
Abstract
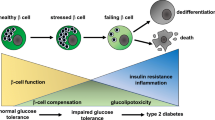
Type 2 diabetes mellitus is a complex disease characterized by β-cell failure in the setting of insulin resistance. In early stages of the disease, pancreatic β-cells adapt to insulin resistance by increasing mass and function. As nutrient excess persists, hyperglycemia and elevated free fatty acids negatively impact β-cell function. This happens by numerous mechanisms, including the generation of reactive oxygen species, alterations in metabolic pathways, increases in intracellular calcium and the activation of endoplasmic reticulum stress. These processes adversely affect β-cells by impairing insulin secretion, decreasing insulin gene expression and ultimately causing apoptosis. In this review, we will first discuss the regulation of β-cell mass during normal conditions. Then, we will discuss the mechanisms of β-cell failure, including glucotoxicity, lipotoxicity and endoplasmic reticulum stress. Further research into mechanisms will reveal the key modulators of β-cell failure and thus identify possible novel therapeutic targets. Type 2 diabetes mellitus is a multifactorial disease that has greatly risen in prevalence in part due to the obesity and inactivity that characterize the modern Western lifestyle. Pancreatic β-cells possess the potential to greatly expand their function and mass in both physiologic and pathologic states of nutrient excess and increased insulin demand. β-cell response to nutrient excess occurs by several mechanisms, including hypertrophy and proliferation of existing β-cells, increased insulin production and secretion, and formation of new β-cells from progenitor cells [1, 2]. Failure of pancreatic β-cells to adequately expand in settings of increased insulin demand results in hyperglycemia and diabetes. In this review, we will first discuss the factors involved in β-cell growth and then discuss the mechanisms by which β-cell expansion fails and leads to β-cell failure and diabetes (Fig. 1).

The mechanisms by which -cell failure and apoptosis occur are complex, not completely unraveled and involve the interplay of numerous factors and conditions. These factors are summarized in this figure. Glucotoxicity and lipotoxicity lead to the production of ROS, which activate JNK. JNK activity leads to a decrease in IRS signaling and may directly be involved in decreased Pdx-1 activity by translocation from the nucleus to the cytoplasm [183]. In addition, glucose and FA have both been found to induce ER stress. Chronic glucose elevation inhibits FA oxidation and favors the generation of ceramide and lipid partitioning, which ultimately results in β-cell dysfunction and apoptosis. AMPK activation promotes fatty acid oxidation by phosphorylation and inhibition of acetyl-CoA carboxylase or via down regulation of the transcription factor sterol-regulatory-element-binding-protein-1c (SREBP1c) and subsequent decreases in acetyl-CoA carboxylase. Glucose and FA also activate the UPR response and induce ER stress. The ER stress response and its effectors are activated in response to misfolded proteins in order to protect β-cells from apoptosis; however, activation of these processes under conditions of long-term elevation of FFA and glucose can lead to β-cell dysfunction and ultimately apoptosis. Activation of ER stress leads to inhibition of insulin mRNA and protein expression and may also be pro-apoptotic. The mechanisms for induction of apoptosis by ER stress are not completely known, but induction of CHOP is an important component. In addition, induction of ATF3 and SREBP can downregulate IRS signaling by repressing IRS2 transcription. One interesting finding is that inhibition of IRS signaling seems to be a common pathway induced by the majority of the mechanisms described for β-cell failure. An additional event is the increase in mTOR signaling by nutrient excess (glucose). This results in negative feedback inhibition on IRS1 and possibly IRS2 by activation of S6K signaling. The decrease in IRS signaling induces GSK3β and Foxo1 function. Activation of these molecules ultimately reduces Pdx1 levels and increases the levels of the cell cycle inhibitor p27
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Importance of proliferation, neogenesis, apoptosis
On the basis of multiple studies performed by several laboratories, it has been shown that the respective contributions of β-cell proliferation, neogenesis and apoptosis to overall β-cell mass varies at different stages of postnatal life as well as in response to stress conditions [1–8]. Maintenance of β-cell mass in adult life results predominantly from proliferation of pre-existing β-cells [9]. However, recent data using lineage tracing demonstrates that neogenesis from ductal progenitors can also contribute to β-cell mass during normal conditions or after β-cell injury [10]. The overall contribution of neogenesis to maintenance of β-cell mass in postnatal life is unclear.
1.1 Response to increased insulin demand—β-cell proliferation
The regulation of pancreatic β-cell mass occurs via the interplay of multiple proteins. The three major classes of cell cycle proteins—cyclins, cyclin-dependent kinases (CDKs), and cyclin-dependent kinase inhibitors (CKIs) have been extensively studied and have been found to govern cell cycle progression in various mammalian cell types [11]. Their role in β-cell proliferation as demonstrated in various rodent models is briefly summarized here. Cyclin D1 and cyclin D2 are both expressed in β-cells. Cyclin D1 knockout mice exhibit normal islet cell size and number. However, cyclin D1 overexpression in mice has been found to increase β-cell proliferation and mass in vivo [12]. Cyclin D2 is essential for regulation of β-cell mass; cyclin D2 knockout mice have decreased β-cell mass and decreased insulin levels [13, 14]. In the class of cyclin-dependent kinases (CDKs), CDK4 and CDK2 are expressed in β-cells. CDK4 knockout mice exhibit normal islets at birth but develop diabetes early in life [15]. The cyclin-dependent kinase inhibitors (CKIs), including the four INK4 proteins and the three members of the Cip/Kip family, are all expressed in pancreatic islets [16]. INK4 proteins complex with CDK4 and prevent the binding of CDK4 or CDK6 to cyclin D1, leading to cell cycle arrest [17]. Mice with a mutant form of CDK4 that is resistant to binding by INK4 proteins exhibit islet hyperplasia [15, 18]. P27Kip1 is particularly interesting because it is regulated by the insulin signaling pathway and is thought to be a major factor in the regulation of β-cell mass. Mice lacking p27 show improved glucose tolerance with increased β-cell mass and proliferation as well as increased serum insulin levels [19, 20]. In contrast, mice overexpressing p27 during the early neonatal period show reduced β-cell mass and glucose intolerance [19]. However, overexpression of p27 in adult mice has no effect on glucose tolerance or β-cell mass. This suggests that p27 functions more in the context of β-cell development or during proliferative conditions and plays a greater role during early postnatal life than during adult life. In addition, β-cells from db/db mice exhibit increased p27 levels and deletion of this inhibitor protein rescues the diabetes from mice deficient in IRS2, indicating that p27 is part of the abnormal adaptation of β-cells to insulin resistance [20]. In summary, maintenance of β-cell mass in adult life during normal conditions results predominantly from proliferation of pre-existing β-cells and this process is dependent on the balance between numerous cell cycle proteins [9, 13, 15, 21].
1.2 Response to increased insulin demand—neogenesis
The contribution of neogenesis to the maintenance of β-cell mass in both normal conditions and during adaptation to stress has been debated. The formation of new β-cells from precursor cells is a process that normally halts after birth. Numerous studies have implicated the pancreatic ducts as a source of new β-cells under regenerative conditions or after exposure to exendin-4 or beta-cellulin [7, 8, 22–25]. Most recently, Bonner-Weir et al. [10] genetically labeled pancreatic ductal cells with carbonic anhydrase II to serve as a duct cell-specific promoter to drive Cre recombinase. This technique allowed for the identification of ductal cells as the source of new islets via lineage tracing experiments.
1.3 Response to increased insulin demand—apoptosis
Apoptosis in β-cells is a highly complex process, governed by pro- and anti-apoptotic genes, extracellular signals and intracellular ATP levels [26]. β-cell apoptosis is a major contributor to the development of type 1 diabetes, with a reduction in β-cell mass of approximately 70% at the time of diagnosis. In contrast, there is a 25–50% reduction in β-cell mass seen at the time of diagnosis of type 2 diabetes, suggesting that β-cell dysfunction also contributes to the initial disease process [27, 28]. However, β-cell apoptosis could be a major contributor to the pathogenesis of β-cell failure in late stages of the development of Type 2 Diabetes [29, 30].
2 Factors that govern β-cell mass
2.1 Glucose and nutrients
Evidence implicating the role of glucose in β-cell mass and proliferation comes from in vitro and in vivo experiments. Glucose induces β-cell replication in fetal islets and adult rat islets [31, 32]. In vivo experiments have shown that rats and mice subjected to glucose infusion demonstrate increased β-cell mass [33–35]. These results have also been demonstrated using insulinoma cells [36]. Some evidence suggests that glucose also increases β-cell mass via anti-apoptotic effects in rats treated with glucose infusion and in insulinoma cells [34]. The contribution of proliferation, cell size and apoptosis in response to glucose infusion is unclear, but the early changes appear to be mediated by increases in proliferation and cell size.
The signaling pathways and mechanism by which glucose stimulates β-cell mass and proliferation are not entirely understood. Several pathways have been implicated: (1) Autocrine effects of secreted insulin. (2) Induction of calcium signaling. (3) Activation of the TSC2/mTOR signaling pathway. Glucose activation of the insulin receptor activates Akt signaling, via autocrine effects of secreted insulin [37, 38]. Activation of the insulin receptor and other receptor tyrosine kinases activates Akt signaling, a major pathway for β-cell proliferation [39, 40]. In addition to activation of Akt signaling, glucose can induce the mTOR signaling pathway directly or indirectly, by increasing the concentration of ATP and subsequent inactivation of AMP kinase. Recent data suggests that the mTOR signaling pathway is an important regulator of β-cell mass and proliferation [41–45]. Finally, an increase in intracellular calcium could also play an important role in growth signals induced by glucose as demonstrated by recent studies addressing the importance of calcineurin, the only calcium-regulated phosphatase. β-cell-specific deletion of the calcineurin phosphatase regulatory subunit, calcineurin b1 (Cnb1), induces age-dependent diabetes characterized by decreased β-cell proliferation and mass. This phenotype was rescued by overexpression of active NFATc1, indicating the importance of this pathway in regulation of β-cell mass by calcium signals [41]. Activation of other transcription factors like CREB and SRF could also participate in growth responses induced by glucose/calcium signaling [42, 43]. It is important to note that while moderate increases in glucose levels induce β-cell proliferation and survival, prolonged exposure of β-cells to significant elevations in blood glucose levels causes impaired proliferation and increased β-cell failure and apoptosis (see glucotoxicity below).
