
Abstract
Cu-ZSM-5 (Cu-Zeolite Socony Mobil-five) catalysts with different Cu loadings were prepared by ion exchange method. Different Ce loadings were coated on the surface of 0.1Cu-ZSM-5 to form CeO2-Cu-ZSM-5 catalysts. Cu could improve the NH3-SCR activity and broaden SCR temperature window. 0.1Cu-ZSM-5 showed the best catalytic performance (above 90%) in 200–400 ℃. 30% CeO2-Cu-ZSM-5 catalyst was able to increase the NO conversion in the low-temperature region and keep the activity unchanged in the high-temperature region. CeO2 that was coated on the catalysts preferentially reacted with SO2, enhancing the excellent anti-sulfur poisoning performance. TPD, TPR, and XPS results showed that the synergistic effect of Ce and Cu increased the acidic sites and improved the redox capacity of catalysts. According to in situ DRIFTS spectra, Cu-ZSM-5 catalyst followed the Langmuir–Hinshelwood mechanism, and 30% CeO2–0.1Cu-ZSM-5 catalyst followed not only the Langmuir–Hinshelwood mechanism but also the Eley–Rideal mechanism.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Nitrogen oxides (NOx) can cause photochemical smog, acid rain, and many other environmental problems, and seriously damage the ecological environment, causing the great concern [1]. The emission of NOx mainly comes from industrial kilns and automobile exhausts. There are many methods for the removal of nitrogen oxides from industrial kilns (denitrification). At present, the selective catalytic reduction with NH3 (NH3-SCR) technology has been widely used in the thermal power industry, glass industry, and cement industry, due to its high efficiency for NOx removal [2, 3]. The core of SCR technology is the catalyst. The temperature window of the commonly used commercial SCR catalyst (V2O5-WO3/TiO2) is 300 ~ 400 ℃ and the vanadium is very harmful to the environment, so the V-based catalyst is not suitable for the treatment of low and medium-temperature exhaust gases containing NOx (below 300 ℃) [4, 5]. At present, the main obstacles to the industrialization of low and medium-temperature SCR technology are the low catalytic activity and the poor resistance to SO2 poisoning. The active components are metal oxides for most SCR catalysts. Currently, the active components used for low-temperature SCR catalysts include metal oxides such as Mn, Cu, Fe, Cr, Mo, and V [6, 7], and carbon materials, inorganic oxides, and molecular sieves are selected as the carriers [8, 9].
Molecular sieves have many pore channels with uniform pore size and neatly arranged pores in their structures, and molecular sieves with different pore sizes separate molecules of different sizes and shapes. As a kind of porous crystal material, molecular sieve has large specific surface area, rich pore structure and strong stability, which is very suitable for SCR catalyst. Among them, ZSM-5, as a typical ultra-microporous material, has been widely used due to its advantages of good adsorption performance, moderate acidity, and flexible reaction temperature window [10, 11]. Brandenberger et al. [12] modified ZSM-5 molecular sieves with transition metals such as Cu and Fe, which substantially improved the catalytic efficiency of ZSM-5. Compared with the zeolite loaded iron, the zeolite loaded copper had better SCR activity at low temperature. Vennestrøm et al. [13] reported that ZSM-5 loaded Cu catalysts with lower Cu contents demonstrated over 80% NOx conversions in the temperature range of 200–500 ℃. Wang et al. prepared Cu-ZSM-5 catalysts with different Cu contents using the ion exchange method. However, molecular sieve catalysts encounter the problem of sulfur poisoning during the practical application, so numerous researchers have focused on enhancing the sulfur resistance of the catalysts. According to the previous study, Ma et al. [14] found that the SAPO-5/34 catalyst doped with Cu and Ce had excellent sulfur resistance. It is well known that catalysts with core–shell or cladding structures have higher thermal stability and anti-poisoning properties [15]. Liu et al. [16] designed loaded catalysts with two metal oxides as the core and CeO2 thin film as the shell, which showed high SO2 and H2O tolerance and denitrification activity. Chen et al. [17] prepared the core–shell structured Fe-ZSM-5@CeO2 catalyst with uniformly distributed active sites and found that the synergistic effect between Fe-ZSM-5 and CeO2 could improve the NH3-SCR performance. Therefore, the introduction of Ce can significantly enhance the sulfur resistance and SCR performance for catalysts, and the core–shell and cladding structures can improve the comprehensive performance of the catalysts. Therefore, the denitrification performance of CeO2-coated Cu-ZSM-5 catalyst will be investigated in this paper.
