
Abstract
While obesity is defined as an excessive fat accumulation conferring a risk to metabolic health, increased adipose mass by itself does not fully explain obesity’s propensity to promote metabolic alterations. Adipose tissue regulates multiple processes critical for energy homeostasis and its dysfunction favors the development and perpetuation of metabolic diseases. Obesity drives inflammatory leucocyte infiltration in adipose tissue and fibrotic transformation of the fat depots. Both features associate with metabolic alterations such as impaired glucose control and resistance to fat mass loss. In this context, adipose progenitors, an heterogenous resident population of mesenchymal stromal cells, display functions important to shape healthy or unhealthy adipose tissue expansion. We, here, outline the current understanding of adipose progenitor biology in the context of obesity-induced adipose tissue remodeling.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Adipose tissue (AT) regulates numerous physiological processes and its dysfunction favors development and perpetuation of metabolic diseases. As a consequence, AT has been extensively studied since acting on this tissue may provide novel therapeutic opportunities. Two morphologically and functionally different types of AT can be distinguished: brown/beige adipose tissue and white adipose tissue (WAT). The brown adipose tissue (BAT) is found subcutaneously in specific locations mostly in newborns and in smaller amounts in adults. Moreover, BAT primarily functions as a thermogenic organ owing to the presence of multilocular adipocytes enriched with mitochondria and uncoupling protein 1 (UCP1) [1,2,3]. The overall morphology of beige adipocytes is similar to the brown adipocytes but beige cells infiltrate diffuse areas within the WAT depot. Beige adipogenesis, considered as a healthy remodeling process in the AT, significantly increases in response to thermogenic stimuli such as decreased temperature [4,5,6], β3-adrenergic receptor activation [7,8,9] or response to some metabolites [10, 11]. With obesity development, both brown and beige fat depots are reduced [12,13,14].
By contrast to brown/beige adipocytes, the white adipocytes display low mitochondrial abundance, are unilocular and function in storing calories from triglycerides rather than dissipating energy in the form of heat. In rodents or in humans, WAT displays functional differences according to their subcutaneous or visceral location. With obesity, both depots can expand and a high deposition of visceral WAT is generally associated with increased risk of developing cardiometabolic diseases. On the contrary predominant subcutaneous WAT storage may reduce the risk for comorbidities in some individuals [15,16,17]. Sex hormones and genetic determinants both influence fat distribution [18, 19]. Despite major progresses in physio-pathological understanding in this field, how depot-specific expansion of fat mass is controlled still remains elusive. In addition to adipose tissue growth, obesity is a chronic condition associated with AT histological alterations, depicting a maladaptive expansion of AT. This pathological remodeling includes adipocyte hypertrophy, inflammatory leucocyte infiltration and perturbed immunity, and eventually fibrosis deposition. These features generally associate with altered AT functions suggested to link obesity to obesity-related metabolic dysregulation [20]. By contrast, healthy adipose tissue growth, uncoupled to these pathological features, can dampen the consequences of obesity on whole-body metabolism [21, 22]. In this context, we here review the current understanding of the progenitor contributions in shaping healthy or unhealthy AT expansion during obesity.
2 Obesity induces fibrosis in white adipose tissue
WAT has the unique capacity to massively expand or shrink in response to nutritional or even temperature challenges. This remarkable plasticity relies on a dynamic and versatile metabolism which is responsive to energy demand. Overfeeding without adapted increased energy expenditure results in fat accretion, a physiological response necessary to prevent the toxic lipid deposition in other organs, such as in the skeletal muscle, liver or the heart. This remarkable ability is closely associated with preserved systemic metabolism. As a consequence, the lack of AT exerts important deleterious effects as exemplified in lipodystrophic condition. Lipodystrophy is indeed an extreme form of adipose tissue depletion that associates with ectopic lipid deposition leading to fatty liver and lipid accumulation in the muscle which result in severe insulin resistance. Interestingly, this phenomenon can be reversed with AT implantation in animal models (see below and [23, 24]).
In chronic obesity, whereas the AT expands, it is generally coupled to pathological remodeling of AT with local inflammation and subsequently fibrosis deposition in the latter stages of the disease. These processes result in AT dysfunctions. The local inflammation relies on the infiltration of leucocytes (CD45 expressing cells, CD45+) in which macrophages represent a large population. Local hypoxia due to suboptimal angiogenesis was proposed as an originating event [25,26,27,28,29]. AT macrophages accumulation coincides with the observation of adipocytes surrounding by macrophages (named crown like structure, CLS) on histological Sects. [30]. Adipocytes engaged in CLS display loss of perilipin expression (lipid droplet protein) and ultrastructural features of stressed cells suggestive of dying adipocytes [31, 32]. In mice, macrophages critically control AT inflammation and favor the onset of insulin resistance, however the kinetic of events in human and their relationships with metabolic deterioration still need understanding [33,34,35,36]. Inflammatory pathway activated by the local production of many cytokines including TNFα, IL1β or IL6, can interact with insulin signaling pathway in adipocytes to precipitate insulin resistance [37]. Beside leucocyte infiltration, adipose tissue remodeling is also characterized with senescence contributing to the altered adipose tissue secretory profile and to the local inflammation status [38, 39].
However, while chronic inflammation and the obesity associated metabolic alterations are closely related, studies have suggested a paradoxical beneficial effect of inflammation on adipose tissue in the context of obesity. The use of transgenic mouse models harboring anti-inflammatory construction showed that constitutive inhibition of inflammation was also damaging for adipose tissue expansion [40]. Similarly, the lack of Il6 in myeloid lineage has detrimental consequences for metabolic fitness [41]. Thus, the remained ability to produce balanced inflammation appears necessary for AT homeostasis.