2.2 Growth factors
2.2.1 Prolactin, placental lactogen, growth hormone, PTHrP and HGF
Numerous growth factors have been shown to regulate β-cell proliferation. The lactogens prolactin, placental lactogen (PL) and growth hormone (GH) increase β-cell mass in response to increased insulin demand. Prolactin receptors are expressed by β-cells and their expression is upregulated during pregnancy [44]. In vitro experiments in islets showed that incubation with prolactin, placental lactogen and growth hormone leads to increased β-cell proliferation [45, 46]. Transgenic mice overexpressing PL in β-cells exhibit increased β-cell mass and hyperinsulinemia [47]. Moreover, mice lacking the prolactin receptor or the growth hormone receptor have reduced β-cell mass, decreased insulin secretion and glucose intolerance [48, 49]. Another factor that has been shown to augment β-cell proliferation in in vitro and in vivo studies is parathyroid hormone-related protein (PTHrP) [50, 51]. However, it is notable that PTHrP knockout mice have normal β-cell mass as compared to controls, suggesting the PTHrP may not be crucial for normal β-cell mass in vivo [52].
Hepatocyte growth factor (HGF) is a mesenchyme-derived growth factor that is involved in cell proliferation, migration and differentiation in a variety of tissues [53]. Numerous studies have demonstrated the role of HGF in the function and proliferation of β-cells [54]. Activated HGF exerts its actions via the transmembrane c-met receptor [55]. HGF and c-met are expressed at high levels during early pancreatic development and are subsequently maintained at low levels in adult rats [56]. Transgenic mice with overexpression of HGF have increased β-cell mass and proliferation and display improved glucose tolerance; isolated β-cells of these animals show increased glucose-stimulated insulin secretion (GSIS) and glucose utilization [54]. Furthermore, mice injected with exogenous HGF gene prior to receiving streptozotocin showed a protective effect on β-cell death and an increase in β-cell proliferation relative to controls [57]. In contrast, mice with deletion of c-met in β-cells are glucose-intolerant and demonstrate impaired GSIS [58]. These glucose intolerant mice have normal β-cell mass and proliferation, suggesting that HGF is essential for normal insulin secretion, but is not crucial for β-cell development. Studies thus far suggest that HGF upregulates β-cell proliferation, decreases apoptosis, and increases β-cell function, making it an attractive potential target for therapy [59].
These growth factors impact β-cell proliferation via diverse signaling pathways. Prolactin and placental lactogen both bind to the prolactin receptor. The prolactin receptor and growth hormone receptor belong to the cytokine family of receptors which act via the Janus Kinase (JAK)/Signal Transduction and Activators of Transcription (STAT) pathway [60]. Activation of this pathway results in upregulation of Cyclin D2 [61]. Cyclin D2, as discussed above, is essential for β-cell proliferation. PTHrP exerts its effects on β-cell proliferation via the adenylate cyclase/PKA and MAP kinase pathways [11, 62]. This leads to inactivation of the JNK/c-Jun pathway by dephosphorylation, leading to upregulation of insulin gene expression [62]. HGF binds to the c-met receptor and causes activation of the MAPK and PI3K/Akt pathways, leading to β-cell proliferation [54]. In addition, in vitro studies in HGF-treated INS-1 cells demonstrated increased expression of Protein Kinase C, suggesting another pathway by which HGF increases β-cell proliferation [63].
2.2.2 Insulin growth factors and insulin
The insulin-like growth factors I (IGF-I) and II (IGF-II) and their receptors are expressed at different stages during pancreatic development. Insulin and IGF-I bind to the insulin and IGF-I receptor respectively, but can each also cross-react with the other receptor. This makes it difficult to separate the specific effect of these peptides in β-cell growth. IGF-I and IGF-II increase β-cell proliferation in rat islets and insulinoma cell lines in vitro [64–66]. Glucose enhances IGF-I mediated proliferation of insulinoma cells in culture and this process is phosphoinositide 3-kinase (PI3K) dependent [36]. Interestingly, overexpression of IGF-I in β-cells is associated with increased β-cell proliferation, but not mass [67]. In contrast, transgenic mice overexpressing IGF-II exhibit an increase in β-cell mass due to augmented proliferation [68]. Taken together, there is clear evidence that the IGF molecules are important factors in β-cell proliferation and mass.
The effect of insulin receptor signaling on the β-cell has been assessed in both in vivo and in vitro models. Insulin infusion in mice induces β-cell proliferation and increases β-cell mass and this effect is augmented by concomitant glucose infusion [33]. Mice deficient in the insulin receptor in β-cells exhibit hyperglycemia and reduced β-cell mass with age [37]. MIN6 cells with 80% knockdown of the insulin receptor exhibit decreased proliferation, suggesting that insulin is a growth factor at least for this insulinoma cell line [69]. It is important to mention that the effects of the insulin receptor in β-cell function are less clear.
The abnormalities demonstrated in animal models deficient in insulin receptor substrate 2 (IRS2) suggest that events downstream of the insulin and IGF receptors are critically important for β-cell mass [70]. IRS2 signaling leads to stimulation of the PI3 kinase/Akt and ERK1/2 pathways [71]. Akt, also known as protein kinase B (PKB), has been proposed to be one of the critical mediators of many IRS2 signals in β-cells. Mice deficient in AKT2 develop insulin resistance and diabetes due to impaired adaptation of β-cells, providing evidence for the importance of Akt signaling in β-cells [72]. Moreover, overexpression of Akt in β-cells induces β-cell mass by augmented proliferation, cell size and resistance to apoptosis [39, 40]. Akt exerts several biological functions by modulation of multiple downstream targets. The current evidence suggests that Foxo1, GSK3β and the mTOR signaling pathway could be critical downstream effectors of Akt signaling [73–77]. The events linking GSK3, Foxo1 and mTOR to regulation of β-cell proliferation and apoptosis are ill defined.
2.3 Incretins
The two most studied incretin hormones are glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP). The incretin hormones have been shown to increase β-cell proliferation and decrease β-cell apoptosis [78]. Glucose-intolerant rats demonstrate increased β-cell mass after infusion with GLP-1 [79]. Mice treated with GLP-1 similarly show increased insulin secretion, β-cell size and neogenesis [80]. GLP-1 has also been shown to cause enhanced β-cell regeneration after partial pancreatectomy and streptozotocin administration [81, 82]. In addition to its effects on β-cell proliferation, β-cell regeneration and insulin secretion, GLP-1 has also been shown to have an anti-apoptotic effect [83]. GIP has been less studied, but also has been shown to induce proliferation in cultured β-cells [84].
GLP-1 binds to the GLP-1 receptor (GLP1R), a G protein-coupled receptor expressed in many tissues, including β-cells [78]. Activation of the GLP1R stimulates cyclic AMP formation and activation of downstream targets such as protein kinase A and cAMP-dependent guanine nucleotide exchange factors [85]. GLP1R activation also leads to an increase in intracellular calcium, which triggers insulin release [86, 87]. An important downstream target of GLP-1 is the transcription factor pancreatic duodenal homeobox-1 (Pdx-1) [88]. Indeed, many of the effects of GLP1R activation seem to be due to effects on Pdx-1 [89]. GLP1R activation has also been shown to activate PI3K/Akt; human islets and MIN-6 cells treated with the GLP-1 agonist exendin-4 demonstrated increased IRS2 expression and stimulated Akt phosphorylation [90]. GLP1R activation causes activation of the PI3K/Akt pathway via the epidermal growth factor receptor (EGFR) [91]. Binding of GLP-1 to its receptor can also transactivate the EGF receptor via production of EGF-like ligands. This is caused by c-SRC activation; inhibitors of c-SRC and the EGFR both cause suppression of PI3K activation by GLP-1 in INS-1 cells [92]. The GIP receptor is another G protein-coupled receptor. Like GLP-1, it also acts via adenylyl cyclase to upregulate cyclic AMP. GIP leads to stimulation of the MAP kinase pathway and the PI3K/Akt pathway [78, 84].
3 Mechanisms of β-cell failure
3.1 Glucotoxicity
Glucose concentration is the major determinant for regulation of β-cell mass and function, as discussed above. Transient increases in glucose levels within physiological range induce insulin secretion and potentially beneficial signals. In contrast, glucotoxicity induced by prolonged hyperglycemia causes β-cell dysfunction and altered β-cell mass [93]. The effects of chronic hyperglycemia in β-cells have been assessed in animal models and in vitro using insulinoma cells and isolated islets. In the setting of chronic exposure to hyperglycemia, rat islets exhibit basal insulin hypersecretion and defective GSIS [94]. In animal models and humans, chronic hyperglycemia is associated with alterations in β-cell mass and function [29]. The β-cell has an incredible ability to adapt and compensate for chronic hyperglycemia, as seen in the Zucker diabetic fatty (ZDF) rat, but ultimately, obesity, chronic hyperglycemia, and worsening insulin resistance lead to increased β-cell apoptosis [95]. Similarly, postmortem studies in human type 2 diabetic patients reveal low frequency of replication and reduced β-cell mass, mainly by increased apoptosis [96].