In this study, a series of Cu-ZSM-5 catalysts were prepared, and the effect of Cu loading on SCR denitrification performance was investigated. Meanwhile, CeO2 was loaded on the surface of Cu-ZSM-5 catalysts to form a CeO2 capping layer, and the SCR performance and sulfur resistance of the CeO2-Cu-ZSM-5 catalysts were investigated.
Experiment
Preparation of Cu-ZSM-5 and CeO2-Cu-ZSM-5 catalysts
Cu-ZSM-5 catalyst was prepared by the ion exchange method. 500 mL of deionized water, a certain mass of Cu(CH3COO)2·H2O and 10.00 g ZSM-5 molecular sieves were mixed together in a beaker, and the temperature was preheated to 50 ℃. The mixed solution was stirred at constant temperature for 2 h. The reacted slurry was filtered and washed repeatedly with deionized water, and then dried in an oven at 60 ℃ for 12 h. The dried samples were placed in a muffle furnace and calcined under the air atmosphere at 500 ℃ for 4 h. After the samples were cooled down, they were ground to obtain the xCu-ZSM-5 catalysts (where x stands for the concentration of the copper acetate solution, and x = 0 mol/L, 0.01 mol/L, 0.05 mol/L, 0.10 mol/L, 0.15 mol/L 0.20 mol/L).
3 g 0.1Cu-ZSM-5, 2 g polyvinylpyrrolidone (PVP), 1 g hexamethylenetetetramine (HMT) and a certain amount of Ce(NO3)3·6H2O were added to the solvent mixture of 100 mL deionized water and 100 mL ethanol, and stirred by refluxing at 60 ℃ for 2 h. The slurry after the reaction was filtered and washed with deionized water for 3 times, before it was placed in an oven and dried at 60 ℃ for 12 h. The dried sample was put into a muffle furnace and calcined under the air atmosphere at 450 ℃ for 3.5 h. And the xCeO2–0.1Cu-ZSM-5 (x = 10 wt%, 20 wt %, 30 wt% and 40 wt%) catalysts were obtained.
Catalytic activity test
The catalytic activity measurements of NH3-SCR were measured using a solid bed quartz reactor. 2 g catalyst was pressed into cake-like blocks with 4 mm diameter and used for the catalytic activity. The feedstock consists of 800 ppm NH3, 720 ppm NO, 3% O2, 150 ppm SO2 (when used), and N2 (as an equilibrium gas). In order to remove impurity gases from the line, N2 was added into the reaction system for half an hour before the test. Then NO and NH3 were added into the reaction system together, while the temperature was increased to 80 ℃ by programmed heating. The SCR activities of the catalysts are calculated by the equation: XNO = ([NO]in–[NO]out)/[NO]in × 100%. [NO]out is the output of NO (ppm), [NO]in is the input of NO (ppm), and XNO is the NO conversion.
Catalyst characterization
The samples were physically analyzed using a Bruker D8 Advance diffractometer (Bruker, German) equipped with the Cu Kɑ radiation source (λ = 0.15406 nm). The scanning speed was 5°/min and the scanning range of 2θ was 10 ~ 70°.
Hydrogen temperature programmed reduction (H2-TPR) experiments were performed on an automated catalyst characterization device (Chemisorb 2720, Micromeritics, USA). 40 mg of the samples were exposed to a H2 (5%)/Ar atmosphere, and the temperature raised from the room temperature to 900 ℃ at a rate of 10 ℃/min. The consumption of H2 was then detected by the TCD.
Ammonia temperature programmed desorption (NH3-TPD) experiments were performed on the same instrument that was used for H2-TPR test. 40 mg of the sample was pretreated in a stream of He gas at 300 ℃ for 1 h. After the sample was cooled down to 110 ℃, it was fed with NH3 (10%)/He gas for 1 h until the adsorption equilibrium was achieved. Finally, the treated catalysts were heated from 30 °C to 600 °C at a rate of 10 ℃/min under a He flow.
A field emission scanning electron microscope (FESEM, Ultra Plus, Zeiss) was used to observe the shape and size of the samples.