By contrast, the persistence of inflammatory stress in tissues is often associated with altered remodeling in a number of pathological states that can progress to fibrosis, as also observed in AT [42, 43].
Fibrosis is a dysfunctional process characterized by excessive extracellular matrix (ECM) component deposition. The ECM is composed of two main classes of macromolecules: the extremely hydrophilic proteoglycans and the fibrous proteins including collagens, elastins, fibronectins and laminins [44]. Collagen is the most abundant fibrous protein of the ECM, and in the physiological context, the ECM provides tensile strength, regulates cell adhesion, supports chemotaxis and migration, and guides tissue development [44, 45]. In pathological context, continuous ECM synthesis with enhanced ECM crosslinking by lysyl oxidase (LOX) enzymes promote the formation of collagen bundles that stiffen the tissue [46]. In human AT, fibrosis forms collagen bundles traversing the parenchyma and also surrounding the adipocytes [47]. Several evidences support that AT fibrosis is an aggravating factor for metabolic condition [20, 48]. Various studies indeed link AT fibrosis to the loss of glycemic control, insulin resistance and liver disease in mouse models but also in human [49,50,51]. Moreover, increased AT fibrosis accumulation in subcutaneous depot is associated with a decreased fat mass loss induced by bariatric surgery in subjects with severe obesity [48]. Thus, targeting AT fibrosis with the aim of maintaining or rescuing AT plasticity could be of interest in the treatment of obesity associated metabolic alterations. In this setting, pathways are being identified to efficiently brake AT fibrosis progression (see sections below) [43, 52]. However, the cellular and molecular mechanisms of AT fibrosis resolution remained to be elucidated. While fibrosis resolution can be observed in various models following the cessation of the profibrotic stimuli [53, 54], AT fibrosis could be an irreversible condition, especially in advanced stages and chronic conditions. In mouse and human, even when the obesogenic trigger (i.e. dietary intervention or bariatric surgery) is abrogated and, despite the metabolic improvement induced by weight loss, there is no evidence of fibrosis resolution as collagen accumulation is maintained in the long term [55, 56].
3 Molecular alterations linking fibrosis to adipose tissue dysfunction
The fibrotic transformation of AT is generally associated with loss of function and, some of the adipocyte failures were attributed to the perturbation of ECM stiffness. Actually, the potential involvement of mechano-sensing pathways, was first suggested following the evaluation of tissue rigidity with a non-invasive prototypic tool [57]. The analysis of human obese abdominal subcutaneous AT (scAT) revealed increased stiffness in scAT with high fibrosis content [57]. Furthermore, modeling the physical constrains applied to adipocytes in ex vivo systems showed that the mechanical compression can lead to increased production and secretion of inflammatory molecules as well as dysregulated lipolysis, adipokine secretion and perturbed insulin responsiveness in adipocytes [58, 59]. The mechanosensitive Integrin β1, FAK and Caveolin activation were proposed to regulate those effects in adipocytes [58].
In addition, some evidences suggest that fibrosis deposition also compromises the adapted expansion capacity of AT. The use of static compression to mirror the fibrosis effects alters adipocyte differentiation as well as lipid accumulation [60, 61]. By contrast, the reduced adipose tensile strength in Collagen VI-knockout mice is associated with abnormally large but healthy adipocytes [62]. Thus, AT fibrosis appears to impede fat expandability in limiting both adipogenesis and adipocyte hypertrophy, suggesting that fatty acids can more easily spill over into ectopic sites. In line with this assumption, increased subcutaneous AT fibrosis was shown to be associated to visceral fat accretion in a cohort of Chinese American men and women [63] or to fatty liver in women [49, 64].
Sustained fibrosis and modified ECM composition may probably promote pathways that amplify alterations of tissue structure and functions. For instance, the soluble cleavage product of collagen VI chain, referred as endotrophin, seems to play an important role in obesity induced systemic insulin resistance by stimulating inflammation and fibrosis in AT [52, 65]. Similar pathological effects were suggested for osteopontin [66]. This matricellular protein is known to mediate diverse biological functions through interactions with integrins [66]. In obesity, AT macrophages express high levels of osteopontin [67] and osteopontin neutralization partially decreases obesity-associated inflammation in AT and, reverses signal transduction related to insulin resistance [8, 68]. Furthermore, increased circulating osteopontin, related to visceral fat production, was shown to mediate cardiac aging in mice [69]. Likewise, Tenascin C (TNC), an ECM glycoprotein, was also recently highlighted for its role in amplifying fibrosis pathway [70]. TNC can interact with several extracellular matrix molecules and cell receptors, including Toll-like receptor 4 (TLR4). The expression levels of TNC are increased in the visceral AT from obese subjects with normal glycemia or type 2 diabetes with nonalcoholic steatohepatitis [57]. Similarly, expression levels of TNC in epididymal AT was increased in obese mice [71], and fibrosis is attenuated in TNC deficient mice [70]. Thus, TNC is suggested to be a relevant mediator of AT fibrosis via a TLR4-dependent activation of fibroblasts.