Proposed mechanisms by which glucotoxicity acts include mitochondrial dysfunction with production of reactive oxygen species (ROS), endoplasmic reticulum (ER) stress and increased levels of intracellular calcium. Although high glucose has been shown to induce these pathways and impair insulin secretion, these processes do not appear to consistently result in β-cell apoptosis [93]. Initial studies showed that exposure of β-cell lines to conditions of prolonged hyperglycemia led to decreased β-cell function with decreased insulin mRNA levels, insulin content, and insulin release [97]. Chronically increased glucose concentrations cause increased glucose metabolism through oxidative phosphorylation. This causes mitochondrial dysfunction and the production of reactive oxygen species (ROS) [98]. Several lines of evidence suggest that this is an important mechanism for the induction of β-cell dysfunction. β-cells have a limited defense against excess ROS production due to low levels of ROS-detoxifying enzymes [99]. Markers of oxidative stress are significantly higher in the islets of type 2 diabetics than of controls, and the levels of these markers correlate with the degree of impairment of GSIS [100]. Overexpression of antioxidant enzymes in isolated islets resulting in decreased levels of ROS prevents islet dysfunction in conditions that mimic prolonged hyperglycemia [100]. Also, improved β-cell function in db/db mice and ZDF rats treated with antioxidant agents such as n-acetylcysteine or aminoguanidine provide further evidence for the role of oxidative stress in the deleterious effects of chronic hyperglycemia [98, 101]. A similar improvement in β-cell function was observed in isolated islets from diabetic patients treated with antioxidant agents [102]. Increases in oxidative stress lead to decreased transcription of the insulin gene by decreasing Pdx1 and Maf A binding [97, 103]. The mechanisms by which ROS decrease β-cell mass and function are not completely understood. However, it is known that the generation of ROS will ultimately activate stress-induced pathways, including nuclear factor kB (NF-kB), c-Jun N-terminal kinase (JNK), and hexosamines [104, 105]. The activation of the JNK signaling pathway after induction of oxidative stress inhibits IRS1 signaling by phosphorylation of IRS-1 on Ser307 [106, 107].
In addition to oxidative stress, chronic hyperglycemia can disrupt β-cell mass and function by inducing ER stress (see below), increasing intracellular calcium and increasing nutrient signaling. Chronic hyperglycemia leads to long-term increases in cytosolic Ca2+ that may be proapoptotic and induce β-cell dysfunction [94, 108]. Another potential mechanism of apoptosis by glucose is the production of interleukin-1 beta [104]. Finally, recent publications support the concept that the nutrient-regulated mTOR/S6K signaling pathway exerts negative feedback to regulate IRS proteins by Ser/Thr phosphorylation [109–111]. Experiments in INS1 cells showed that IGFI signaling downregulates IRS2 protein levels by activation of mTOR signaling, suggesting that a similar mechanism is operating in β-cells. The biological and physiological effects of this feedback regulation are not clear, but modulation of IRS signaling by mTOR/S6K could be implicated in the adaptive responses of β-cells to nutrient excess. However there is no direct evidence suggesting that this mechanism is part of signaling events induced by glucotoxicity. In summary, there is some knowledge of the signaling pathways induced by chronic hyperglycemia. However, the downstream events and targets governing the effects of glucotoxicity have not been completely elucidated.
3.2 Lipotoxicity
Dyslipidemia characterized by an increase in circulating free fatty acids (FFA) is one of the major abnormalities in the lipid profile of diabetics. Experiments in humans suggest that elevation of FFA in healthy individuals has stimulatory effects on insulin secretion, but may contribute to progressive β-cell failure in some individuals with a genetic predisposition to diabetes [112–114]. In vitro experiments using isolated islets demonstrated toxic effects of FFA on insulin secretion and apoptosis [115]. It is important to note that several in vitro experiments have been performed using concomitant high glucose concentrations. The current evidence suggests that the deleterious effects of lipids are observed predominantly in the presence of high glucose. Therefore, we will expand on the different mechanisms in the next section.
3.3 Glucolipotoxicity
In the process of glucolipotoxicity, toxic actions of FFA on tissues become apparent in the context of hyperglycemia as described by Prentki et al [116]. Studies in humans reveal that lipid infusion in type 2 diabetic patients causes impaired insulin secretion [117]. Long-term exposure of islets or insulin-secreting cells to increased levels of fatty acids is associated with inhibition of GSIS in vitro [118, 121], impairment of insulin gene expression [122–127], and induction of cell death by apoptosis [115, 117, 128–135]. Notably, reducing plasma FFA concentrations in type 2 diabetics with the niacin derivative acipimox was associated with enhanced insulin sensitivity and improvement in oral glucose tolerance tests [93]. These and other experiments support the concept that FFA alters β-cell function and survival.
Several mechanisms by which glucolipotoxicity impairs GSIS have been postulated. One of the proposed mechanisms for glucolipotoxicity is the inhibition of FFA oxidation by elevated glucose [116]. In the setting of hyperglycemia and elevated FFA, glucose metabolism results in elevated levels of malonyl CoA, a known inhibitor of carnitine palmitoyl transferase-1 (CPT1). The inhibition of CPT1 decreases fatty acid oxidation, which causes accumulation of elevated cytosolic long-chain acyl-CoA esters, generation of ceramide and lipid partitioning [93]. Previous experiments have shown that long–chain acyl-CoA esters result in β-cell dysfunction [136]. Studies also suggest that AMP-activated protein kinase (AMPK) activity may play a role in glucolipotoxicity. AMPK activation promotes fatty acid oxidation by phosphorylation and inhibition of acetyl-CoA carboxylase or via down regulation of the transcription factor sterol-regulatory-element-binding-protein-1c (SREBP1c) [99, 137]. In addition to affecting insulin secretion, glucolipotoxicity can decrease insulin gene expression by alterations in Pdx1 and MafA binding to the insulin promoter [127, 138]. Ceramide generation and activation of JNK with subsequent decrease in IRS signaling have been postulated to relate the signals on the insulin promoter [93, 126]. In summary, high glucose inhibits detoxification of fat and promotes partitioning of FFA to toxic complex lipids, which in turn induces β-cell dysfunction, inhibits insulin gene expression and causes apoptosis. This concept was supported by the findings that treatment with an inhibitor of fat oxidation promoted β-cell death, while treatment with an AMPK activator, which leads to increased FFA oxidation, protected β-cells from glucolipotoxicity [117]. In addition, alteration of the mitochondrial pathway of pyruvate metabolism, with a reduction in the glucose-induced exchange of pyruvate with intermediates of the Krebs cycle has also been proposed as a mechanism by which an increased lipid supply blunts normal GSIS [139].
Another possible mechanism by which FFA may impair β-cell function involves the expression of uncoupling protein-2 (UCP2), part of the UCP family of proteins, which act to regulate cellular ATP production. Previous studies have shown that chronic exposure of islets or insulinoma lines to elevated FFA cause increased UCP2 expression and UCP−/− mice are protected from impaired β-cell function [140]. The mechanisms by which UCP2 may play a role in β-cell failure are unclear. Some studies have suggested increased UCP2 expression leading to increased ROS production as a possible mechanism, but this has not been reproduced nor have antioxidants been shown to cause any benefit in restoring impaired GSIS in lipid-exposed islets [141]. Recently, the ATP-binding cassette transporter subfamily A member 1 (ABCA1), a mediator of reverse cholesterol efflux, was shown to be an important mediator of the effects of FFA on insulin secretion. Conditional deletion of ABCA1 results in increased cellular cholesterol content and impaired insulin secretion at the level of exocytosis [142].
As discussed above, multiple studies have shown that fatty acids can induce β-cell death by apoptosis and that this effect is potentiated by glucose [117, 130, 134, 143]. Several mechanisms have been proposed to mediate fatty acid induced apoptosis in β-cells, including ceramide formation leading to altered lipid partitioning, and the generation of ROS (for review [144]). More recently, ER stress and the unfolded protein response have received experimental support [138, 145, 146]. In addition to these processes, apoptosis after fatty acid administration can result from the activation of the JNK pathway and decreased Akt signaling with subsequent activation of Foxo1-dependent gene expression [132, 143, 145]. While the exact mechanisms are not delineated, it is clear that elevated FFA play a role in impaired β-cell function and apoptosis. It is conceivable that the combination of elevated FFA and chronic hyperglycemia synergize to create a milieu conducive to β-cell dysfunction and failure.
3.4 ER stress
Recent evidence suggests that ER stress links obesity with insulin resistance (reviewed in [147, 148]). Studies in humans and rodent models also implicate this mechanism in β-cell adaptation to the diabetic milieu. Evidence for ER stress in islets from type 2 diabetics has been demonstrated by increased staining for ER chaperones and CHOP along with increased ER size [146, 149, 150]. In rodent models, increased ER stress markers have been demonstrated in mouse islets from db/db mice [146]. Insulin-2 mutations in Akita mice induce accumulation of misfolded insulin and progressive β-cell loss caused by ER stress, demonstrating the importance of this pathway for β-cell survival [151]. Similarly, mice with deletion of wfs1, the affected gene in Wolfram Syndrome, which is characterized by juvenile-onset diabetes mellitus, also exhibit decreased β-cell mass and impaired GSIS [151, 152]. Taken together, these data suggest that ER stress is present in human β-cells and that this could be a common mechanism for the two major pathophysiological events in type 2 diabetes, insulin resistance and β-cell failure.
ER stress has been postulated to result from increased biosynthetic demand induced by chronic hyperglycemia, elevated FFA, and chronic over-nutrition in the β-cell. This pathway is best understood in the context of the Unfolded Protein Response (UPR). The UPR alleviates ER stress, restores homeostasis, and prevents cell death by inducing a number of downstream responses that: 1) decrease new protein arrival to the ER; 2) increase the amount of ER chaperones to improve folding capacity; and 3) increase the cell’s capacity to dispose of misfolded proteins. If unable to successfully perform these tasks, the UPR will trigger the apoptosis cascade [152]. The three primary modulators of the UPR are: inositol requiring protein 1-alpha (IRE1-alpha), activating transcription factor 6 (ATF6), and protein kinase RNA (PRK)-like ER associated kinase (PERK) [153]. These sensors remain inactive via interaction with the ER chaperone BiP until activated by increased ER stress [154].
