
Abstract
The hydrogen adsorption energies for nitrogen-containing carbon nanotubes (N-CNTs) and for bare carbon nanotubes were calculated using the density functional theory methods at the B3LYP/6–31-G(d) level, including dispersion force corrections. The N-CNTs were finite saturated and non-saturated single-walled carbon nanotubes that contained one or more pyrimidine units, the relative positions of which defined the different configurations of the nanotube. The chemisorption of atomic hydrogen to a full exocyclic monolayer of zigzag, armchair, and chiral N-CNTs was studied as a function of the structural parameters. Zigzag N-CNTs of any configuration, with a larger number of nitrogen atoms, a small diameter and a small length, are more reactive compared to chiral and armchair N-CNTs. The presence of nitrogen in the carbon nanotubes enhances their reactivity to chemisorb atomic hydrogen, showing exothermic energy values. In contrast, the physisorption of molecular hydrogen was endothermic for most of the studied saturated N-CNTs, even when including corrections for van der Waals interactions. The endothermicity was greatest for zigzag nanotubes, then decreased for chiral nanotubes and decreased again for armchair nanotubes. In general, the endothermicity decreased for longer nanotubes, which have larger diameters, and a small number of nitrogen atoms. The results of this study suggest that, with saturated bare carbon nanotubes, saturated, and unsaturated N-CNTs could potentially have a higher capacity as hydrogen-storage media than the corresponding unsaturated carbon nanotubes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Carbon nanotubes likely represent one of the most versatile structures of our time, and recent research has been conducted to find new applications for these promising compounds [1–4]. One application is hydrogen storage [5, 6], which is necessary to safely use hydrogen, for example, as a renewable source of clean energy. A material is expected to fulfill the requirement proposed by the US Department of Energy, that is, a gravimetric target of 5.5 wt% by 2015 [7], to be suitable as a hydrogen-storage medium.
Carbon nanotubes have been extensively investigated as hydrogen uptake media, with reported values between 0.2 and 10 wt% [8–10]. Many theoretical and experimental critical analyses have been published considering molecular and atomic hydrogen adsorption [6, 11–17]. The general consensus is that bare carbon nanotubes, as such, are not a promising hydrogen-storage media.
An important point to consider for hydrogen adsorption is the presence of heteroatoms, with the goal of modifying the electronic density of the nanotube surface. For example, it is known that carbon nanotubes with a small nitrogen content have increased conductivity [18, 19]. The presence of nitrogen in the carbon nanotubes decreases their toxicity [20] and adds a number of useful properties [21–23], possibly from the induced charge that forms in the vicinity of the nitrogen. With respect to the synthesis of nitrogen-doped carbon nanotubes, chemical vapor deposition with pyridine as the nitrogen source and acetylene as the carbon source can be selectively performed with either sp 2 or sp 3 nitrogen atoms and with different nitrogen levels [24–27].
Research on the interaction of N-CNTs with atomic and molecular hydrogen using density functional theory (DFT) [28–30] and molecular dynamics [30, 31] has arrived at contradictory results because the comparisons of these methods were conducted in completely different systems. For example, the physisorption of one molecule of hydrogen on the external surface of zigzag and armchair N-CNTs exhibits a hydrogen molecular adsorption energy that is significantly smaller for a pristine carbon nanotube [29] using DFT. The same unfavorable situation is found when evaluating the chemisorption of one atom of hydrogen on the external surface of the N-CNTs compared with the pristine nanotube [29]. However, the opposite results are found when calculating the hydrogen physisorption energy for N-decorated nanotubes (as opposed to N-substituted nanotubes) using DFT and molecular dynamics, resulting in a calculated adsorption energy of −80 meV [30]. This energy level indicates a potentially high hydrogen-storage capacity. Alternatively, studies of molecular hydrogen dissociative adsorption using density functional methods indicate a reduced barrier when an (8,0) carbon nanotube is doped with nitrogen [28]. Studies using DFT to evaluate the hydrogen chemisorption energy of N-CNTs with a high nitrogen content at 100 % hydrogen coverage suggest that saturated N-CNTs are stable and that N-CNTs could be used as effective media for the storage of hydrogen [32]. However, there are no systematic studies concerning the effect of the structural parameters on the hydrogen-storage capacity of these nitrogenated carbon nanotubes. Hydrogen physisorption has not yet been reported for saturated N-CNTs.
In this work, we theoretically investigated both the relative reactivity of N-CNTs against atomic hydrogen chemisorption and the relative reactivity of saturated N-CNTs against molecular hydrogen physisorption to evaluate how structural parameters cooperate with the goal of optimizing the hydrogen-storage capacity of these N-CNTs.