4 Cellular origin of adipose tissue fibrosis
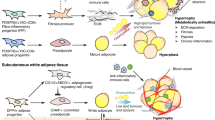
In fibrotic organs, the excessive deposition of extracellular matrix (ECM) starts with the local accumulation of cells producing high level of ECM components. In AT, the fibrosis producing cells originate from resident cells exhibiting features of mesenchymal progenitor cells. In the stroma-vascular fraction, these progenitors are non-hematopoietic cells and display multipotentiality allowing them to become adipocytes, chondrocytes or even osteoblasts among other cell lineages [72, 73]. In AT, they delineate a cell population with a strong adipogenic potential with surface epitope including CD44, CD34, CD29, PDGFRα and PDGFRβ expression. In C3H mice prone to AT fibrosis development [43, 74], PDGFRα+ CD45− CD31− progenitors were isolated as a main contributors to ECM production [74]. In response to fibrogenic stimuli, these cells can differentiate into myofibroblast and start to express αSMA forming cellular stress fibers, high amount of ECM proteins together with autocrine growth factor maintaining cell proliferation and survival [74]. In fibrotic AT, PDGFRα+ cells express the highest levels of the fibrosis markers, such as collagens, as compared to other predominant cells in AT (i.e. adipocytes, endothelial cells, macrophages) [74]. The PDGFRα+ progenitors are not homogeneous populations and, although they need better investigation in AT, lineage tracing experiments suggested that only a subset of the PDGFRα+ cell population originates the pro-fibrotic cells. These progenitors were identified as ADAM12 or GLI1 expressing cells in injured heart, kidney, lung, and liver [75, 76]. In the AT, our team identified the pro-fibrotic cells thanks to the expression level of the tetraspanin CD9 among PDGFRα+ progenitor populations. PDGFRα+ CD9high cells were driven toward a myofibroblastic phenotype, whereas PDGFRα+ CD9low cells were committed to adipogenesis [74]. In the fibrotic AT, PDGFRα+ CD9high progenitor population expands while their PDGFRα+ CD9low counterparts were rapidly lost. In human AT, CD9high and CD9low PDGFRα+ progenitors were equally observed. However, PDGFRα + CD9high cell frequency positively correlated with the degree of fibrosis, and with the deterioration of the glycemic control in patients with obesity. Indeed, significant positive associations were observed between the amount of PDGFRα+ CD9high cells in AT and glycated hemoglobin, fasting glycemia and insulinemia and HOMA-IR, a surrogate of insulin resistance. Thus, an imbalance favoring WAT CD9high over CD9low PDGFRα+ progenitors appears to promote AT fibrotic transformation associated with altered glucose control [74]. More recently, unbiased analysis using single cell RNA sequencing of progenitors from visceral fat depot narrowed the definition of the profibrotic and proinflammatory progenitors (FIP) as CD9high LY6C+ progenitors in mice [77]. In addition to their ability for fibrosis production, the FIP exert strong inhibitory effects on adipogenesis. Such regulatory activity was also described for the progenitor subsets defined by CD142 expression in subcutaneous WAT with adipogenesis-regulatory properties [78]. Furthermore, FIP display important proinflammatory activity as illustrated by their contribution to chemokines and cytokines production in obese AT [74, 77, 79, 80]. Thus, in obesity, the cell progenitors harbor functions that can be highly detrimental for AT homeostasis. Importantly, the interplay between adipogenic and fibrogenic pathways regulate progenitor fates during obesity (Fig. 1). Profibrotic signaling, indeed, also acts as anti-adipogenic pathway as shown with PDGFRα signaling that drives AT fibrosis by limiting progenitor cell adipogenic capacity [74, 81, 82]. Accordingly, PPARγ activity is pivotal in progenitor fate and the bidirectional manipulation of PPARγ expression induced reciprocal changes in driving adipogenic or myofibroblastic fate decision [83].

The interplay between Adipogenic and fibrogenic pathways to shape progenitor fate in adipose tissue. Various signals and transcription factors found to promote beige or white adipogenesis can also limit fibrogenic pathways, and conversely
The interplay between the pro-adipogenic transcription factor ZFP423 (C2H2 zinc finger protein 423) and the TLR4 signaling in the progenitors also controls macrophage accumulation in the AT in response to high fat feeding. Mechanistically, ZFP423 suppresses the DNA-binding capacity of the p65 subunit of NF-κB activated through TLR4 signaling [80]. The immunoregulatory potential of the progenitors not only affects AT macrophage accumulation, but also other immune cells. For example, as a main producer of IL33 in AT [84], the IL33+ PDGFRα+ progenitor subset can control both the accumulation of the regulatory T cell and ILC2 in the AT [84, 85]. With obesity, IL33 is significantly downregulated while the administration of IL33 was associated with a healthy remodeling with increased AT expression of UCP1 [86]. Thus, the progenitors most probably exert critical regulatory functions that can either participate in healthy or unhealthy AT remodeling.
5 Adipogenesis in white adipose tissue and metabolic health
When tipped into storage mode, fat pad growth is driven by both adipocyte hypertrophy (enlarged adipocytes) and hyperplasia (increased cell number). Evidences support that the maintenance of metabolic health involves the increased number of white adipocytes rather than enlargement of adipocytes knowing that bigger cells are more dysfunctional [83, 87]. Oversized adipocytes indeed experience hypoxia and higher mechanical stress that promote a reoriented secretome associated with increased inflammation which promotes insulin resistance. These enlarged adipocytes indeed display induced secretion of tumor necrosis factor α (TNFα), interleukin (IL)-6, IL-8, monocyte chemoattractant protein-1 (MCP-1) and acute-phase serum amyloid A proteins amongst others [88], thus sustaining low grade inflammation in AT. In addition, lower adiponectin secretion and elevated basal lipolysis by adipocytes [89, 90], also favor inflammation [91]. Overall, unaltered adipogenic capacity per se may accompany healthy AT. As such, better understanding of in vivo adipogenesis in human may lead to strategies to uncouple obesity from metabolic diseases.