IRE1-alpha possesses critical endoribonuclease activity that induces the splicing of X-box binding protein 1 (XBP1). The spliced form of XBP1, XBP1s, regulates the transcription of genes involved in ER expansion as well as protein maturation, folding and export. In addition, XBP1 modulates the expression of genes that regulate the degradation of misfolded proteins and ER-targeted mRNAs in order to decrease protein synthesis [152]. These actions may be critical in β-cell adaptation to ER stress. Previous evaluation has shown that short-term glucose exposure of isolated islets induces IRE1-alpha signaling but not XBP1 splicing and this was found to be important for pro-insulin biosynthesis [155]. In contrast, long-term exposure of islets to hyperglycemia is associated with XBP1 splicing and progressive inhibition of insulin mRNA and protein expression [155]. Recent work demonstrates that activation of IRE1-alpha caused by chronic high glucose leads to insulin mRNA degradation [156, 157]. In addition to causing the degradation of mRNA, IRE1-alpha may also signal apoptosis via activation of the JNK signaling pathway by interaction with TNF receptor-associated factor (TRAF) 2 and the activation of procaspase 12 [158, 159]. Palmitate has also been shown to activate IRE1-alpha signaling, suggesting that this pathway could be relating some of the ER stress signals induced by fatty acids (for review [152]). The potentiation of ER stress markers by glucose has not always been reproduced in primary β-cells and human islets [160].
ATF6, another essential modulator of the UPR, is released from BiP in response to accumulation of unfolded proteins in the ER lumen. ATF6 acts as a co-activator of the UPR by binding to the promoters of UPR-responsive genes, including those controlling ER chaperones [161–163]. ATF6 augments the expression of XBP1 mRNA, providing more substrate for IRE1-induced generation of XBP1s [164]. Combined null mutations in both isoforms of ATF6 in mice result in an early developmental lethal phenotype, but loss of function of ATF6-alpha alone in mice results in impaired recovery from acute stress and inability to tolerate chronic stress [165]. Studies of Dutch and Pima Indians reveal that missense mutations and polymorphisms within ATF6-alpha may be linked to type 2 diabetes [166, 167]. Further evidence that this signaling pathway is important is demonstrated by induction of ATF6 signaling by palmitate [145]. Recent evidence showed that ATF6 levels were increased in the pancreatic islets of diabetic OLETF rats [168]. In the same study, induction of ATF6 by ER stress was associated with repression of the insulin gene via the up regulation of SHP. This suggests that this pathway could also be implicated in ER stress induced β-cell dysfunction.
Finally, the activation of PERK causes phosphorylation of eukaryotic translation initiation factor-2a (eIF2a), resulting in an overall decrease in mRNA translation but increased translation of select proteins such as ATF4 [169–171]. ATF4 induces the transcription of genes involved in amino acid import, glutathione biosynthesis, protein synthesis (inducing of 4EBP) and resistance to oxidative stress as well as the proapoptotic gene CHOP [172–176]. Palmitate has been shown to activate PERK and eIF2a phosphorylation, leading to inhibition of protein synthesis and induction of ATF4 and CHOP in islets and insulinoma cells [138, 143, 177]. CHOP induction by FFA is mediated by ATF4 binding to the C/EBP-ATF binding site in the CHOP promoter, as well as by c-Fos and Jun-B dimer binding to the activator protein-1 (AP-1) binding site and possibly Foxo1 [145, 178]. The importance of the PERK pathway in β-cell survival has been assessed in genetically modified animals. Deficiency in the PERK-eIF2a pathway leads to progressive loss of β-cell mass and severe diabetes in both humans and mice [153, 179]. It would then follow that activation of the PERK-eIF2a pathway would provide “a respite” for the ER and prevent further damage. However, further study into the function of the PERK/eIF2a pathway reveals that chronic induction of the PERK-eIF2a pathway may also lead to cell apoptosis at least in part by upregulation of CHOP [174]. Indication that CHOP is a critical component in apoptosis induced by this pathway was demonstrated by delayed onset of diabetes and ameliorated β-cell apoptosis in Akita mice with targeted disruption of CHOP [180].
Current evidence suggests that ER stress is an important contributor to β-cell failure in Type 2 Diabetes. The molecular mechanisms by which FFA- and glucolipotoxicity-induced ER stress cause β-cell apoptosis are not well understood. In addition to induction of CHOP, ER stress can induce apoptosis by JNK, ATF-3 and inhibition of Bcl-2 and/or activation of proapoptotic members of the Bcl-2 family (for review [152]). In particular, activation of JNK could lead to suppression of IRS/Akt signaling through serine phosphorylation of IRS-1 in liver and β-cells [143, 181]. Inhibition of IRS/Akt signaling reduces survival signals and ultimately leads to apoptosis. Inhibition of Foxo1 using a dominant-negative mutant reduces ER stress markers and promotes β-cell survival at least in part by modulation of CHOP [143]. Recently, a potential important link between ER stress and IRS2 signaling was demonstrated by transcriptional repression of the IRS2 promoter by ATF3 [182]. Inhibition of IRS2 signaling was also observed by ER stress-induced activation of SREBP in insulinoma cells treated with high levels of glucose, suggesting that this pathway could also play a role [183]. While the exact mechanisms of ER stress mediated apoptosis are not completely understood, there is no question that chronic hyperglycemia and nutrient excess lead to activation of the UPR and its cascade of downstream responses. It is likely that cumulative damage from hyperglycemia, over-nutrition, and elevated FFA levels overwhelm the ER of the β-cell, resulting in activation of the UPR and eventual apoptosis and β-cell failure.
4 Concluding remarks
Type 2 diabetes mellitus is a disease with devastating complications that is increasing in prevalence at an alarming rate, at great cost to the lives of patients and to society as a whole. The inherent defect in this disease is β-cell failure in the setting of insulin resistance. Pancreatic β-cells possess the ability to greatly increase their mass in response to stress conditions such as insulin resistance. Elegant studies have identified some of the cell cycle machinery governing β-cell proliferation. The proliferation of β-cells is regulated by a multitude of nutrient signals and growth factors. Further research into the machinery of β-cell proliferation may identify potential therapeutic strategies in the treatment of diabetes. The findings that certain factors, such as incretins, can upregulate β-cell mass is an exciting prospect for possible methods of increasing β-cell mass or improving the adaptation of β-cells to insulin resistance. Furthermore, recent lineage-tracing experiments provide insight into the origins of new β-cells. Delineation of the processes and conditions required for β-cell neogenesis are of great relevance to the understanding and possible treatment of diabetes. Understanding the mechanisms involved in β-cell dysfunction and failure observed in late stages of diabetes is also a topic of major importance. Research has begun to unravel how excess glucose and lipids lead to impaired β-cell function and apoptosis. The generation of reactive oxygen species, ER stress, alterations in β-cell metabolism, decreased IRS signaling and induction of pro-apoptotic signals have been found to be key players in β-cell failure. Further research into the process of ER stress may reveal how a response designed to protect the β-cell can ultimately lead to its demise. As the pathways of β-cell expansion and β-cell failure are further clarified, the essential modulators of these processes will be identified, providing potential novel therapeutic targets to investigate. For now, it is conceivable that pharmacological agents that decrease oxidative stress, modulate ER stress or sensitize β-cells to the action of insulin could have major implications to delay or prevent the development of diabetes. Finally, it is important to note that current evidence indicates that diet and exercise are the most effective interventions to prevent or delay type 2 diabetes.
References
Bonner-Weir S, Deery D, Leahy JL, Weir GC. Compensatory growth of pancreatic beta-cells in adult rats after short-term glucose infusion. Diabetes 1989;38:49–53.
Bruning JC, Winnay J, Bonner-Weir S, Taylor SI, Accili D, Kahn CR. Development of a novel polygenic model of NIDDM in mice heterozygous for IR and IRS-1 null alleles. Cell 1997;88:561–72.
Parsons JA, Brelje TC, Sorenson RL. Adaptation of islets of Langerhans to pregnancy: increased islet cell proliferation and insulin secretion correlates with the onset of placental lactogen secretion. Endocrinology 1992;130:1459–66.
Scaglia L, Smith FE, Bonner-Weir S. Apoptosis contributes to the involution of beta cell mass in the post partum rat pancreas. Endocrinology 1995;136:5461–8.
Withers DJ, Gutierrez JS, Towery H, Burks DJ, Ren J-M, Previs S, et al. Disruption of IRS-2 causes type 2 diabetes in mice. Nature 1998;391:900–3.
Kido Y, Burks DJ, Withers D, Bruning JC, Kahn CR, White MF, et al. Tissue-specific insulin resistance in mice with mutations in the insulin receptor, IRS-1, and IRS-2. J Clin Invest 2000;105:199–205.
Xu G, Stoffers DA, Habener JF, Bonner-Weir S. Exendin-4 stimulates both beta-cell replication and neogenesis, resulting in increased beta-cell mass and improved glucose tolerance in diabetic rats. Diabetes 1999;48:2270–6.
Bonner-Weir S, Baxter LA, Schuppin GT, Smith FE. A second pathway for regeneration of adult exocrine and endocrine pancreas. A possible recapitulation of embryonic development. Diabetes 1993;42:1715–20.
Dor Y, Brown J, Martinez OI, Melton DA. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature 2004;429:41–6.
Bonner-Weir S, Inada A, Yatoh S, Li WC, Aye T, Toschi E, Sharma A. Transdifferentiation of pancreatic ductal cells to endocrine beta-cells. Biochem Soc Trans 2008;36:353–6.
Cozar-Castellano I, Fiaschi-Taesch N, Bigatel TA, Takane KK, Garcia-Ocana A, Vasavada R, et al. Molecular Control of Cell Cycle Progression in the Pancreatic {beta}-Cell. Endocr Rev 2006;27:356–70.
Zhang X, Gaspard JP, Mizukami Y, Li J, Graeme-Cook F, Chung DC. Overexpression of cyclin D1 in pancreatic beta-cells in vivo results in islet hyperplasia without hypoglycemia. Diabetes 2005;54:712–9.
Georgia S, Bhushan A. Beta cell replication is the primary mechanism for maintaining postnatal beta cell mass. J Clin Invest 2004;114:963–8.
Kushner JA, Ciemerych MA, Sicinska E, Wartschow LM, Teta M, Long SY, et al. Cyclins D2 and D1 Are Essential for Postnatal Pancreatic {beta}-Cell Growth. Mol Cell Biol 2005;25:3752–62.