Methods
For all simulations, finite open nanotubes were used, and the dangling bonds at the edges were saturated with hydrogen. The N-CNT structures were built using Hamada indices with HyperTube [33], a specific algorithm implemented as a script embedded in HyperChem [34]. Saturated and non-saturated carbon nanotubes were created by choosing the appropriate option in the HyperTube interface. Nitrogen atoms were added by replacing either a C atom (for non-saturated structures) or a CH unit (for saturated tubes) so that after two substitutions (following the pattern –N1–C2–N3–C4–C5–C6–), a pyrimidine-like ring was formed. The next two substitutions, forming a second pyrimidine ring, were located in the opposite position of the same carbon layer. Different configurations were characterized according to the rotation angle between opposite pyrimidine rings to yield S, O, M, and P configurations for rotation angles (θ) of 0°, 60°, 120°, and 180°, respectively. (The O, M, and P names were chosen related to the ortho, meta, and para positions of disubstituted benzene rings, with the pyrimidine carbon atom C2 as a reference, as shown in Fig. 1). All of the structures were first optimized with AM1 (Austin Model 1, semi empirical, as installed in HyperChem) and were then further optimized using the DFT method with Jaguar v7.8 [35] at the B3LYP/6–31G(d) theoretical level [36, 37]. DFT calculations have been commonly used for studying nanotube structures [12, 13, 32, 38–40]. They have also been widely used for the study of non-covalent stacking [41], producing good results for the structure and the relative energies for different isomer conformations (even without considering the typical DFT underestimation) [42]. DFT calculations have also been used to explain conductivities for wide-band-gap semiconductors and oxides [43]. To accurately account for the van der Waals (vdW) interactions, self-consistent field (SCF) energies of fully exo-hydrogenated N-CNTs and their corresponding inside-hydrogen physisorption systems were corrected for dispersion forces using the DFT-D3 method developed by Grimme et al. [44] and implemented in the electronic structure program ORCA 2.9 for the B3LYP functional [45] (B3LYP-D3 functional). Some of the N-CNTs-hydrogen molecules systems were randomly chosen to perform a dispersion force correction for the SCF energies using the DFT-non-local method [46, 47] (a more expensive method than DFT-D3 because the DFT-non-local method needs the converged electron density to compute the dispersion energy of the non-local term), with results that differ very little from the DFT-D3 correction [vdW term in expression (2); see Table 1], thus validating the calculation method and the use of the corrected B3LYP energies. The evaluation of the participation weight of the hydrogen molecules’ interaction alone over the total vdW correction resulted in a low value of approximately 6 kcal/mol for an 8-carbon layer zigzag (8,0) N-CNT with four nitrogen atoms and nine interior hydrogen molecules. For the verification and characterization of energy minima, harmonic vibrational frequency calculations for optimized geometries at the same level of DFT were performed and they all resulted in real values. No symmetry constraints were applied.

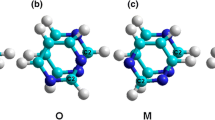
Examples of the S, O, M and P configurations of the N-CNTs are shown in (a), (b), (c), and (d), respectively, where the disposition of the C2 carbon atom for the posterior pyrimidine ring is obtained after a rotation of 0°, 60°, 120°, and 180°. N atom blue, C atom gray, H atom white (Color figure online)
The hydrogen chemisorption energy, Er, was calculated as follows:
where EN-CNT(ch) and EN-CNT are the total energies for N-CNT with and without the chemisorbed hydrogen, respectively; n is the number of chemisorbed hydrogen atoms; and EH is the hydrogen atom energy. Full coverage exo-hydrogenation of a hydrogen monolayer was considered. A negative value of Er indicates that hydrogen chemisorption is a favorable process, that is, the corresponding nanotube shows a good reactivity for chemisorbing hydrogen and that the fully saturated nanotube is stable.
Hydrogen physisorption energy, Eph, was calculated according to expression (2) as follows:
where E(ch + ph) and E(ch) are the total energies for the saturated N-CNTs with and without physisorbed molecular hydrogen, respectively; h is the number of physisorbed hydrogen molecules; \({\text{E}}_{{{\text{H}}_{ 2} }}\) is the total energy of a hydrogen molecule, all of which are calculated at the same level of DFT; and, vdW corresponds to the contribution of dispersion energy from van der Waals interactions among hydrogen molecules and nanotube systems, as calculated by the DFT-D3 method. A positive value of Eph indicates that the hydrogen physisorption process is endothermic.
The notation used in this work is the following. First, the type of nanotube is specified according to the chirality indices (n, m). Second, the configuration according to the rotation angle between opposite pyrimidine groups is indicated by the corresponding symbol (S, O, M, or P). Third, the number of nitrogen atoms present in the nanotube (4N, 8N, etc.) is indicated by a hyphen. When necessary, a character H separated by a hyphen is added to indicate that a saturated nanotube is present. Finally, the number of carbon layers (cl) added, when needed, is indicated by 8cl, 12cl, 16cl, and so on. For example, a chiral carbon nanotube with a P configuration having 4 nitrogen atoms that is fully exo-hydrogenated is denoted as (6,3)P-4N–H. If the nanotube contained five hydrogen molecules physisorbed inside, then, instead of the final H, the characters 5H2 are added, and the compound is written as (6,3)P-4N–5H2.
Results and discussion
Hydrogen adsorption is a process that takes place through the interaction of the hydrogen atom and the adsorbent material to form covalent bonds (chemisorption) or the interaction of molecular hydrogen and the adsorbent material to form non-covalent bonds (physisorption). In this section, we present the results for both hydrogen atom chemisorption energies and hydrogen molecule physisorption energies for N-CNTs as well as the effects on the energy values of structural parameters, such as the diameter, length, chirality, configuration, and the number of nitrogen atoms in the nanotubes. In the first part of the section, the hydrogen chemisorption energy results for bare carbon nanotubes and for N-CNTs are presented to characterize the effect of each of the named parameters, although most of the parameters are interrelated. At the end of the section, the results for hydrogen physisorption energies are presented.