The generation of new adipocytes requires the proliferation and differentiation of progenitors that reside within the AT stromal cell reservoir. Most of the current knowledge about adipocyte differentiation derived from in vitro study examining heterogenous cell populations including 3T3-L1 cell line, mouse embryonic fibroblast (MEF) and plastic adherent stroma vascular cell fraction of AT. Although very informative, it remained to elucidate how the associated molecular pathways are relevant to in vivo progenitor biology.
The use of markers allowing the specific tracking of these progenitors within the AT combined to single cell RNA sequencing highlight a high diversity of progenitors. Initially, the tracing of PPARγ (peroxisome proliferator-activated receptor gamma)-expressing cells revealed an adipocyte lineage tightly associated with the adipose vasculature [92]. Concomitantly, with multiparameter flow cytometry the use of various antibodies targeting cell surface epitopes, previously reported as mesenchymal stem cells antigens Sca1, CD34, CD29 and PDGFRα delineate a cell population with a strong adipogenic potential [93, 94]. CD24 expressing precursors exhibit stem cell-like properties, which play a role in the maintenance or the growth of local adipocyte precursors [19, 93]. Indeed, sorted CD24+ cells, but not the CD24− cells, transplanted in the residual fat depot of lipodystrophic mice, provided a favorable adipogenic microenvironment enabling the generation of a functional WAT depot. Interestingly, this transplantation led to major metabolic improvement with the rescue of a diabetic phenotype that develops in lipodystrophic animals [93]. In many models of obesity, the activation of the precursors is dependent on the phosphoinositide 3-kinase (PI3K)-AKT2 pathway [19]. Moreover, the coexpression of the pro-adipogenic transcription factors PPARγ and ZFP423 defined a sub-set of progenitors with a strong commitment in the adipocyte lineage [95, 96].
Other studies also identified a preadipocyte factor 1, Pref-1,-expressing progenitors as cells with high proliferative capacity, being early adipose cell precursors prior to cells with the expression of ZFP423 or PPARγ. Upon high-fat feeding stimulation, Pref1+ cells are engaged in adipogenesis. However, upon adipogenesis, Pref1 (also called Dlk1/FA1) expression is downregulated as it prevents adipocyte differentiation to maintain progenitor stemness [97].
Interestingly, Merrick et al. examined the progenitor cell hierarchy in subcutaneous inguinal WAT [98]. The analysis of cellular trajectory in the adipogenic fate pointed out dipeptidyl peptidase–4 (DPP4 +) cells as multipotent progenitors giving rise to both CD54 + and CD142 + cells, which further differentiate into differentiated adipocytes. In this work, the adipogenesis-regulatory properties of CD142+ subset is however not recapitulated. In obesity, the depletion of DPP4+ progenitors leads to reduced precursor differentiation that may contribute to pathological remodeling and metabolic disease progression [98]. Overall, single cell RNA sequencing studies evidenced that progenitor subsets, that may delineate functional differences, are rearranged with AT remodeling [99, 100]. Further investigations are still needed to appreciate subcutaneous versus visceral depot peculiarities. In addition, it remains to clarify whether progenitor clusters represent distinct states of adipogenic differentiation or whether they are independent cell subsets in AT.
6 Interplay between beige adipogenic and fibrogenic pathways
Upon thermogenic or some metabolic stimuli, beige adipocytes can arise in specific regions inside the WAT depot. Depending on the stimulus, beige adipocytes can emerge from preexisting white adipocytes or from AT progenitors [4, 6, 7, 101, 102]. From a metabolic point of view, in obesogenic environment, activating beige adipocytes display therapeutic potential due to their ability to improve glucose and lipid homeostasis [2]. Those beneficial effects were initially attributed to energy burning capacity achieved through non-shivering thermogenesis, during which these cells dissipate chemical energy as heat notably by increasing UCP1 activity. However, recent evidences highlight that pro-beigeing pathways potently repress AT fibrosis (Fig. 1), independently of UCP1 uncoupling function [103]. As such, the PRDM16 transcriptional complex not only activates brown/beige fat development [104], but also potently represses AT fibrosis through its direct interaction with GTF2IRD1 [103]. In addition, PRDM16 dependent metabolic signals arising from adipocytes regulates the progenitor fate blocking fibrosis together with enhancing beige adipogenesis [11]. In this reciprocal relationship between fibrogenesis and beige adipogenesis, the highly conserved canonical TGF-β/BMP (bone morphogenetic proteins) signaling cascade is of particular interest, since members have been shown to produce beige adipogenesis from AT progenitors. The BMP7-ROCK signaling axis regulates the formation of beige adipocytes via controlling the G-actin-regulated transcriptional coactivator myocardin related transcription factor A (MRTFA) [105]. WAT from mice deficient for MRTFA contains more multilocular adipocytes and expresses enhanced levels of UCP1 [105]. Conversely, MRTFA was highlighted as an inducer of progenitor fibrotic fate [106]. Similarly, in AT, BMP4 signaling is known to induce commitment of pluripotent stem cells to the adipocyte lineage by producing cells that possess the characteristics of preadipocytes. As such, the overexpression of a BMP4 transgene promotes a healthy WAT remodeling with reduced AT mass and white adipocyte size along with an increased number of beige, thermogenic adipocytes (i.e. adipocytes enriched with mitochondria and uncoupling protein 1) [107, 108]. Most interestingly, adding BMP in a profibrotic environmental promotes the resolution of fibrosis driving myofibroblast dedifferentiation to regenerate the adipocyte pool [109]. The transcriptional landscape of TGF-β/BMP family can be regulated by the progenitor in a cell autonomous dependent manner [110], as shown in mice harboring autophagy deficient progenitors. In these mice, the emergence of beige adipocyte features in the white fat depot was coincident with lower fibrosis expression (110).
