameters (run no. 27–41). The main trend observed for all systems confirms that Eph/H2 increases when the number of physisorbed H2 molecules is higher.
The nitrogen-doped bumpy defected nanotubes exhibited slightly higher hydrogen physisorption energies compared to the regular nanotubes as observed from Fig. 9. When analyzing data from Figs. 9, 10 and Table 4 for saturated nanotubes, it is important to note that the following systems are all predicted to have good hydrogen desorption behavior because they exhibit Eph/H2 values in the range 0.1–0.4 eV, recognized as an ideal range for reversible hydrogen storage at room temperature and low pressure [31, 43]: (i) regular doped armchair (4,4) nanotubes with 12 carbon-atom layers; (ii) bumpy doped armchair (4,4) nanotubes with 12, 16 and 20 carbon-atom layers (run 23, 25 and 26), (iii) bumpy undoped armchair (4,4) nanotubes with 8 (run 4 and 5) and 12 carbon-atom layers (run 16 and 17), with 7, 9, 15 and 18 physisorbed H2 molecules together with undoped armchair (5,5) nanotubes with 9–15 physisorbed H2 molecules (run 30–32 in Table 4) and (iv) bumpy zigzag doped and undoped nanotubes with seven physisorbed H2 molecules (run 40–41 in Table 4). Therefore, several saturated bumpy nanotubes exhibit low endothermic hydrogen physisorption energies within the reported ideal range for reversible hydrogen storage. The hydrogen content for all of the nanotubes is approximately 11 %. The dispersion force correction values are important in the evaluation of the hydrogen physisorption energies for bumpy saturated CNTs, as they were for similar regular nanostructures [30].
Conclusions
Hydrogen physisorption energies were calculated for bumpy CNTs using DFT-D3 methods at the B3LYP/6-31G(d) level of theory and considering dispersion forces. Finite open nanotubes terminated with hydrogen were considered. Armchair, chiral and zigzag CNTs that were undoped or nitrogen doped with non-saturated or saturated structures were studied with the aim of understanding how structural features can favor hydrogen storage behavior.
The calculation results predict that bumpy nanotubes can adsorb a much higher number of hydrogen molecules in the interior of the nanotubes than similar regular nanotubes, probably because the specific geometry of bumpy nanotubes increases the internal volume.
The van der Waals energy correction is an important component of the hydrogen physisorption energy for both saturated and non-saturated bumpy CNTs.
The results of both the calculated hydrogen physisorption energies for a single hydrogen molecule, Eph/H2 and the frontier orbital analysis suggest that the bumpy defect is indeed an important nanotube structural feature that together with nanotube chirality, can improve the value of Eph/H2 to the level of 0.1–0.5 eV/molecule considered ideal for reversible hydrogen storage.
The Eph/H2 value increased with the number of physisorbed hydrogen molecules for all studied structures.
In armchair nanotube structures, the bumpy defect caused a significant decrease in the Eph/H2 values that was even more pronounced for nitrogen-doped nanotubes. The Eph/H2 value decreased significantly as the number of carbon-atom layers in the nanostructures increased.
Both smaller diameters and larger lengths decrease the hydrogen physisorption energies of saturated bumpy N-CNTs.
The final results indicate that it is possible to design saturated bumpy defected armchair and zigzag nanotubes (with or without nitrogen doping) that can behave as an ideal system for reversible hydrogen storage. Chiral counterparts need to be further investigated to better understand how to predict their behavior as a hydrogen storage material.
References
Kim EK, Kim T, Paik S, Haam S, Huh YM, Lee K (2015) Chem Rev 115:327–394
Yoosefian M, Barzgari Z, Yoosefian J (2014) Struct Chem 25(1):9
Abdelhalim A, Abdellah A, Scarpa G, Lugli P (2014) Nanotechnology 25(5):5208. doi:10.1088/0957-4484/25/5/055208
Bareket-Keren L, Hanein Y (2013) Front Neural Circuits 6:112. doi:10.3389/fncir.2012.00122
De Volder MFL, Tawfick SH, Baughman RH, Hart AJ (2013) Science 339:535
Bianco S (2011) Carbon nanotubes. From research to applications. InTech, Rijeka
Marulanda JM (2010) Carbon nanotubes. InTech, Rijeka