Hydrogen chemisorption energy
Chemisorption energy, Er, measures the energy involved in the adsorption of hydrogen atoms to the sp 2 C atoms to form covalent C–H bonds. In that process, the hybridization of the nanotube C atoms is changed from sp 2 to sp 3. The C–N bond lengths exhibited for the C2 atom that were in the range of 1.37–1.42 Å for the N-CNTs (Fig. 2; in agreement with the 1.42 Å reported for the average C–N bond length for nitrogen-doped armchair carbon nanotubes [48]) were changed to be in the range of 1.45–1.48 Å for the saturated N-CNTs. An exothermic Er value [see expression (1)] indicates that energy is released during hydrogen chemisorption. The value of Er measures how easily such a reaction occurs and the stability of the product of the reaction. In general, an exothermic Er indicates that the formation of the saturated nanotube is a favorable process. A larger exothermic value indicates that the corresponding non-saturated nanotube is a good hydrogen adsorbent.

Bond lengths obtained for one of the pyrimidine rings of an optimized (5,5)S-4N nanotube at the B3LYP/6–31G(d) level
In this work, a single layer of 100 % hydrogen external coverage (which is energetically more favorable [49] than internal hydrogenation) was considered for chemisorption. Larger nanotubes will adsorb a greater number of hydrogen atoms. To make them comparable, a value of Er/H was used to represent the energy associated with the chemisorption per hydrogen atom for the analyzed nanotube.
Nanotube diameter effect
The diameter has been reported as a parameter that strongly affects the hydrogen adsorption energy of carbon nanotubes [12, 13, 50–52]. The hydrogen chemisorption energy generally decreases as the nanotube diameter decreases, such that small-diameter carbon nanotubes are more reactive to chemisorb hydrogen than those with larger diameters. This phenomenon can be understood in terms of the higher curvature of the small-diameter nanotubes. The calculations in this study for bare carbon nanotubes at 100 % coverage show the same trend, with endothermic values for the nanotubes with diameters larger than 7.0 Å, such as (5,5), (6,6), and (6,4) nanotubes, and exothermic values for nanotubes with diameters smaller than 6.5 Å, as shown in Fig. 3 (for 0 nitrogen atoms). All of the nanotubes tested for the data presented in Fig. 3 have an S configuration and are listed in order of increasing diameter, from 3.7 to 9.2 Å. For example, for a (6,6) armchair carbon nanotube with a diameter of 8.2 Å, the Er/H value is 0.122 eV, and for a (3,3) carbon nanotube with a diameter of 4.3 Å, the Er/H value is −0.957 eV. The same reactivity trend was reported previously with an unfavorable endothermic chemisorption energy value of 0.965 eV for a (6,6) armchair carbon nanotube at full exo-hydrogenation using gradient-corrected DFT [11]. Our results indicate the same hydrogen chemisorption energy trend for armchair, chiral, and zigzag carbon nanotubes, with greater exothermic values observed for nanotubes with smaller diameters. Different results have been reported for the chemisorption of one H atom on the sidewall of the nanotube. Even though armchair carbon nanotubes present the same trend, with values of Er/H (calculated at the B3LYP/6–31G(d) level of DFT) from −53.5 to −38.8 kcal/mol (−2.32 to −1.68 eV) for (3,3) and (6,6) nanotubes of nine carbon layers, respectively, [12], zigzag carbon nanotubes show a different trend, with values of −85 kcal/mol (−3.7 eV) and −95 kcal/mol (−4.1 eV) for (5,0) and (9,0) nanotubes, respectively [51].

Hydrogen chemisorption energy per chemisorbed hydrogen atom calculated at the B3LYP/6–31G(d) level versus the number of nitrogen atoms in the corresponding zigzag, chiral, and armchair carbon nanotube represented in green, red and blue, respectively, with symbols that indicate a decreasing diameter order: triangle > diamond > square > circle (Color figure online)
The Er/H for N-CNTs also depends on the diameter, with exothermic values observed for all of the nanotubes studied. The smaller-diameter N-CNTs are the most reactive to atomic hydrogen chemisorption. The results presented in Fig. 3 indicate that this trend is valid for armchair, chiral, and zigzag N-CNTs. For example, the Er/H values, for (4,0), (6,0), and (8,0) zigzag N-CNTs with four nitrogen atoms that have diameters of 3.7, 5.5, and 7.5 Å, are −0.96, −0.5, and −0.4 eV, respectively. The results show that a global diameter effect on atomic hydrogen chemisorption energies remains the same, regardless of either the nanotube chirality or the presence of nitrogen. An Er/H value of −1.34 eV for N-CNTs with smaller diameters (~3 Å), calculated at the same level of DFT [32], confirms the observed trend described above.
Effect of the number of nitrogen atoms in the nanotube
The results of this study indicate that, in general, the Er/H decreases (i.e., it is more favorable) when the carbon nanotube contains nitrogen, presenting smaller values as the number of nitrogen atoms in the N-CNTs increases (the Er/H decreases by approximately 0.03 eV for each nitrogen atom added to the structure), with the exception of those nanotubes that have the smallest diameters (3.7 Å), which also have the most exothermic Er/H values (−0.98 eV) and are almost insensitive to the presence of nitrogen (see Fig. 3).