In human, the ability to develop beige adipocytes is observed in limited situations such as burn trauma victims and pheochromocytoma patients [111, 112]. However, in vitro experimentation revealed that progenitors isolated from human AT can undergo beige adipogenesis [113]. Interestingly, progenitors defined with high or low expression of CD34 appeared to have similar adipogenic properties but are characterized by unique molecular profiles with different potential for adaptive thermogenesis [114]. However, the development of a pro-inflammatory microenvironment in the obese WAT seems to restrict the beige adipogenic potential of the progenitors [113].
7 Conclusions
AT progenitors are a highly heterogenous population of stromal cells. Subsets are defined through not only their degree of commitment toward white or beige adipogenesis but also through their immunoregulatory or fibrogenic potential. The AT exhibits a complex lobular architecture that is suggested to provide a local environment influencing the progenitor phenotype and functionality [115]. Therefore, the functional heterogeneity of the progenitor can also be explained by a spatial and temporal heterogeneity in addition to specific depot microenvironments [116]. Given the pivotal role of progenitors in maintaining AT homeostasis, a better understanding of their biology is certainly of interest in a therapeutic perspective. Future studies will aim to identify molecular and surface markers allowing the discrimination of the various progenitor sub-populations to understand how they crosstalk with adipocytes and other stromal cells in the adipose tissue.
References
Cannon B, Nedergaard J. Brown Adipose Tissue: Function and Physiological Significance. Physiol Rev. 2004;84(1):277–359.
Chouchani ET, Kajimura S. Metabolic adaptation and maladaptation in adipose tissue. Nat Metab. 2019;1(2):189–200.
Wu J, Cohen P, Spiegelman BM. Adaptive thermogenesis in adipocytes: Is beige the new brown? Genes Dev. 2013;27(3):234–50.
Vishvanath L, et al. Pdgfrβ+ mural preadipocytes contribute to adipocyte hyperplasia induced by high-fat-diet feeding and prolonged cold exposure in adult mice. Cell Metab. 2016;23(2):350–9.
Wang QA, Scherer PE. The AdipoChaser mouse. Adipocyte. 2014;3(2):146–50.
Wang QA, Tao C, Gupta RK, Scherer PE. Tracking adipogenesis during white adipose tissue development, expansion and regeneration. Nat Med. 2013;19(10):1338–44.
Lee Y-H, Petkova AP, Mottillo EP, Granneman JG. In vivo identification of bipotential adipocyte progenitors recruited by β3-adrenoceptor activation and high-fat feeding. Cell Metab. 2012;15(4):480–91.
Lee Y-H, Petkova AP, Granneman JG. Identification of an adipogenic niche for adipose tissue remodeling and restoration. Cell Metab. 2013;18(3):355–67.
Burl RB, et al. Deconstructing adipogenesis induced by β3-adrenergic receptor activation with single-cell expression profiling. Cell Metab. 2018;28(2):300-309.e4.
Carrière A, et al. Browning of white adipose cells by intermediate metabolites: an adaptive mechanism to alleviate redox pressure. Diabetes. 2014;63(10):3253–65.
Wang W, et al. A PRDM16-driven metabolic signal from adipocytes regulates precursor cell fate. Cell Metab. 2019;30(1):174-189.e5.
Kajimura S, Spiegelman BM, Seale P. Brown and beige fat: Physiological roles beyond heat-generation. Cell Metab. 2015;22(4):546–59.
Wang W, Seale P. Control of brown and beige fat development. Nat Rev Mol Cell Biol. 2016;17(11):691–702.
Leitner BP, et al. Mapping of human brown adipose tissue in lean and obese young men. Proc Natl Acad Sci U S A. 2017;114(32):8649–54.
Kissebah AH, Krakower GR. Regional adiposity and morbidity. Physiol Rev. 1994;74(4):761–811.
Wajchenberg BL. Subcutaneous and Visceral Adipose Tissue: Their Relation to the Metabolic Syndrome. Endocr Rev. 2000;21(6):697–738.
Ibrahim MM. Subcutaneous and visceral adipose tissue: structural and functional differences. Obes Rev. 2010;11(1):11–8.
Schleinitz D, Böttcher Y, Blüher M, Kovacs P. The genetics of fat distribution. Diabetologia. 2014;57(7):1276–86.
Jeffery E, et al. The adipose tissue microenvironment regulates depot-specific adipogenesis in obesity. Cell Metab. 2016;24(1):142–50.
Marcelin G, Silveira ALM, Martins LB, Ferreira AVM, Clément K. Deciphering the cellular interplays underlying obesity-induced adipose tissue fibrosis. J Clin Invest. 2019;129(10):4032–40.
Kim J-Y, et al. Obesity-associated improvements in metabolic profile through expansion of adipose tissue. J Clin Invest. 2007;117(9):2621–37.
Kusminski CM, Bickel PE, Scherer PE. Targeting adipose tissue in the treatment of obesity-associated diabetes. Nat Rev Drug Discovery. 2016;15(9):639–60.
Gao H, et al. Lipoatrophy and metabolic disturbance in mice with adipose-specific deletion of kindlin-2 [Internet]. JCI Insight 4(13). https://doi.org/10.1172/jci.insight.128405.
Gavrilova O, et al. Surgical implantation of adipose tissue reverses diabetes in lipoatrophic mice. J Clin Invest. 2000;105(3):271–8.
Sung H-K, et al. Adipose vascular endothelial growth factor regulates metabolic homeostasis through angiogenesis. Cell Metab. 2013;17(1):61–72.