Rane SG, Dubus P, Mettus RV, Galbreath EJ, Boden G, Reddy EP, et al. Loss of Cdk4 expression causes insulin-deficient diabetes and Cdk4 activation results in beta-islet cell hyperplasia. Nat Genet 1999;22:44–52.
Cozar-Castellano I, Weinstock M, Haught M, Velazquez-Garcia S, Sipula D, Stewart AF. Evaluation of beta-cell replication in mice transgenic for hepatocyte growth factor and placental lactogen: comprehensive characterization of the G1/S regulatory proteins reveals unique involvement of p21cip. Diabetes 2006;55:70–7.
Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature 1993;366:704–7.
Marzo N, Mora C, Fabregat ME, Martin J, Usac EF, Franco C, et al. Pancreatic islets from cyclin-dependent kinase 4/R24C (Cdk4) knockin mice have significantly increased beta cell mass and are physiologically functional, indicating that Cdk4 is a potential target for pancreatic beta cell mass regeneration in Type 1 diabetes. Diabetologia 2004;47(4):686–94.
Rachdi L, Balcazar N, Elghazi L, Barker DJ, Krits I, Kiyokawa H, et al. Differential effects of p27 in regulation of beta-cell mass during development, neonatal period, and adult life. Diabetes 2006;55:3520–8.
Uchida T, Nakamura T, Hashimoto N, Matsuda T, Kotani K, Sakaue H, et al. Deletion of Cdkn1b ameliorates hyperglycemia by maintaining compensatory hyperinsulinemia in diabetic mice. Nat Med 2005;11:175–82.
Tsutsui T, Hesabi B, Moons DS, Pandolfi PP, Hansel KS, Koff A, et al. Targeted disruption of CDK4 delays cell cycle entry with enhanced p27(Kip1) activity. Mol Cell Biol 1999;19:7011–9.
Bonner-Weir S, Taneja M, Weir GC, Tatarkiewicz K, Song KH, Sharma A, et al. In vitro cultivation of human islets from expanded ductal tissue. Proc Natl Acad Sci U S A 2000;97:7999–8004.
Ramiya VK, Maraist M, Arfors KE, Schatz DA, Peck AB, Cornelius JG. Reversal of insulin-dependent diabetes using islets generated in vitro from pancreatic stem cells. Nat Med 2000;6:278–82.
Rooman I, Lardon J, Bouwens L. Gastrin stimulates beta-cell neogenesis and increases islet mass from transdifferentiated but not from normal exocrine pancreas tissue. Diabetes 2002;51:686–90.
Li L, Seno M, Yamada H, Kojima I. Betacellulin improves glucose metabolism by promoting conversion of intraislet precursor cells to beta-cells in streptozotocin-treated mice. Am J Physiol Endocrinol Metab 2003;285:E577–83.
Cnop M, Welsh N, Jonas JC, Jorns A, Lenzen S, Eizirik DL. Mechanisms of pancreatic beta-cell death in type 1 and type 2 diabetes: many differences, few similarities. Diabetes 2005;54(Suppl 2):S97–107.
Kloppel G, Lohr M, Habich K, Oberholzer M, Heitz PU. Islet pathology and the pathogenesis of type 1 and type 2 diabetes mellitus revisited. Surv Synth Pathol Res 1985;4:110–25.
Clark A, Wells CA, Buley ID, Cruickshank JK, Vanhegan RI, Matthews DR, et al. Islet amyloid, increased A-cells, reduced B-cells and exocrine fibrosis: quantitative changes in the pancreas in type 2 diabetes. Diabetes Res 1988;9:151–9.
Wajchenberg BL. Beta-cell failure in diabetes and preservation by clinical treatment. Endocr Rev 2007;28:187–218.
Donath MY, Halban PA. Decreased beta-cell mass in diabetes: significance, mechanisms and therapeutic implications. Diabetologia 2004;47:581–9.
Swenne I. The role of glucose in the in vitro regulation of cell cycle kinetics and proliferation of fetal pancreatic B-cells. Diabetes 1982;31:754–60.
Chick WL. Beta cell replication in rat pancreatic monolayer cultures. Effects of glucose, tolbutamide, glucocorticoid, growth hormone and glucagon. Diabetes 1973;22:687–93.
Paris M, Bernard-Kargar C, Berthault MF, Bouwens L, Ktorza A. Specific and combined effects of insulin and glucose on functional pancreatic beta-cell mass in vivo in adult rats. Endocrinology 2003;144:2717–27.
Bernard C, Berthault MF, Saulnier C, Ktorza A. Neogenesis vs. apoptosis As main components of pancreatic beta cell ass changes in glucose-infused normal and mildly diabetic adult rats. FasebJ 1999;13:1195–205.
Alonso LC, Yokoe T, Zhang P, Scott DK, Kim SK, O’Donnell CP, et al. Glucose infusion in mice: a new model to induce beta-cell replication. Diabetes 2007;56:1792–801.
Hugl SR, White MF, Rhodes CJ. Insulin-like growth factor I (IGF-I)-stimulated pancreatic beta-cell growth is glucose-dependent. Synergistic activation of insulin receptor substrate-mediated signal transduction pathways by glucose and IGF-I in INS-1 cells. J Biol Chem 1998;273:17771–9.
Kulkarni RN, Bruning JC, Winnay JN, Postic C, Magnuson MA, Kahn CR. Tissue-specific knockout of the insulin receptor in pancreatic beta cells creates an insulin secretory defect similar to that in type 2 diabetes. Cell 1999;96:329–39.
Ohsugi M, Cras-Meneur C, Zhou Y, Warren W, Bernal-Mizrachi E, Permutt MA. Glucose and insulin treatment of insulinoma cells results in transcriptional regulation of a common set of genes. Diabetes 2004;53:1496–508.
Tuttle RL, Gill NS, Pugh W, Lee JP, Koeberlein B, Furth EE, et al. Regulation of pancreatic beta-cell growth and survival by the serine/threonine protein kinase Akt1/PKBalpha. Nat Med 2001;7:1133–7.
Bernal-Mizrachi E, Wen W, Stahlhut S, Welling CM, Permutt MA. Islet beta cell expression of constitutively active Akt1/PKB alpha induces striking hypertrophy, hyperplasia, and hyperinsulinemia. J Clin Invest 2001;108:1631–8.
Heit JJ, Apelqvist AA, Gu X, Winslow MM, Neilson JR, Crabtree GR, et al. Calcineurin/NFAT signalling regulates pancreatic beta-cell growth and function. Nature 2006;443:345–9.
Jhala US, Canettieri G, Screaton RA, Kulkarni RN, Krajewski S, Reed J, et al. cAMP promotes pancreatic beta-cell survival via CREB-mediated induction of IRS2. Genes Dev 2003;17:1575–80.
Bernal-Mizrachi E, Wice B, Inoue H, Permutt MA. Activation of serum response factor in the depolarization induction of Egr-1 transcription in pancreatic islet beta-cells. J Biol Chem 2000;275:25681–9.
Sorenson RL, Stout LE. Prolactin receptors and JAK2 in islets of Langerhans: an immunohistochemical analysis. Endocrinology 1995;136:4092–8.
Brelje TC, Sorenson RL. Role of prolactin versus growth hormone on islet B-cell proliferation in vitro: implications for pregnancy. Endocrinology 1991;128:45–57.
Brelje TC, Scharp DW, Lacy PE, Ogren L, Talamantes F, Robertson M, et al. Effect of homologous placental lactogens, prolactins, and growth hormones on islet B-cell division and insulin secretion in rat, mouse, and human islets: implication for placental lactogen regulation of islet function during pregnancy. Endocrinology 1993;132:879–87.
Vasavada RC, Garcia-Ocana A, Zawalich WS, Sorenson RL, Dann P, Syed M, et al. Targeted expression of placental lactogen in the beta cells of transgenic mice results in beta cell proliferation, islet mass augmentation, and hypoglycemia. J Biol Chem 2000;275:15399–406.
Freemark M, Avril I, Fleenor D, Driscoll P, Petro A, Opara E, et al. Targeted deletion of the PRL receptor: effects on islet development, insulin production, and glucose tolerance. Endocrinology 2002;143:1378–85.
Liu JL, Coschigano KT, Robertson K, Lipsett M, Guo Y, Kopchick JJ, et al. Disruption of growth hormone receptor gene causes diminished pancreatic islet size and increased insulin sensitivity in mice. Am J Physiol Endocrinol Metab 2004;287:E405–13.
Villanueva-Penacarrillo ML, Cancelas J, de Miguel F, Redondo A, Valin A, Valverde I, et al. Parathyroid hormone-related peptide stimulates DNA synthesis and insulin secretion in pancreatic islets. J Endocrinol 1999;163:403–8.
Fujinaka Y, Sipula D, Garcia-Ocana A, Vasavada RC. Characterization of mice doubly transgenic for parathyroid hormone-related protein and murine placental lactogen: a novel role for placental lactogen in pancreatic beta-cell survival. Diabetes 2004;53:3120–30.
Vasavada RC, Gonzalez-Pertusa JA, Fujinaka Y, Fiaschi-Taesch N, Cozar-Castellano I, Garcia-Ocana A. Growth factors and beta cell replication. Int J Biochem Cell Biol. 2005;
Matsumoto K, Nakamura T. Emerging multipotent aspects of hepatocyte growth factor. J Biochem 1996;119:591–600.
Garcia-Ocana A, Vasavada RC, Cebrian A, Reddy V, Takane KK, Lopez-Talavera JC, et al. Transgenic overexpression of hepatocyte growth factor in the beta-cell markedly improves islet function and islet transplant outcomes in mice. Diabetes 2001;50:2752–62.
Furge KA, Zhang YW, Vande Woude GF. Met receptor tyrosine kinase: enhanced signaling through adapter proteins. Oncogene 2000;19:5582–9.
Calvo EL, Boucher C, Pelletier G, Morisset J. Ontogeny of hepatocyte growth factor and c-met/hgf receptor in rat pancreas. Biochem Biophys Res Commun 1996;229:257–63.