N-CNT configuration effect
In Fig. 4, the hydrogen chemisorption energy results for zigzag N-CNTs with diameters greater than 3.7 Å and containing four and eight nitrogen atoms are presented. These results exhibit no strong dependence on the configuration of S, O, M, or P (different rotation angles). For (6,0) and (4,0) N-CNTs with four nitrogen atoms, Er/H is −0.56 and −0.97 eV, respectively, for any configuration. For (8,0) N-CNTs that have four and eight nitrogen atoms, Er/H is approximately −0.37 and −0.5 eV, respectively, regardless of configuration. The data in Fig. 4 also confirm that hydrogen chemisorption is an energetically more favorable process when the N-CNT diameter decreases and when the number of nitrogen atoms in the N-CNT increases, consistent with the reported DFT calculations for zigzag N-CNTs with diameters of ~3 Å and a nitrogen content >30 % [32].

Hydrogen chemisorption energies per hydrogen atom at the B3LYP/6–31G(d) level for zigzag N-CNTs with four and eight nitrogen atoms versus the rotation angle that represents the different configurations (i.e., 0°, 60°, 120°, and 180° represent S, O, M, and P configurations, respectively)
N-CNT chirality effect
A chirality effect on the hydrogen chemisorption energies for N-CNTs was illustrated by the data. Zigzag N-CNTs exhibit the most exothermic Er/H values and are, therefore, the most reactive to hydrogen chemisorption, as shown in Fig. 3. For example, the Er/H values for the (6,0), (6,2), and (4,4) nanotubes with four nitrogen atoms (and a diameter of 5.5 Å) are −0.5, −0.38, and −0.32 eV, respectively. The same trend is found for the larger diameter N-CNTs, that is, the Er/H values for (8,0), (6,3), and (5,5) N-CNTs with four nitrogen atoms (and a diameter of ~7.5 Å) are −0.4, −0.23, and −0.16 eV, respectively. Therefore, the predicted order of the reactivity of N-CNTs for hydrogen chemisorption is zigzag > chiral > armchair. The same chirality effect was also observed for bare carbon nanotubes, as shown in Fig. 3.
N-CNT length effect
The N-CNT length depends on the number of carbon layers constituting the nanotube. However, for N-CNTs with the same number of carbon layers, the same number of nitrogen atoms and a similar diameter, their lengths depend upon nanotube chirality. For example, the (8,0), (6,3), and (5,5) N-CNTs with molecular formulae C60N4H16, C78N4H20, and C76N4H20, respectively, all of which have eight carbon layers, four nitrogen atoms and 7.5 Å diameters [structures (a–c) in Fig. 5], have 7.1, 7.4, and 8.5 Å lengths, respectively (measured from C to C). When these nanotubes are completely saturated [structures (d–f) in Fig. 5], their lengths are 6.9, 7.2, and 8.8 Å, respectively. Maintaining most of the N-CNT characteristics constant, it appears that the N-CNT length increases in the order of zigzag < chiral < armchair. Interestingly, this order is the inverse of the N-CNT reactivity to hydrogen chemisorption, as was established above.

N-CNT structures optimized at the B3LYP/6–31G(d) level for a (8,0)S-4N-8cl; b (6,3)S-4N-8cl; and c (5,5)S-4N–8cl; and their saturated structures d (8,0)S-4N–H-8cl; e (6,3)S-4N-H-8cl; and f (5,5)S-4N-H-8cl. N atom blue, C atom gray. For the sake of simplicity, hydrogen atoms are not shown (Color figure online)
In Fig. 6, the hydrogen chemisorption energies for armchair N-CNTs that only differ in their lengths [(4,4)S-8cl with an 8.5 Å length and (4,4)S-12cl with a 13.3 Å length] as a function of the number of nitrogen atoms are compared. The data show that the number of nitrogen atoms has a significant effect on the exothermicity of the hydrogen chemisorption reaction for armchair N-CNTs and that the effect is more pronounced for shorter nanotubes. Er/H decreases as the number of nitrogen atoms increases from 0 to 8; the exothermicity is nearly 0.15 eV higher for the N-CNTs, which are approximately 5 Å shorter (8 carbon layer vs 12 carbon layers). This difference is better appreciated in N-CNTs with a higher number of nitrogen atoms. That is, the shorter N-CNTs appear to be more favorable for full hydrogen chemisorption than the longer ones, regardless of the number of nitrogen atoms contained in the nanotube. This trend coincides with the reported results computed at the B3LYP/6–31G(d) level for the chemisorption of one hydrogen atom on the sidewalls of (4,4) single-walled carbon nanotubes [12]. The tendency for shorter N-CNTs to be more favorable for chemisorption could explain in part why the zigzag N-CNTs are the most reactive. A further confirmation of the greater reactivity of zigzag N-CNTs compared with the other chiralities is shown in Fig. 6 for (6,0)S-12cl N-CNTs (which have the same configuration and the same number of carbon layers as well as the similar diameters as the (4,4)S-12cl nanotubes), which show nearly 0.25 eV higher exothermicity than the (4,4)S-12cl N-CNTs. Therefore, chirality appears to affect hydrogen chemisorption energies for N-CNTs more than length does.