An YA et al. Angiopoietin-2 in white adipose tissue improves metabolic homeostasis through enhanced angiogenesis. Elife 2017;6. https://doi.org/10.7554/eLife.24071.
Robciuc MR, et al. VEGFB/VEGFR1-nduced expansion of adipose vasculature counteracts obesity and related metabolic complications. Cell Metab. 2016;23(4):712–24.
Wree A, et al. Adipokine expression in brown and white adipocytes in response to hypoxia. J Endocrinol Invest. 2012;35(5):522–7.
Michailidou Z, et al. Increased angiogenesis protects against adipose hypoxia and fibrosis in metabolic disease-resistant 11β-hydroxysteroid dehydrogenase type 1 (HSD1)-deficient mice. J Biol Chem. 2012;287(6):4188–97.
Hill DA, et al. Distinct macrophage populations direct inflammatory versus physiological changes in adipose tissue. Proc Natl Acad Sci USA. 2018;115(22):E5096–105.
Giordano A, et al. Obese adipocytes show ultrastructural features of stressed cells and die of pyroptosis. J Lipid Res. 2013;54(9):2423–36.
Cinti S, et al. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J Lipid Res. 2005;46(11):2347–55.
Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259(5091):87–91.
Ferrante AW. The immune cells in adipose tissue. Diabetes Obes Metab. 2013;15(Suppl 3):34–8.
Weisberg SP, et al. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112(12):1796–808.
Cancello R, et al. Increased infiltration of macrophages in omental adipose tissue is associated with marked hepatic lesions in morbid human obesity. Diabetes. 2006;55(6):1554–61.
de Luca C, Olefsky JM. Inflammation and Insulin Resistance. FEBS Lett. 2008;582(1):97–105.
Villaret A, et al. Adipose tissue endothelial cells from obese human subjects: differences among depots in angiogenic, metabolic, and inflammatory gene expression and cellular senescence. Diabetes. 2010;59(11):2755–63.
Rouault C, et al. Senescence-associated β-galactosidase in subcutaneous adipose tissue associates with altered glycaemic status and truncal fat in severe obesity. Diabetologia. 2021;64(1):240–54.
Wernstedt Asterholm I, et al. Adipocyte inflammation is essential for healthy adipose tissue expansion and remodeling. Cell Metab. 2014;20(1):103–18.
Mauer J, et al. Signaling by IL-6 promotes alternative activation of macrophages to limit endotoxemia and obesity-associated resistance to insulin. Nat Immunol. 2014;15(5):11.
Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. 2012;18(7):1028–40.
Vila IK, et al. Immune cell Toll-like receptor 4 mediates the development of obesity- and endotoxemia-associated adipose tissue fibrosis. Cell Rep. 2014;7(4):1116–29.
Frantz C, Stewart KM, Weaver VM. The extracellular matrix at a glance. J Cell Sci. 2010;123(Pt 24):4195–200.
Datta R, Podolsky MJ, Atabai K. Fat fibrosis: friend or foe? [Internet]. JCI Insight 2018;3(19). https://doi.org/10.1172/jci.insight.122289.
Tschumperlin DJ, Ligresti G, Hilscher MB, Shah VH. Mechanosensing and fibrosis. J Clin Invest. 2018;128(1):74–84.
Divoux A. Fibrosis in human adipose tissue: composition, distribution, and link with lipid metabolism and fat mass loss. - PubMed - NCBI [Internet]2010;https://www.ncbi.nlm.nih.gov/pubmed/?term=Divoux+A%2C+et+al.+(2010).+Diabetes+59(11)%3A2817-2825. cited September 26, 2018.
Bel Lassen P, et al. The FAT score, a fibrosis score of adipose tissue: predicting weight-loss outcome after gastric bypass. J Clin Endocrinol Metab. 2017;102(7):2443–53.
Abdennour M, et al. Association of adipose tissue and liver fibrosis with tissue stiffness in morbid obesity: links with diabetes and BMI loss after gastric bypass. J Clin Endocrinol Metab. 2014;99(3):898–907.
Sun K, Tordjman J, Clément K, Scherer PE. Fibrosis and adipose tissue dysfunction. Cell Metab. 2013;18(4):470–7.
Guglielmi V, et al. Omental adipose tissue fibrosis and insulin resistance in severe obesity. Nutr Diabetes 2015;5:e175.
Sun K, et al. Endotrophin triggers adipose tissue fibrosis and metabolic dysfunction. Nat Commun. 2014;5(1):3485.
Jun JI, Lau LF. Resolution of organ fibrosis. J Clin Invest 128(1):97–107.
Tacke F, Trautwein C. Mechanisms of liver fibrosis resolution. J Hepatol. 2015;63(4):1038–9.
Liu Y, et al. Accumulation and changes in composition of collagens in subcutaneous adipose tissue after bariatric surgery. J Clin Endocrinol Metab. 2016;101(1):293–304.
Zamarron BF, et al. Macrophage proliferation sustains adipose tissue inflammation in formerly obese mice. Diabetes. 2017;66(2):392–406.
Sasso M, et al. AdipoScan: a novel transient elastography-based tool used to non-invasively assess subcutaneous adipose tissue shear wave speed in obesity. Ultrasound Med Biol. 2016;42(10):2401–13.
Pellegrinelli V, et al. Human adipocyte function is impacted by mechanical cues: Human adipocytes as mechanosensitive cells. J Pathol. 2014;233(2):183–95.
Li Q, Hosaka T, Jambaldorj B, Nakaya Y, Funaki M. Extracellular matrix with the rigidity of adipose tissue helps 3T3-L1 adipocytes maintain insulin responsiveness. J Medic Investigation 2009;56(3,4):142–149.