Dai C, Li Y, Yang J, Liu Y. Hepatocyte growth factor preserves beta cell mass and mitigates hyperglycemia in streptozotocin-induced diabetic mice. J Biol Chem 2003;278:27080–7.
Roccisana J, Reddy V, Vasavada RC, Gonzalez-Pertusa JA, Magnuson MA, Garcia-Ocana A. Targeted inactivation of hepatocyte growth factor receptor c-met in beta-cells leads to defective insulin secretion and GLUT-2 downregulation without alteration of beta-cell mass. Diabetes 2005;54:2090–102.
Vasavada RC, Gonzalez-Pertusa JA, Fujinaka Y, Fiaschi-Taesch N, Cozar-Castellano I, Garcia-Ocana A. Growth factors and beta cell replication. Int J Biochem Cell Biol 2006;38:931–50.
Nielsen JHGE, Møldrup A, Friedrichsen BN, Billestrup N, Hansen JA, Lee YC, et al. Regulation of beta-cell mass by hormones and growth factors. Diabetes 2001;50:S25–9.
Friedrichsen BN, Richter HE, Hansen JA, Rhodes CJ, Nielsen JH, Billestrup N, et al. Signal transducer and activator of transcription 5 activation is sufficient to drive transcriptional induction of cyclin D2 gene and proliferation of rat pancreatic beta-cells. Mol Endocrinol 2003;17:945–58.
Zhang B, Hosaka M, Sawada Y, Torii S, Mizutani S, Ogata M, et al. Parathyroid hormone-related protein induces insulin expression through activation of MAP kinase-specific phosphatase-1 that dephosphorylates c-Jun NH2-terminal kinase in pancreatic beta-cells. Diabetes 2003;52:2720–30.
Vasavada RC, Wang L, Fujinaka Y, Takane KK, Rosa TC, Mellado-Gil JM, et al. Protein kinase C-zeta activation markedly enhances beta-cell proliferation: an essential role in growth factor mediated beta-cell mitogenesis. Diabetes 2007;56:2732–43.
Hogg J, Han VK, Clemmons DR, Hill DJ. Interactions of nutrients, insulin-like growth factors (IGFs) and IGF-binding proteins in the regulation of DNA synthesis by isolated fetal rat islets of Langerhans. J Endocrinol 1993;138:401–12.
Sieradzki J, Fleck H, Chatterjee AK, Schatz H. Stimulatory effect of insulin-like growth factor-I on [3H]thymidine incorporation, DNA content and insulin biosynthesis and secretion of isolated pancreatic rat islets. J Endocrinol 1988;117:59–62.
Swenne I, Hill DJ, Strain AJ, Milner RD. Growth hormone regulation of somatomedin C/insulin-like growth factor I production and DNA replication in fetal rat islets in tissue culture. Diabetes 1987;36:288–94.
George M, Ayuso E, Casellas A, Costa C, Devedjian JC, Bosch F. Beta cell expression of IGF-I leads to recovery from type 1 diabetes. J Clin Invest 2002;109:1153–63.
Petrik J, Pell JM, Arany E, McDonald TJ, Dean WL, Reik W, et al. Overexpression of insulin-like growth factor-II in transgenic mice is associated with pancreatic islet cell hyperplasia. Endocrinology 1999;140:2353–63.
Ohsugi M, Cras-Meneur C, Zhou Y, Bernal-Mizrachi E, Johnson JD, Luciani DS, et al. Reduced expression of the insulin receptor in mouse insulinoma (MIN6) cells reveals multiple roles of insulin signaling in gene expression, proliferation, insulin content, and secretion. J Biol Chem 2005;280:4992–5003.
Withers DJ, Gutierrez JS, Towery H, Burks DJ, Ren JM, Previs S, et al. Disruption of IRS-2 causes type 2 diabetes in mice. Nature 1998;391:900–4.
Kulkarni RN. New insights into the roles of insulin/IGF-I in the development and maintenance of beta-cell mass. Rev Endocr Metab Disord 2005;6:199–210.
Garofalo RS, Orena SJ, Rafidi K, Torchia AJ, Stock JL, Hildebrandt AL, et al. Severe diabetes, age-dependent loss of adipose tissue, and mild growth deficiency in mice lacking Akt2/PKB beta. J Clin Invest 2003;112:197–208.
Nakae J, Biggs WH 3rd, Kitamura T, Cavenee WK, Wright CV, Arden KC, et al. Regulation of insulin action and pancreatic beta-cell function by mutated alleles of the gene encoding forkhead transcription factor Foxo1. Nat Genet 2002;32:245–53.
Tanabe K, Liu Z, Patel S, Doble BW, Li L, Cras-Meneur C, et al. Genetic deficiency of glycogen synthase kinase-3beta corrects diabetes in mouse models of insulin resistance. PLoS Biol 2008;6:e37.
Pende M, Kozma SC, Jaquet M, Oorschot V, Burcelin R, Le Marchand-Brustel Y, et al. Hypoinsulinaemia, glucose intolerance and diminished beta-cell size in S6K1-deficient mice. Nature 2000;408:994–7.
Ruvinsky I, Sharon N, Lerer T, Cohen H, Stolovich-Rain M, Nir T, et al. Ribosomal protein S6 phosphorylation is a determinant of cell size and glucose homeostasis. Genes Dev 2005;19:2199–211.
Rachdi L. BN, Osorio F., Elghazi L., Weiss A., Gould A., Chang-Chen K., et la. Disruption of Tsc2 in pancreatic β-cells induces β-cell mass expansion and improved glucose tolerance in a TORC1-dependent manner. Proc Natl Acad Sci U S A 2008;In Press
Drucker DJ. The biology of incretin hormones. Cell Metab 2006;3:153–65.
Perfetti R, Zhou J, Doyle ME, Egan JM. Glucagon-like peptide-1 induces cell proliferation and pancreatic-duodenum homeobox-1 expression and increases endocrine cell mass in the pancreas of old, glucose-intolerant rats. Endocrinology 2000;141:4600–5.
Stoffers DA, Kieffer TJ, Hussain MA, Drucker DJ, Bonner-Weir S, Habener JF, et al. Insulinotropic glucagon-like peptide 1 agonists stimulate expression of homeodomain protein IDX-1 and increase islet size in mouse pancreas. Diabetes 2000;49:741–8.
De Leon DD, Deng S, Madani R, Ahima RS, Drucker DJ, Stoffers DA. Role of endogenous glucagon-like peptide-1 in islet regeneration after partial pancreatectomy. Diabetes 2003;52:365–71.
Tourrel C, Bailbe D, Meile MJ, Kergoat M, Portha B. Glucagon-like peptide-1 and exendin-4 stimulate beta-cell neogenesis in streptozotocin-treated newborn rats resulting in persistently improved glucose homeostasis at adult age. Diabetes 2001;50:1562–70.
Farilla L, Bulotta A, Hirshberg B, Li Calzi S, Khoury N, Noushmehr H, et al. Glucagon-like peptide 1 inhibits cell apoptosis and improves glucose responsiveness of freshly isolated human islets. Endocrinology 2003;144:5149–58.
Trumper A, Trumper K, Trusheim H, Arnold R, Goke B, Horsch D. Glucose-dependent insulinotropic polypeptide is a growth factor for beta (INS-1) cells by pleiotropic signaling. Mol Endocrinol 2001;15:1559–70.
Holz GG, Leech CA, Heller RS, Castonguay M, Habener JF. cAMP-dependent mobilization of intracellular Ca2+ stores by activation of ryanodine receptors in pancreatic beta-cells. A Ca2+ signaling system stimulated by the insulinotropic hormone glucagon-like peptide-1-(7–37). J Biol Chem 1999;274:14147–56.
Gromada J, Dissing S, Bokvist K, Renstrom E, Frokjaer-Jensen J, Wulff BS, et al. Glucagon-like peptide I increases cytoplasmic calcium in insulin-secreting beta TC3-cells by enhancement of intracellular calcium mobilization. Diabetes 1995;44:767–74.
Tsuboi T, da Silva Xavier G, Holz GG, Jouaville LS, Thomas AP, Rutter GA. Glucagon-like peptide-1 mobilizes intracellular Ca2+ and stimulates mitochondrial ATP synthesis in pancreatic MIN6 beta-cells. Biochem J 2003;369:287–99.
Wang X, Cahill CM, Pineyro MA, Zhou J, Doyle ME, Egan JM. Glucagon-like peptide-1 regulates the beta cell transcription factor, PDX-1, in insulinoma cells. Endocrinology 1999;140:4904–7.
Li Y, Cao X, Li LX, Brubaker PL, Edlund H, Drucker DJ. beta-Cell Pdx1 expression is essential for the glucoregulatory, proliferative, and cytoprotective actions of glucagon-like peptide-1. Diabetes 2005;54:482–91.
Park S, Dong X, Fisher TL, Dunn SL, Omer AK, Weir G, et al. Exendin-4 promotes IRS2 signaling to mediate pancreatic beta-cell growth and function. J Biol Chem 2005;281:1159–68.
Miettinen PJ, Ustinov J, Ormio P, Gao R, Palgi J, Hakonen E, et al. Downregulation of EGF receptor signaling in pancreatic islets causes diabetes due to impaired postnatal beta-cell growth. Diabetes 2006;55:3299–308.
Buteau J, Foisy S, Joly E, Prentki M. Glucagon-like peptide 1 induces pancreatic beta-cell proliferation via transactivation of the epidermal growth factor receptor. Diabetes 2003;52:124–32.
Poitout V, Robertson RP. Glucolipotoxicity: fuel excess and beta-cell dysfunction. Endocr Rev 2008;29:351–66.
Khaldi MZ, Guiot Y, Gilon P, Henquin JC, Jonas JC. Increased glucose sensitivity of both triggering and amplifying pathways of insulin secretion in rat islets cultured for 1 wk in high glucose. Am J Physiol Endocrinol Metab 2004;287:E207–17.