Variation of the chemisorption energies per adsorbed H atom at the B3LYP/6–31G(d) level for (4,4) armchair N-CNTs of different lengths and for (6,0) zigzag N-CNTs versus the number of nitrogen atoms
Hydrogen physisorption energy
In the case of physisorption, a non-covalent interaction between the nanotube and the molecular hydrogen inside the nanostructure occurs. The energy involved in the physisorption of molecular hydrogen inside the nanotube, Eph, is calculated using equation (2), which incorporates a correction for dispersion forces, as explained above. The energy associated with the physisorption of a single hydrogen molecule is called Eph/H2. Hydrogen physisorption is performed over fully saturated N-CNTs (i.e., N-CNTs that have been submitted to exo-hydrogenation at 100 % coverage) without involving any change of the hybridization of the nanotube C atoms. All of the calculated values of the hydrogen physisorption energies for N-CNTs are endothermic (except for the small-diameter (4,4) armchair nanostructures containing one or two hydrogen molecules). A high value of Eph/H2 indicates that hydrogen physisorption requires more energy and that the resulting (nanotube-H2) system is less stable. A good hydrogen-storing material should be able to release hydrogen in a controlled manner, without requiring much added energy. For this reason, such a material should have an exothermic Eph/H2 that is not far from 0 eV. An ideal range for the hydrogen binding energy to carbon nanotubes that allows for reversible hydrogen storage at ambient conditions has been reported as 0.1–0.5 eV/H2 [6, 53]. Several thermodynamic estimations of the binding energy of H2 molecules for reversible storage are almost in the same range, that is, 0.16–0.4 eV/molecule [54–56].
In Table 2, a comparative summary of the hydrogen chemisorption energies and the hydrogen physisorption energies for a number of different systems that were studied, with the corresponding diameters and vdW corrections, are presented including the total hydrogen content. As the data in Table 2 illustrates, non-covalent interactions contribute significantly to the hydrogen physisorption energies. The B3LYP-D3 method used in this work was recently found to be one of the most accurate methods available for treating intermolecular interactions when tested on several databases of non-covalently interacting dimers with the C-DCP, which is a new dispersion-correcting potential developed by DiLabio et al. [57] for the modeling of C–C bond-making/bond-breaking chemistry.
The data in Table 2 show a significant difference in physisorption energies between the small-diameter (4,0) and the larger-diameter (8,0) N-CNTs. The physisorption endothermicity is substantially higher in the (4,0) N-CNTs compared with the (8,0) structures. In the case of the (8,0) N-CNTs, a marked deviation in the physisorption endothermicity was observed in the S configuration (rotation angle equal to zero) compared with the rest of the configurations. However, the smaller-diameter (4,0) N-CNTs do not follow the same trend as the larger-diameter nanotubes, and the deviations in the physisorption endothermicities in this case are less significant between configurations. The effect of configuration is more evident when comparing the Eph/H2 values for (8,0) N-CNTs when different numbers of H2 molecules are absorbed. For the O and P configurations, the endothermicity of hydrogen physisorption increases as the number of physisorbed hydrogen molecules increases, which is the opposite of the situation for the S configuration. This finding can be attributed to the different polarization of the nanotube structure from the effect caused by the disposition of the nitrogen atoms in the structure, which in turn affects the electrostatic interactions between hydrogen and the nanotube.
The number of nitrogen atoms has a uniform influence on the hydrogen physisorption energy of the smaller-diameter (4,0) zigzag N-CNTs, with smaller values of endothermicity for nanotubes with a smaller number of nitrogen atoms (i.e., the (4,0)-4N–3H2 nanotube compared with the (4,0)-8N–3H2 nanotube in Table 2). In contrast, the number of physisorbed hydrogen molecules inside the nanotube primarily influences the larger-diameter (8,0) N-CNTs.
Data in Fig. 7 indicate that the nanotube configuration does not significantly affect the hydrogen physisorption energy for smaller-diameter armchair N-CNTs and is in agreement with the results found for the zigzag N-CNTs. In addition, the data in Fig. 7 for the (4,4)S-4N–H-12cl nanotubes are within the ideal range mentioned above for hydrogen storage and when compared with shorter (4,4)S-4N–H-8cl nanotubes, indicate that nanotube length has a significant effect on the endothermicity of hydrogen physisorption, with smaller endothermic Eph/H2 values for longer N-CNTs. In Fig. 8 optimized structures of (4,4)S-4N–8H2-12cl and (8,0)S-4N–20H2-12cl are presented. These findings seem reasonable because longer nanotubes have more room to allocate the hydrogen molecules inside the tubes and thus repulsion between the hydrogen molecules remains at a low level. This behavior differs from that observed in the case of hydrogen chemisorption energies for N-CNTs of comparable size, where hydrogen remains bonded to the outside of the nanotube and many hybridization changes take place.