Hossain MdG, et al. Compressive force inhibits adipogenesis through COX-2-mediated down-regulation of PPARγ2 and C/EBPα. J Biosci Bioeng. 2010;109(3):297–303.
Chun T-H, et al. A pericellular collagenase directs the 3-dimensional development of white adipose tissue. Cell. 2006;125(3):577–91.
Khan T, et al. Metabolic dysregulation and adipose tissue fibrosis: role of collagen VI. Mol Cell Biol. 2009;29(6):1575–91.
Alba DL, et al. Subcutaneous fat fibrosis links obesity to insulin resistance in chinese americans. J Clin Endocrinol Metab. 2018;103(9):3194–204.
Walker RW, et al. Macrophage accumulation and fibrosis in adipose tissue is linked to liver damage and metabolic risk in obese children. Obesity (Silver Spring). 2014;22(6):1512–9.
Park J, Morley TS, Scherer PE. Inhibition of endotrophin, a cleavage product of collagen VI, confers cisplatin sensitivity to tumours. EMBO Mol Med. 2013;5(6):935–48.
Denhardt DT, Noda M, O’Regan AW, Pavlin D, Berman JS. Osteopontin as a means to cope with environmental insults: regulation of inflammation, tissue remodeling, and cell survival. J Clin Invest. 2001;107(9):1055–61.
Kiefer FW, et al. Osteopontin expression in human and murine obesity: extensive local up-regulation in adipose tissue but minimal systemic alterations. Endocrinology. 2008;149(3):1350–7.
Kiefer FW, et al. Neutralization of osteopontin inhibits obesity-induced inflammation and insulin resistance. Diabetes. 2010;59(4):935–46.
Sawaki D, et al. Visceral adipose tissue drives cardiac aging through modulation of fibroblast senescence by Osteopontin Production 51.
Bhattacharyya S, et al. Tenascin-C drives persistence of organ fibrosis. Nat Commun. 2016;7(1):11703.
Catalán V, et al. Increased tenascin C and Toll-like receptor 4 levels in visceral adipose tissue as a link between inflammation and extracellular matrix remodeling in obesity. J Clin Endocrinol Metab. 2012;97(10):E1880-1889.
Farahani RM, Xaymardan M. Platelet-derived growth factor receptor alpha as a marker of mesenchymal stem cells in development and stem cell biology [Internet]. Stem Cells Intern. 2015;2015:e362753.
Santini MP, et al. Tissue-resident PDGFRα+ progenitor cells contribute to fibrosis versus healing in a context- and spatiotemporally dependent manner. Cell Rep. 2020;30(2):555-570.e7.
Marcelin G, et al. A PDGFRα-mediated switch toward CD9(high) adipocyte progenitors controls obesity-induced adipose tissue fibrosis. Cell Metab. 2017;25(3):673–85.
Dulauroy S, Di Carlo SE, Langa F, Eberl G, Peduto L. Lineage tracing and genetic ablation of ADAM12+ perivascular cells identify a major source of profibrotic cells during acute tissue injury. Nat Med. 2012;18(8):1262–70.
Kramann R, et al. Perivascular Gli1+ progenitors are key contributors to injury-induced organ vibrosis. Cell Stem Cell. 2015;16(1):51–66.
Hepler C, et al. Identification of functionally distinct fibro-inflammatory and adipogenic stromal subpopulations in visceral adipose tissue of adult mice. eLife 2018;7:e39636.
Schwalie PC, et al. A stromal cell population that inhibits adipogenesis in mammalian fat depots. Nature. 2018;559(7712):103.
Kaplan JL, et al. Adipocyte progenitor cells initiate monocyte chemoattractant protein-1-mediated macrophage accumulation in visceral adipose tissue. Mol Metab. 2015;4(11):779–94.
Shan B, et al. Perivascular mesenchymal cells control adipose-tissue macrophage accrual in obesity [Internet]. Nat Metab [published online ahead of print: November 2, 2020] https://doi.org/10.1038/s42255-020-00301-7.
Olson LE, Soriano P. Increased PDGFRalpha activation disrupts connective tissue development and drives systemic fibrosis. Dev Cell. 2009;16(2):303–13.
Iwayama T, et al. PDGFRα signaling drives adipose tissue fibrosis by targeting progenitor cell plasticity. Genes Dev. 2015;29(11):1106–19.
Shao M, et al. De novo adipocyte differentiation from Pdgfrβ + preadipocytes protects against pathologic visceral adipose expansion in obesity. Nat Commun. 2018;9(1):890.
Spallanzani RG, et al. Distinct immunocyte-promoting and adipocyte-generating stromal components coordinate adipose tissue immune and metabolic tenors. Sci Immunol. 2019;4(35):eaaw3658.
Brestoff JR, et al. Group 2 innate lymphoid cells promote beiging of adipose and limit obesity. Nature. 2015;519(7542):242–6.
Ding X, et al. IL-33-driven ILC2/eosinophil axis in fat is induced by sympathetic tone and suppressed by obesity. J Endocrinol. 2016;231(1):35–48.
Kim SM, et al. Loss of white adipose hyperplastic potential is associated with enhanced susceptibility to insulin resistance. Cell Metab. 2014;20(6):1049–58.
Jernås M, et al. Separation of human adipocytes by size: hypertrophic fat cells display distinct gene expression. FASEB J. 2006;20(9):1540–2.
Berger JJ, Barnard RJ. Effect of diet on fat cell size and hormone-sensitive lipase activity. J Appl Physiol. 1999;87(1):227–32.