Finegood DT, McArthur MD, Kojwang D, Thomas MJ, Topp BG, Leonard T, et al. Beta-cell mass dynamics in Zucker diabetic fatty rats. Rosiglitazone prevents the rise in net cell death. Diabetes 2001;50:1021–9.
Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 2003;52:102–10.
Olson LK, Redmon JB, Towle HC, Robertson RP. Chronic exposure of HIT cells to high glucose concentrations paradoxically decreases insulin gene transcription and alters binding of insulin gene regulatory protein. J Clin Invest 1993;92:514–9.
Tanaka Y, Gleason CE, Tran PO, Harmon JS, Robertson RP. Prevention of glucose toxicity in HIT-T15 cells and Zucker diabetic fatty rats by antioxidants. Proc Natl Acad Sci U S A 1999;96:10857–62.
Prentki M, Nolan CJ. Islet beta cell failure in type 2 diabetes. J Clin Invest 2006;116:1802–12.
Tanaka Y, Tran PO, Harmon J, Robertson RP. A role for glutathione peroxidase in protecting pancreatic beta cells against oxidative stress in a model of glucose toxicity. Proc Natl Acad Sci U S A 2002;99:12363–8.
Kaneto H, Kajimoto Y, Miyagawa J, Matsuoka T, Fujitani Y, Umayahara Y, et al. Beneficial effects of antioxidants in diabetes: possible protection of pancreatic beta-cells against glucose toxicity. Diabetes 1999;48:2398–406.
Del Guerra S, Lupi R, Marselli L, Masini M, Bugliani M, Sbrana S, et al. Functional and molecular defects of pancreatic islets in human type 2 diabetes. Diabetes 2005;54:727–35.
Olson LK, Sharma A, Peshavaria M, Wright CV, Towle HC, Rodertson RP, et al. Reduction of insulin gene transcription in HIT-T15 beta cells chronically exposed to a supraphysiologic glucose concentration is associated with loss of STF-1 transcription factor expression. Proc Natl Acad Sci U S A 1995;92:9127–31.
Maedler K, Sergeev P, Ris F, Oberholzer J, Joller-Jemelka HI, Spinas GA, et al. Glucose-induced beta cell production of IL-1beta contributes to glucotoxicity in human pancreatic islets. J Clin Invest 2002;110:851–60.
Kaneto H, Xu G, Fujii N, Kim S, Bonner-Weir S, Weir GC. Involvement of c-Jun N-terminal kinase in oxidative stress-mediated suppression of insulin gene expression. J Biol Chem 2002;277:30010–8.
Aguirre V, Uchida T, Yenush L, Davis R, White MF. The c-Jun NH(2)-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser(307). J Biol Chem 2000;275:9047–54.
Aguirre V, Werner ED, Giraud J, Lee YH, Shoelson SE, White MF. Phosphorylation of Ser307 in insulin receptor substrate-1 blocks interactions with the insulin receptor and inhibits insulin action. J Biol Chem 2002;277:1531–7.
Grill V, Bjorklund A. Overstimulation and beta-cell function. Diabetes 2001;50(Suppl 1):S122–4.
Um SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M, et al. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature 2004;431:200–5.
Shah OJ, Wang Z, Hunter T. Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance, and cell survival deficiencies. Curr Biol 2004;14:1650–6.
Harrington LS, Findlay GM, Gray A, Tolkacheva T, Wigfield S, Rebholz H, et al. The TSC1–2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J Cell Biol 2004;166:213–23.
Kashyap S, Belfort R, Gastaldelli A, Pratipanawatr T, Berria R, Pratipanawatr W, et al. A sustained increase in plasma free fatty acids impairs insulin secretion in nondiabetic subjects genetically predisposed to develop type 2 diabetes. Diabetes 2003;52:2461–74.
Santomauro AT, Boden G, Silva ME, Rocha DM, Santos RF, Ursich MJ, et al. Overnight lowering of free fatty acids with Acipimox improves insulin resistance and glucose tolerance in obese diabetic and nondiabetic subjects. Diabetes 1999;48:1836–41.
Boden G, Shulman GI. Free fatty acids in obesity and type 2 diabetes: defining their role in the development of insulin resistance and beta-cell dysfunction. Eur J Clin Invest 2002;32(Suppl 3):14–23.
Shimabukuro M, Zhou YT, Levi M, Unger RH. Fatty acid-induced beta cell apoptosis: a link between obesity and diabetes. Proc Natl Acad Sci U S A 1998;95:2498–502.
Prentki M, Joly E, El-Assaad W, Roduit R. Malonyl-CoA signaling, lipid partitioning, and glucolipotoxicity: role in beta-cell adaptation and failure in the etiology of diabetes. Diabetes 2002;51(Suppl 3):S405–13.
El-Assaad W, Buteau J, Peyot ML, Nolan C, Roduit R, Hardy S, et al. Saturated fatty acids synergize with elevated glucose to cause pancreatic beta-cell death. Endocrinology 2003;144:4154–63.
Sako Y, Grill VE. A 48-hour lipid infusion in the rat time-dependently inhibits glucose-induced insulin secretion and B cell oxidation through a process likely coupled to fatty acid oxidation. Endocrinology 1990;127:1580–9.
Elks ML. Chronic perifusion of rat islets with palmitate suppresses glucose-stimulated insulin release. Endocrinology 1993;133:208–14.
Zhou YP, Grill V. Long term exposure to fatty acids and ketones inhibits B-cell functions in human pancreatic islets of Langerhans. J Clin Endocrinol Metab 1995;80:1584–90.
Zhou YP, Grill VE. Long-term exposure of rat pancreatic islets to fatty acids inhibits glucose-induced insulin secretion and biosynthesis through a glucose fatty acid cycle. J Clin Invest 1994;93:870–6.
Gremlich S, Bonny C, Waeber G, Thorens B. Fatty acids decrease IDX-1 expression in rat pancreatic islets and reduce GLUT2, glucokinase, insulin, and somatostatin levels. J Biol Chem 1997;272:30261–9.
Ritz-Laser B, Meda P, Constant I, Klages N, Charollais A, Morales A, et al. Glucose-induced preproinsulin gene expression is inhibited by the free fatty acid palmitate. Endocrinology 1999;140:4005–14.
Jacqueminet S, Briaud I, Rouault C, Reach G, Poitout V. Inhibition of insulin gene expression by long-term exposure of pancreatic beta cells to palmitate is dependent on the presence of a stimulatory glucose concentration. Metabolism 2000;49:532–6.
Briaud I, Harmon JS, Kelpe CL, Segu VB, Poitout V. Lipotoxicity of the pancreatic beta-cell is associated with glucose-dependent esterification of fatty acids into neutral lipids. Diabetes 2001;50:315–21.
Kelpe CL, Moore PC, Parazzoli SD, Wicksteed B, Rhodes CJ, Poitout V. Palmitate inhibition of insulin gene expression is mediated at the transcriptional level via ceramide synthesis. J Biol Chem 2003;278:30015–21.
Hagman DK, Hays LB, Parazzoli SD, Poitout V. Palmitate inhibits insulin gene expression by altering PDX-1 nuclear localization and reducing MafA expression in isolated rat islets of Langerhans. J Biol Chem 2005;280:32413–8.
Pick A, Clark J, Kubstrup C, Levisetti M, Pugh W, Bonner-Weir S, et al. Role of apoptosis in failure of beta-cell mass compensation for insulin resistance and beta-cell defects in the male Zucker diabetic fatty rat. Diabetes 1998;47:358–64.
Cnop M, Hannaert JC, Hoorens A, Eizirik DL, Pipeleers DG. Inverse relationship between cytotoxicity of free fatty acids in pancreatic islet cells and cellular triglyceride accumulation. Diabetes 2001;50:1771–7.
Maedler K, Spinas GA, Dyntar D, Moritz W, Kaiser N, Donath MY. Distinct effects of saturated and monounsaturated fatty acids on beta-cell turnover and function. Diabetes 2001;50:69–76.
Lupi R, Dotta F, Marselli L, Del Guerra S, Masini M, Santangelo C, et al. Prolonged exposure to free fatty acids has cytostatic and pro-apoptotic effects on human pancreatic islets: evidence that beta-cell death is caspase mediated, partially dependent on ceramide pathway, and Bcl-2 regulated. Diabetes 2002;51:1437–42.
Wrede CE, Dickson LM, Lingohr MK, Briaud I, Rhodes CJ. Protein kinase B/Akt prevents fatty acid-induced apoptosis in pancreatic beta-cells (INS-1). J Biol Chem 2002;277:49676–84.
Piro S, Anello M, Di Pietro C, Lizzio MN, Patane G, Rabuazzo AM, et al. Chronic exposure to free fatty acids or high glucose induces apoptosis in rat pancreatic islets: possible role of oxidative stress. Metabolism 2002;51:1340–7.
Maedler K, Oberholzer J, Bucher P, Spinas GA, Donath MY. Monounsaturated fatty acids prevent the deleterious effects of palmitate and high glucose on human pancreatic beta-cell turnover and function. Diabetes 2003;52:726–33.
Maestre I, Jordan J, Calvo S, Reig JA, Cena V, Soria B, et al. Mitochondrial dysfunction is involved in apoptosis induced by serum withdrawal and fatty acids in the beta-cell line INS-1. Endocrinology 2003;144:335–45.
Poitout V, Robertson RP. Minireview: Secondary beta-cell failure in type 2 diabetes–a convergence of glucotoxicity and lipotoxicity. Endocrinology 2002;143:339–42.
Foufelle F, Ferre P. New perspectives in the regulation of hepatic glycolytic and lipogenic genes by insulin and glucose: a role for the transcription factor sterol regulatory element binding protein-1c. Biochem J 2002;366:377–91.
Karaskov E, Scott C, Zhang L, Teodoro T, Ravazzola M, Volchuk A. Chronic palmitate but not oleate exposure induces endoplasmic reticulum stress, which may contribute to INS-1 pancreatic beta-cell apoptosis. Endocrinology 2006;147:3398–407.
Boucher A, Lu D, Burgess SC, Telemaque-Potts S, Jensen MV, Mulder H, et al. Biochemical mechanism of lipid-induced impairment of glucose-stimulated insulin secretion and reversal with a malate analogue. J Biol Chem 2004;279:27263–71.