Variation of the physisorption energies per adsorbed H2 calculated by DFT-D3 methods at the B3LYP/6–31G(d) level for saturated (4,4) armchair N-CNTs of different configurations and different lengths, as a function of the number of adsorbed H2 molecules

N-CNT structures optimized at the B3LYP/6–31G(d) level for (a) (4,4)S-4N–8H2-12cl and (b) (8,0)S-4N–20H2-12cl (nanotubes had both stoichiometry C124N4H140 before the hydrogen physisorption process took place). Lateral and front views are presented. N atom blue, C atom gray, H atom white (Color figure online)
In Table 2, the hydrogen physisorption energy results for (5,5), (6,3), and (8,0) N-CNTs with the same characteristics (i.e., S configuration, four nitrogen atoms, 7.5 Å diameter, and eight carbon layers), except for topological chirality, are compared. These results show that chirality has a strong effect on physisorption endothermicity. For three physisorbed H2 molecules or more, the armchair N-CNTs exhibit Eph/H2 values within the ideal range for a reversible hydrogen-storage medium [6, 53, 55, 56]. For all of the configurations, it is observed that endothermicity increases in the order of armchair < chiral < zigzag and also increases when the number of physisorbed H2 molecules increases. The polarizability of armchair N-CNTs on one hand and the repulsion between H2 molecules on the other hand could explain this behavior. These findings for N-CNTs are in excellent agreement with the DFT calculations for (5,5), (6,4), and (8,2) carbon nanotubes that employed the local density approximation [14]. However, for one or two physisorbed H2 molecules, the N-CNTs show a completely different behavior. Armchair and zigzag N-CNTs exhibit Eph/H2 values for one physisorbed H2 molecule that are much more endothermic than for the rest of the cases. This result is not unusual and has been observed for hydrogen chemisorption energies calculated at the B3LYP/6–31G(d) level for (5,5) armchair carbon nanotubes with 9 and 15 carbon layers [12]. Chiral N-CNTs exhibit a more conservative variation of the Eph/H2 values for the entire range of physisorbed molecular hydrogen. A different situation has been reported for the hydrogen physisorption energy of a single hydrogen molecule on the outer surface of carbon nanotubes where no chirality effect was detected using DFT calculations [52].
Therefore, the best energy values for the inside-hydrogen physisorption of saturated N-CNTs are exhibited by longer armchair N-CNTs with larger diameters and a small number of nitrogen atoms. Bare saturated carbon nanotubes have better hydrogen physisorption energy values than saturated N-CNTs. Furthermore, the wt% total content of hydrogen for saturated carbon nanotubes decreases with nitrogen substitution (see Table 2). For example, a saturated (5,5) carbon nanostructure with five physisorbed H2 molecules exhibits Eph/H2 = −0.068 eV and 10.3 wt% hydrogen for a bare carbon nanotube, but changes to Eph/H2 = 0.337 eV and 9.9 wt% hydrogen for N-CNTs with four nitrogen atoms (both structures have the same configuration and length).
Conclusions
The reactivity of the finite N-CNTs of different chiralities for the adsorption of hydrogen was investigated using the DFT and DFT-D3 methodology at the B3LYP/6–31G(d) level. Smaller diameter carbon nanotubes with or without the presence of nitrogen are more reactive for chemisorbing atomic hydrogen, regardless of chirality. The presence of nitrogen enhances this trend and, accordingly, the Er/H decreases by approximately 0.03 eV for each nitrogen atom added to the structure, except for the most reactive nanotubes with diameters of 3.7 Å, the reactivity of which is not dependent on nitrogen content. The configuration of the N-CNTs had no significant influence on the N-CNT reactivity. In contrast, the chirality of N-CNTs was found to be an important factor that affects their reactivity to hydrogen chemisorption in the order of zigzag > chiral > armchair. The same trend was found for the hydrogen chemisorption of bare carbon nanotubes. The nanotube length was also found to affect the reactivity of N-CNTs, with shorter structures being the most reactive to hydrogen chemisorption.
Therefore, the structures that have higher exothermic values for hydrogen chemisorption energies per hydrogen atom are zigzag N-CNTs, which have more favorable values for nanotubes with smaller diameters and lengths and with an increased number of nitrogen atoms. This finding is in agreement with reported values for even smaller diameter N-CNTs [32].
In contrast, hydrogen physisorption energies for N-CNTs exhibit endothermic values for most of the studied N-CNTs, with nanotubes of larger diameters having lower endothermic values of Eph/H2. The vdW correction term represented an important contribution to the hydrogen physisorption energies. In addition, the diameter has a significant effect on hydrogen physisorption energies. For example, the N-CNTs with diameters of less than 4 Å showed no dependence on the configuration of the N-CNTs, the number of nitrogen atoms, or the number of physisorbed H2 molecules. The hydrogen physisorption energies for larger-diameter N-CNTs are less endothermic and depend on the length, the number of nitrogen atoms, the nanotube configuration and the number of physisorbed H2 molecules inside the N-CNTs, with more favorable values for longer N-CNTs that have a smaller number of nitrogen atoms and a reduced number of physisorbed H2 molecules. For three or more physisorbed H2 molecules, the endothermicity of the hydrogen physisorption for saturated N-CNTs decreases in the chirality order of zigzag > chiral > armchair. Therefore, for saturated N-CNTs, the longer armchair nanotubes with large diameters and a small number of nitrogen atoms are the most reactive to hydrogen physisorption and, according to their energy values, have a high potential capacity as hydrogen-storage media along with the fully saturated bare carbon nanotubes.