Wueest S, Rapold RA, Rytka JM, Schoenle EJ, Konrad D. Basal lipolysis, not the degree of insulin resistance, differentiates large from small isolated adipocytes in high-fat fed mice. Diabetologia. 2009;52(3):541–6.
Lancaster GI, et al. Evidence that TLR4 Is Not a Receptor for Saturated Fatty Acids but Mediates Lipid-Induced Inflammation by Reprogramming Macrophage Metabolism. Cell Metab. 2018;27(5):1096-1110.e5.
Tang W, et al. White fat progenitor cells reside in the adipose vasculature. Science. 2008;322(5901):583–6.
Rodeheffer MS, Birsoy K, Friedman JM. Identification of white adipocyte progenitor cells in vivo. Cell. 2008;135(2):240–9.
Berry R, Jeffery E, Rodeheffer MS. Weighing in on adipocyte precursors. Cell Metab. 2014;19(1):8–20.
Gupta RK, et al. Zfp423 expression identifies committed preadipocytes and localizes to adipose endothelial and perivascular cells. Cell Metab. 2012;15(2):230–9.
Gupta RK, et al. Transcriptional control of preadipocyte determination by Zfp423. Nature. 2010;464(7288):619–23.
Hudak CS, Sul HS. Pref-1, a Gatekeeper of Adipogenesis [Internet]. Front Endocrinol. 2013;4. https://doi.org/10.3389/fendo.2013.00079.
Merrick D,, et al. Identification of a mesenchymal progenitor cell hierarchy in adipose tissue. Dev Biol. 2019;13.
Vijay J, et al. Single-cell analysis of human adipose tissue identifies depot- and disease-specific cell types. Nat Metab. 2020;2(1):97–109.
Cho DS, Lee B, Doles JD. Refining the adipose progenitor cell landscape in healthy and obese visceral adipose tissue using single-cell gene expression profiling. Life Sci. Alliance 2019;2(6):e201900561.
Berry DC, Jiang Y, Graff JM. Mouse strains to study cold-inducible beige progenitors and beige adipocyte formation and function [Internet]. Nat Commun. 2016;7. https://doi.org/10.1038/ncomms10184.
Shao M, et al. Cellular origins of beige fat cells revisited. Diabetes. 2019;68(10):1874–85.
Hasegawa Y, et al. Repression of adipose tissue fibrosis through a PRDM16-GTF2IRD1 complex improves systemic glucose homeostasis. Cell Metab. 2018;27(1):180-194.e6.
Shapira SN, Seale P. Transcriptional Control of Brown and Beige Fat Development and Function. Obesity. 2019;27(1):13–21.
McDonald ME, et al. Myocardin-related transcription factor a regulates conversion of progenitors to beige adipocytes. Cell. 2015;160(1):105–18.
Lin JZ, Rabhi N, Farmer SR. Myocardin-related transcription factor a promotes recruitment of ITGA5+ Profibrotic progenitors during obesity-induced adipose tissue fibrosis. Cell Rep. 2018;23(7):1977–87.
Qian S-W, et al. BMP4-mediated brown fat-like changes in white adipose tissue alter glucose and energy homeostasis. Proc Natl Acad Sci. 2013;110(9):E798–807.
Hoffmann JM, et al. BMP4 gene therapy enhances insulin sensitivity but not adipose tissue browning in obese mice. Molecular Metabolism. 2020;32:15–26.
Plikus MV, et al. Regeneration of fat cells from myofibroblasts during wound healing. Science. 2017;355(6326):748–52.
Marcelin G, et al. Autophagy inhibition blunts PDGFRα+ adipose progenitors’ cell-autonomous fibrogenic response to high fat diet. Autophagy (in press). 2020
Frontini A, et al. White-to-brown transdifferentiation of omental adipocytes in patients affected by pheochromocytoma. Biochimica et Biophysica Acta (BBA) - Mol and Cell Biol Lipids 2013;1831(5):950–959.
Sidossis LS, et al. Browning of subcutaneous white adipose tissue in humans after severe adrenergic stress. Cell Metab. 2015;22(2):219–27.
Estève D, et al. Human white and brite adipogenesis is supported by MSCA1 and is impaired by immune cells. Stem Cells. 2015;33(4):1277–91.
Raajendiran A, et al. Identification of metabolically distinct adipocyte progenitor cells in human adipose tissues. Cell Rep. 2019;27(5):1528-1540.e7.
Estève D, et al. Lobular architecture of human adipose tissue defines the niche and fate of progenitor cells. Nat Commun. 2019;10(1):2549.
Sebo ZL, Jeffery E, Holtrup B, Rodeheffer MS. A mesodermal fate map for adipose tissue. Development 2018;145(17). https://doi.org/10.1242/dev.166801.
Acknowledgments
We acknowledge supports from the Fondation pour la Recherche Medicale (“équipe FRM”), the French National Agency of Research (Adipofib and Captor programs), the research programs CAPES (Coordenação de Aperfeiçooamento de Pessoal de Nível Superior) / COFECUB (Comité Français d'Évaluation de la Coopération Universitaire et Scientifique avec le Brésil), the french clinical program (CRC-fibroTA), the European Foundation for the Study of Diabetes (EFSD), the SFN (Société Française de Nutrition), the AFERO (Association Française d'Etude et de Recherche sur l'Obésité) and the Benjamin Delessert institute.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
No conflict of interest to declare for this present work.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Marcelin, G., Clément, K. The multifaceted progenitor fates in healthy or unhealthy adipose tissue during obesity. Rev Endocr Metab Disord 22, 1111–1119 (2021). https://doi.org/10.1007/s11154-021-09662-0
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11154-021-09662-0