Arsawang U, Saengsawang O, Rungrotmongkol T, Sornmee P, Wittayanarakul K, Remsungnen T, Hannongbua S (2011) J Mol Graph Model 29:591–596
Meng L, Zhang X, Lu Q, Fei Z, Dyson PJ (2012) Biomaterials 33:1689–1698
Tada K, Furuya S, Watanabe K (2001) Phys Rev B 63:155405
Krishna R, Titus E, Salimian M, Okhay O, Rajendran S, Rajkumar A, Sousa JMG, Ferreira ALC, Campos-Gil J, Gracio J (2012) In: J Liu (ed) Hydrogen storage, chap 10. doi:10.5772/51238. ISBN 978-953-51-0731-6
Li J, Furuta T, Goto H, Ohashi T, Fujiwara Y, Yip S (2003) J Chem Phys 119:2376
Bhatia SK, Myers AL (2006) Langmuir 22:1688–1700
Dillon AC, Jones KM, Bekkedahl TA, Kiang CH, Bethune DS, Heben MJ (1997) Nature 386(6623):377–379
Becher M, Haluska M, Hirscher M, Quintel A, Skakalova V, Dettlaff-Weglikovska U, Chen X, Hulman M, Choi Y, Roth S, Meregalli V, Parrinello M, Ströbel R, Jörissen L, Kappes MM, Fink J, Züttel A, Stepanek I, Bernier P (2003) C R Phys 4:1055–1062
Züttel A (2003) Mater Today 6:24
Alonso JA, Arellano JS, Molina LM, Rubio A, López MJ (2004) IEEE Trans Nanotechnol 3:304–310
Ross DK (2006) Vacuum 80:1084–1089
Zhang G, Qi P, Wang X, Lu Y, Mann D, Li X, Dai H (2006) J Am Chem Soc 128:6026–6027
Bilic A, Gale JD (2008) J Phys Chem C 112:12568–12575
Dinadayalane TC, Kaczmarek A, Lukaszewicz J, Leszczynski J (2007) J Phys Chem C 111:7376–7383
Kaczmarek A, Dinadayalane TC, Lukaszewicz J, Leszczynski J (2007) Int J Quantum Chem 107:2211–2219
Baughman RH, Zakhidov AA, de Heer WA (2002) Science 297(5582):787–792
Orimo S, Züttel A, Schlapbach L, Majer G, Fukunaga T, Fujii H (2003) J Alloys Compd 356–357:716–719
Han SS, Lee HM (2004) Carbon 42(11):2169–2177
DOE targets for onboard hydrogen storage systems for light-duty vehicles. http://energy.gov/sites/prod/files/2015/01/f19/fcto_myrdd_table_onboard_h2_storage_systems_doe_targets_ldv.pdf. Accessed 27 Jan 2016; http://www.eere.energy.gov/hydrogenandfuelcells/storage/pdfs/targets_onboard_hydro_storage_explanation.pdf. Accessed 27 Jan 2016; http://www1.eere.energy.gov/hydrogenandfuelcells/storage/current_technology.html. Accessed 27 Jan 2016
Charlier JC (2002) Acc Chem Res 35:1063–1069
Contreras ML, Avila D, Alvarez J, Rozas R (2012) J Mol Graphs Model 38:389
Contreras ML, Rozas R (2011) In: Bianco S (ed) Carbon nanotubes. From research to applications. Intech, Rijeka. http://www.intech.open.com/books/carbon-nanotubes-from-research-to-applications/nitrogen-containing-carbon-nanotubes-a-theoretical-approach
Contreras ML, Cortés-Arriagada D, Villarroel I, Alvarez J, Rozas R (2014) Struct Chem 25:1045–1056
Singh AK, Yakobson BI (2012) J Mater Sci 47:7356–7366
Hamada N, Sawada S, Oshiyama A (1992) Phys Rev Lett 68(10):1579–1581
HyperChem release 7.5. Hypercube Inc., Gainesville
Jaguar version 8.1. Schrödinger LLC, New York, 2013
Grimme S, Antony J, Ehrlich S, Krieg H (2010) J Chem Phys 132(15):154104
Hujo W, Grimme S (2011) J Chem Theory Comput 7(12):3866–3871
DiLabio GA, Koleini M, Torres E (2013) Theor Chem Acc 132:1389
Dinadayalane TC, Leszczynski J (2010) Struct Chem 21:1155
Contreras ML, Villarroel I, Rozas R (2015) Struct Chem 26(3):761–771. doi:10.1007/s11224-014-0535-y
Zhao M, Xia Y, Lewis JP, Zhang RJ (2003) Appl Phys 94:2398
Ho YW, Suen MC (2013) J Chem 2013:765243. doi:10.1155/2013/765243
Amaral SS, Campos PT, dos Santos JM, Fernandes LS, Martins MAP, Bonacorso HG, Zanatta N (2010) Open Crystallogr J 3:59–66
Yao Y (2010) In: Marulanda JM (ed) Carbon nanotubes. InTech, Rijeka
Lochan RC, Head-Gordon M (2006) Phys Chem Chem Phys 8:1357–1370
Acknowledgments
This work was partially supported by the Direction of Scientific and Technological Research DICYT-USACH Project no. 061642CF and by the Sociedad de Desarrollo Tecnológico SDT-USACH Project no. CIA 2981. In addition, the central cluster of the Faculty of Chemistry and Biology and the VRIDeI of the University of Santiago de Chile are acknowledged for allocating computational resources.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Contreras, M.L., Villarroel, I. & Rozas, R. Hydrogen physisorption energies for bumpy, saturated, nitrogen-doped single-walled carbon nanotubes. Struct Chem 27, 1479–1490 (2016). https://doi.org/10.1007/s11224-016-0767-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-016-0767-0