References
De Volder MFL, Tawfick SH, Baughman RH, Hart AJ (2013) Carbon nanotubes: present and future commercial applications. Science 339:535–539
Bareket-Keren L, Hanein Y (2013) Carbon nanotube-based multielectrode arrays for neuronal interfacing: progress and prospects. Front Neural Circuits 6:122
Bianco S (2011) Carbon nanotubes. From research to applications. Intech, Croatia
Marulanda JM (2010) Carbon nanotubes. In-The, India
Kovalev V, Yakunchikov A, Li F (2011) Simulation of hydrogen adsorption in carbon nanotube arrays. Acta Astronaut 68:681–685
Yao Y (2010) In: Marulanda JM (ed) Carbon nanotubes. In-The, India
Targets for onboard hydrogen storage systems for light-duty vehicles http://www.eere.energy.gov/hydrogenandfuelcells/storage/pdfs/targets_onboard_hydro_storage_explanation.pdf. Accessed 12 Sept 2013 http://www1.eere.energy.gov/hydrogenandfuelcells/storage/current_technology.html. Accessed 12 Sept 2013
Dillon AC, Jones KM, Bekkedahl TA, Kiang CH, Bethune DS, Heben MJ (1997) Storage of hydrogen in single-walled carbon nanotubes. Nature 386(6623):377–379
Zhang G, Qi P, Wang X, Lu Y, Mann D, Li X, Dai H (2006) Hydrogenation and hydrocarbonation and etching of single-walled carbon nanotubes. J Am Chem Soc 128:6026–6027
Baughman RH, Zakhidov AA, de Heer WA (2002) Carbon nanotubes-the route toward applications. Science 297(5582):787–792
Bilic A, Gale JD (2008) Chemisorption of molecular hydrogen on carbon nanotubes: a route to effective hydrogen storage? J Phys Chem C 112:12568–12575
Dinadayalane TC, Kaczmarek A, Lukaszewicz J, Leszczynski J (2007) Chemisorption of hydrogen atoms on the sidewalls of armchair single-walled carbon nanotubes. J Phys Chem C 111:7376–7383
Kaczmarek A, Dinadayalane TC, Lukaszewicz J, Leszczynski J (2007) Effect of tube length on the chemisorptions of one and two hydrogen atoms on the sidewalls of (3,3) and (4,4) single-walled carbon nanotubes: a theoretical study. Int J Quantum Chem 107:2211–2219
Alonso JA, Arellano JS, Molina LM, Rubio A, López MJ (2004) Interaction of molecular and atomic hydrogen with single-wall carbon nanotubes. IEEE Trans Nanotech 3:304–310
Orimo S, Züttel A, Schlapbach L, Majer G, Fukunaga T, Fujii H (2003) Hydrogen interaction with carbon nanostructures: current situation and future prospects. J Alloys Compd 356–357:716–719
Becher M, Haluska M, Hirscher M, Quintel A, Skakalova V, Dettlaff-Weglikovska U, Chen X, Hulman M, Choi Y, Roth S, Meregalli V, Parrinello M, Ströbel R, Jörissen L, Kappes MM, Fink J, Züttel A, Stepanek I, Bernier P (2003) Hydrogen storage in carbon nanotubes. C. R. Physique 4:1055–1062
Ross DK (2006) Hydrogen storage: the major technological barrier to the development of hydrogen fuel cell cars. Vacuum 80:1084–1089
Charlier JC (2002) Defects in carbon nanotubes. Acc Chem Res 35:1063–1069
Czerw R, Terrones M, Charlier JC, Blase X, Foley B, Kamalakaran R, Grobert N, Terrones H, Ajayan PM, Blau W, Tekleab D, Rühle M, Carroll DL (2001) Identification of electron donor states in N-doped carbon nanotubes. Nano Lett 1:457–460
Terrones M (2007) Synthesis toxicity and applications of doped carbon nanotubes. Acta Microsc 16(1–2):33–34
Hu X, Zhou Z, Lin Q, Wu Y, Zhang Z (2011) High reactivity of metal-free nitrogen-doped carbon nanotube for the C-H activation. Chem Phys Lett 503:287–291
Xiong W, Du F, Liu Y, Perez A Jr, Supp M, Ramakrishnan TS, Dai L, Jiang L (2010) 3-D carbon nanotube structures used as high performance catalyst for oxygen reduction reaction. J Am Chem Soc 132:15839–15841
Gong KP, Du ZH, Xia ZH, Durstock M, Dai LM (2009) Nitrogen-doped carbon nanotube arrays with high electrocatalytic activity for oxygen reduction. Science 323:760–764
Zhang Y, Wen B, Song XY, Li TJ (2010) Synthesis and bonding properties of carbon nanotubes with different nitrogen contents. Acta Phys Sin 59(5):3583–3588
Yang SH, Shin WH, Kang JK (2008) The nature of graphite- and pyridinelike nitrogen configurations in carbon nitride nanotubes: dependence on diameter and helicity. Small 4:437–441
Zhong Z, Lee GI, Mo CB, Hong SH, Kang JK (2007) Tailored field-emission property of patterned carbon nitride nanotubes by a selective doping of substitutional N(sN) and pyridine-like N(pN) atoms. Chem Mater 19:2918–2920
Trasobares S, Stephan O, Colliex C, Hsu WK, Kroto HW, Walton DRM (2002) Compartmentalized CNx nanotubes: chemistry morphology and growth. J Chem Phys 116:8966–8972
Zhang ZY, Cho K (2007) Ab initio study of hydrogen interaction with pure and nitrogen-doped carbon nanotubes. Phys Rev B 75(7):075420
Zhou Z, Gao XP, Yan J, Song DY (2006) Doping effects of B and N on hydrogen adsorption in single-walled carbon nanotubes through density functional calculations. Carbon 44:939–947