Joseph JW, Koshkin V, Saleh MC, Sivitz WI, Zhang CY, et al. Free fatty acid-induced beta-cell defects are dependent on uncoupling protein 2 expression. J Biol Chem 2004;279:51049–56.
Moore PC, Ugas MA, Hagman DK, Parazzoli SD, Poitout V. Evidence against the involvement of oxidative stress in fatty acid inhibition of insulin secretion. Diabetes 2004;53:2610–6.
Brunham LR, Kruit JK, Pape TD, Timmins JM, Reuwer AQ, Vasanji Z, et al. Beta-cell ABCA1 influences insulin secretion, glucose homeostasis and response to thiazolidinedione treatment. Nat Med 2007;13:340–7.
Martinez SC, Tanabe K, Cras-Meneur C, Abumrad NA, Bernal-Mizrachi E, Permutt MA. Inhibition of Foxo1 protects pancreatic islet beta-cells against fatty acid and endoplasmic reticulum stress-induced apoptosis. Diabetes 2008;57:846–59.
Robinson KA, Buse MG. Mechanisms of high-glucose/insulin-mediated desensitization of acute insulin-stimulated glucose transport and Akt activation. Am J Physiol Endocrinol Metab 2008;294:E870–81.
Kharroubi I, Ladriere L, Cardozo AK, Dogusan Z, Cnop M, Eizirik DL. Free fatty acids and cytokines induce pancreatic beta-cell apoptosis by different mechanisms: role of nuclear factor-kappaB and endoplasmic reticulum stress. Endocrinology 2004;145:5087–96.
Laybutt DR, Preston AM, Akerfeldt MC, Kench JG, Busch AK, Biankin AV, et al. Endoplasmic reticulum stress contributes to beta cell apoptosis in type 2 diabetes. Diabetologia 2007;50:752–63.
Hotamisligil GS. Role of endoplasmic reticulum stress and c-Jun NH2-terminal kinase pathways in inflammation and origin of obesity and diabetes. Diabetes 2005;54(Suppl 2):S73–8.
Nakatani Y, Kaneto H, Kawamori D, Yoshiuchi K, Hatazaki M, Matsuoka TA, et al. Involvement of endoplasmic reticulum stress in insulin resistance and diabetes. J Biol Chem 2005;280:847–51.
Huang CJ, Lin CY, Haataja L, Gurlo T, Butler AE, Rizza RA, et al. High expression rates of human islet amyloid polypeptide induce endoplasmic reticulum stress mediated beta-cell apoptosis, a characteristic of humans with type 2 but not type 1 diabetes. Diabetes 2007;56:2016–27.
Marchetti P, Bugliani M, Lupi R, Marselli L, Masini M, Boggi U, et al. The endoplasmic reticulum in pancreatic beta cells of type 2 diabetes patients. Diabetologia 2007;50:2486–94.
Wang J, Takeuchi T, Tanaka S, Kubo SK, Kayo T, Lu D, et al. A mutation in the insulin 2 gene induces diabetes with severe pancreatic beta-cell dysfunction in the Mody mouse. J Clin Invest 1999;103:27–37.
Eizirik DL, Cardozo AK, Cnop M. The role for endoplasmic reticulum stress in diabetes mellitus. Endocr Rev 2008;29:42–61.
Kaufman RJ, Scheuner D, Schroder M, Shen X, Lee K, Liu CY, et al. The unfolded protein response in nutrient sensing and differentiation. Nat Rev Mol Cell Biol 2002;3:411–21.
Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol 2000;2:326–32.
Lipson K, Fonseca S, Ishigaki S, Nguyen L, Foss E, Bortell R, et al. Regulation of insulin biosynthesis in pancreatic beta cells by anendoplasmic reticulum-resident protein kinase IRE1. Cell Metabolism 2006;4:245–54.
Lipson KL, Ghosh R, Urano F. The role of IRE1alpha in the degradation of insulin mRNA in pancreatic beta-cells. PLoS ONE 2008;3:e1648.
Pirot P, Naamane N, Libert F, Magnusson NE, Orntoft TF, Cardozo AK, et al. Global profiling of genes modified by endoplasmic reticulum stress in pancreatic beta cells reveals the early degradation of insulin mRNAs. Diabetologia 2007;50:1006–14.
Yoneda T, Imaizumi K, Oono K, Yui D, Gomi F, Katayama T, et al. Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. J Biol Chem 2001;276:13935–40.
Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000;287:664–6.
Cunha DA, Hekerman P, Ladriere L, Bazarra-Castro A, Ortis F, Wakeham MC, et al. Initiation and execution of lipotoxic ER stress in pancreatic {beta}-cells. J Cell Sci 2008;121:2308–18.
Yoshida H, Haze K, Yanagi H, Yura T, Mori K. Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. Involvement of basic leucine zipper transcription factors. J Biol Chem 1998;273:33741–9.
Yamamoto K, Yoshida H, Kokame K, Kaufman RJ, Mori K. Differential contributions of ATF6 and XBP1 to the activation of endoplasmic reticulum stress-responsive cis-acting elements ERSE, UPRE and ERSE-II. J Biochem 2004;136:343–50.
Okada T, Yoshida H, Akazawa R, Negishi M, Mori K. Distinct roles of activating transcription factor 6 (ATF6) and double-stranded RNA-activated protein kinase-like endoplasmic reticulum kinase (PERK) in transcription during the mammalian unfolded protein response. Biochem J 2002;366:585–94.
Lee K, Tirasophon W, Shen X, Michalak M, Prywes R, Okada T, et al. IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev 2002;16:452–66.
Wu J, Rutkowski DT, Dubois M, Swathirajan J, Saunders T, Wang J, et al. ATF6alpha optimizes long-term endoplasmic reticulum function to protect cells from chronic stress. Dev Cell 2007;13:351–64.
Thameem F, Farook VS, Bogardus C, Prochazka M. Association of amino acid variants in the activating transcription factor 6 gene (ATF6) on 1q21-q23 with type 2 diabetes in Pima Indians. Diabetes 2006;55:839–42.
Meex SJ, van Greevenbroek MM, Ayoubi TA, Vlietinck R, van Vliet-Ostaptchouk JV, Hofker MH, et al. Activating transcription factor 6 polymorphisms and haplotypes are associated with impaired glucose homeostasis and type 2 diabetes in Dutch Caucasians. J Clin Endocrinol Metab 2007;92:2720–5.
Seo HY, Kim YD, Lee KM, Min AK, Kim MK, Kim HS, et al. ER stress-induced Activation of ATF6 Decreases Insulin Gene Expression via Upregulation of Orphan Nuclear Receptor SHP. Endocrinology. 2008;149–3832–41.
Hinnebusch AG. Gene-specific translational control of the yeast GCN4 gene by phosphorylation of eukaryotic initiation factor 2. Mol Microbiol 1993;10:215–23.
Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, et al. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell 2000;6:1099–108.
Vattem KM, Wek RC. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc Natl Acad Sci U S A 2004;101:11269–74.
Wek RC, Cavener DR. Translational control and the unfolded protein response. Antioxid Redox Signal 2007;9:2357–71.
Tajiri S, Oyadomari S, Yano S, Morioka M, Gotoh T, Hamada JI, et al. Ischemia-induced neuronal cell death is mediated by the endoplasmic reticulum stress pathway involving CHOP. Cell Death Differ 2004;11:403–15.
Ma Y, Brewer JW, Diehl JA, Hendershot LM. Two distinct stress signaling pathways converge upon the CHOP promoter during the mammalian unfolded protein response. J Mol Biol 2002;318:1351–65.
Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell 2003;11:619–33.
Yamaguchi S, Ishihara H, Yamada T, Tamura A, Usui M, Tominaga R, et al. ATF4-mediated induction of 4E-BP1 contributes to pancreatic beta cell survival under endoplasmic reticulum stress. Cell Metab 2008;7:269–76.
Cnop M, Ladriere L, Hekerman P, Ortis F, Cardozo AK, Dogusan Z, et al. Selective inhibition of eukaryotic translation initiation factor 2 alpha dephosphorylation potentiates fatty acid-induced endoplasmic reticulum stress and causes pancreatic beta-cell dysfunction and apoptosis. J Biol Chem 2007;282:3989–97.
Pirot P, Ortis F, Cnop M, Ma Y, Hendershot LM, Eizirik DL, et al. Transcriptional regulation of the endoplasmic reticulum stress gene chop in pancreatic insulin-producing cells. Diabetes 2007;56:1069–77.
Harding H, Zeng H, Zhang Y, Jungreis R, Chung P, Plesken H, et al. Diabetes Mellitus and Exocrine Pancreatic Dysfunction in Perk/Mice Reveals a Role for Translational Control in Secretory Cell Survival. Mol Cell 2001;7:1153–63.
Oyadomari S, Koizumi A, Takeda K, Gotoh T, Akira S, Araki E, et al. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J Clin Invest 2002;109:525–32.
Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004;306:457–61.
Li D, Yin X, Zmuda EJ, Wolford CC, Dong X, White MF, et al. The repression of IRS2 gene by ATF3, a stress-inducible gene, contributes to pancreatic beta-cell apoptosis. Diabetes 2008;57:635–44.
Wang H, Kouri G, Wollheim CB. ER stress and SREBP-1 activation are implicated in beta -cell glucolipotoxicity. J Cell Sci 2005;118:3905–15.
Kawamori D, Kajimoto Y, Kaneto H, Umayahara Y, Fujitani Y, Miyatsuka T, et al. Oxidative stress induces nucleo-cytoplasmic translocation of pancreatic transcription factor PDX-1 through activation of c-Jun NH(2)-terminal kinase. Diabetes 2003;52:2896–904.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Chang-Chen, K.J., Mullur, R. & Bernal-Mizrachi, E. β-cell failure as a complication of diabetes. Rev Endocr Metab Disord 9, 329–343 (2008). https://doi.org/10.1007/s11154-008-9101-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11154-008-9101-5