Rangel E, Ruiz-Chavarria G, Magana LF, Arellano JS (2009) Hydrogen adsorption on N-decorated single wall carbon nanotubes. Phys Lett A 373:2588–2591
Oh KS, Kim DH, Park S, Lee JS, Kwon O, Choi YK (2008) Movement of hydrogen molecules in pristine hydrogenated and nitrogen-doped single-walled carbon nanotubes. Mol Simul 34:1245–1252
Contreras ML, Rozas R (2011) In: Bianco S (ed) Carbon nanotubes. From research to applications. Intech, Croatia. http://www.intech.open.com/books/carbon-nanotubes-from-research-to-applications/nitrogen-containing-carbon-nanotubes-a-theoretical-approach
Contreras ML, Avila D, Alvarez J, Rozas R (2012) Computational algorithms for a fast building of 3-D carbon nanotube models having different defects. J Mol Graph Mod 38:389–395
HyperChem release 7.5 Hypercube Inc 1115 NW 4th Street Gainesville Florida 32601 USA
Jaguar version 7.8 Schrödinger LLC New York NY 2011
Becke AD (1993) Density-functional thermochemistry. The role of exact exchange. J Chem Phys 98:5648–5652
Lee C, Yang W, Parr RG (1988) Development of the colle-salvetti correlation-energy formula into a functional of the electron-density. Phys Rev B 37:785–789
Ahmadi A, Beheshtian J, Hadipour NL (2011) Chemisorption of NH3 at the open ends of boron nitride nanotubes: a DFT study. Struct Chem 22:183–188
Contreras ML, Avila D, Alvarez J, Rozas R (2010) Exploring the structural and electronic properties of nitrogen-containing exohydrogenated carbon nanotubes: a quantum chemistry study. Struct Chem 21(3):573–581
Beheshtian J, Peyghan AA, Bagheri Z (2013) Hydrogen dissociation on diene-functionalized carbon nanotubes. J Mol Model 19:255–261
Felice RD, Calzolari A, Varsano D, Rubio A (2005) Electronic structure calculations for nanomolecular systems. Lect Notes Phys 680:77
Dreizler RM, Gross EKU (1990) Density functional theory, an approach to the quantum many body problem. Springer, Berlin
Van de Walle CG (2008) Computational studies of conductivity in wide-band-gap semiconductors and oxides. J Phys: Condens Matter 20:064230
Grimme S, Antony J, Ehrlich S, Krieg H (2010) A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J Chem Phys 132(15):154104
Neese F (2012) The ORCA program system. WIREs Comput Mol Sci 2(1):73–78
Vydrov OA, Van Voorhis TJ (2010) Nonlocal van der Waals density functional: the simpler the better. Chem Phys 133:244103 (9 pp)
Hujo W, Grimme S (2011) Performance of the van der Waals density functional VV10 and (hybrid)GGA variants for thermochemistry and noncovalent interactions. J Chem Theory Comput 7(12):3866–3871
Zhao M, Xia Y, Lewis JP, Zhang R (2003) First-principles calculations for nitrogen-containing single-walled carbon nanotubes. J Appl Phys 94(4):2398–2402
Tada K, Furuya S, Watanabe K (2001) Ab initio study of hydrogen adsorption to single-walled carbon nanotubes. Phys Rev B 63:155405 (4 pp)
Shamina EN, Lebedev NG (2012) The chiral effect of adsorption of univalent atoms and diatomic molecules on the surface of carbon nanotubes. Russ J Phys Chem B 6:448–454
Yang FH, Lachawiec AJ Jr, Yang RT (2006) Adsorption of spillover hydrogen atoms on single-wall carbon nanotubes. J Phys Chem B 110:6236–6244
Gayathri V, Geetha R (2007) Hydrogen adsorption in defected carbon nanotubes. Adsorpt 13:53–59
Singh AK, Yakobson BI (2012) First principles calculations of H-storage in sorption materials. J Mater Sci 47:7356–7366
Lochan RC, Head-Gordon M (2006) Computational studies of molecular hydrogen binding affinities: the role of dispersion forces electrostatics and orbital interactions. Phys Chem Chem Phys 8:1357–1370
Li J, Furuta T, Goto H, Ohashi T, Fujiwara Y, Yip S (2003) Theoretical evaluation of hydrogen storage capacity in pure carbon nanostructures. J Chem Phys 119:2376
Bhatia SK, Myers AL (2006) Optimum conditions for adsorptive storage. Langmuir 22:1688–1700
DiLabio GA, Koleini M, Torres E (2013) Extension of the B3LYP–dispersion-correcting potential approach to the accurate treatment of both inter- and intra-molecular interactions. Theor Chem Acc 132:1389
Acknowledgments
This work was partially supported by the Direction of Scientific and Technological Research DICYT-USACH Project Nr. 061342CF and by the Sociedad de Desarrollo Tecnológico SDT-USACH project Nr. CIA 2981. In addition, the central cluster of the Faculty of Chemistry and Biology and the VRID of the University of Santiago de Chile are acknowledged for allocating computational resources.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Contreras, M.L., Cortés-Arriagada, D., Villarroel, I. et al. Evaluating the hydrogen chemisorption and physisorption energies for nitrogen-containing single-walled carbon nanotubes with different chiralities: a density functional theory study. Struct Chem 25, 1045–1056 (2014). https://doi.org/10.1007/s11224-013-0377-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-013-0377-z