All X-ray photoelectron spectroscopy (XPS) experiments were implemented on the ESCALAB 250Xi spectrometer with the mono Al anode. Operation details and analysis methods were used according to previous research [18]. The C1s binding energy of air-adsorbed carbon (284.8 eV) was used as a reference peak to correct the binding energies of other elements in the spectrum [19].
A Nicolet 6700 IR spectrometer (Thermo Fisher) equipped with an MCT detector and a high-temperature reaction chamber was used to test the in situ DRIFTS spectra. Prior to the experiments, the catalysts were purged with the samples in N2 atmosphere at 300 ℃ to remove impurity gases from the experimental lines as well as impurities adsorbed on the catalyst surfaces. NH3 (NO + O2) was adsorbed at this temperature for 30 min. Then the pre-adsorbed gas was disconnected and NO + O2 (NH3) was added in reaction system for 30 min, and the changes for the catalysts were recorded [20, 21].
Experimental results and discussion
Cu-ZSM-5 catalyst
SCR performance
NO conversions with temperatures for xCu-ZSM-5 and ZSM-5 catalysts are shown in Fig. 1. For pure ZSM-5, the NO conversion was less than 30% below 300 ℃. When the temperature increased to 400 ℃, the NO conversion was less than 70%. The NO conversions of xCu-ZSM-5 catalysts were significantly increased. Although the SCR activities of Cu-loaded samples slightly decreased in the high temperature region, they still far exceeded that of ZSM-5, indicating that the introduction of Cu increased the SCR activity and widened the SCR temperature window. 0.1Cu-ZSM-5 catalyst could maintain the NO conversion above 90% in the temperature range of 200–400 ℃ and the performance remained stable, illustrating the best catalytic performance. Compared with 0.1Cu-ZSM-5, 0.05Cu-ZSM-5 had similar maximum NO conversion. However, the NO conversion of 0.05Cu-ZSM-5 catalyst showed a significant decrease in the high temperature region, and the temperature window corresponding to T90 (NO conversion of 90% and above) was narrowed. Compared with 0.1Cu-ZSM-5, the temperature windows of 0.15Cu-ZSM-5 and 0.2Cu-ZSM-5 catalysts corresponding to T90 were shifted from 200–400 ℃ to 160–300 ℃. This might be due to the fact that the high content of copper leaded to the agglomeration of copper species and the formation of larger CuO particles, enhancing the oxidation of NH3 to narrow the temperature window. xCu-ZSM-5 catalysts showed slight decreases in catalytic activity at high temperatures, which was attributed to the non-selective oxidation of NH3.

NH3-SCR performances of xCu-ZSM-5 catalysts
XRD analysis
XRD patterns of pure ZSM-5 and xCu-ZSM-5 catalysts are shown in Fig. 2. Diffraction peaks at 2θ diffraction angles of 23.1°, 23.9°, and 24.4° were matched with ZSM-5, indicating that the microstructure of ZSM-5 molecular sieves was still retained after the addition of Cu. Faint CuO diffraction peaks were observed for the xCu-ZSM-5 catalysts, indicating that the copper ions were highly dispersed and the particle size of CuO formed on the surface of the ZSM-5 molecular sieve was very small.

XRD spectra of xCu-ZSM-5 catalysts
Surface morphologies of xCu-ZSM-5 catalysts
The surface morphologies of ZSM-5 molecular sieves and xCu-ZSM-5 catalysts are investigated by FESEM. The surface morphologies of all samples magnified at 20.0 kx are shown in Fig. S1. From the figures, it could be seen that ZSM-5 molecular sieves showed regular geometry and good dispersion. After copper ions were loaded on ZSM-5 molecular sieves, the morphologies of ZSM-5 were still clearly visible, but the gap between xCu-ZSM-5 particles decreased and the particle size became larger. And the corners of ZSM-5 became smooth after the addition of Cu, which might be caused by the ion exchange of copper ions on the surface of ZSM-5 catalysts.
In situ DRIFTS results of 0.1Cu-ZSM-5 catalyst
The in situ DRIFTS spectra of pre-adsorbed NH3 on 0.1Cu-ZSM-5 catalyst are shown in Fig. 3a. Peaks at 1180, 1320, and 1330 cm−1 were corresponded to the coordination NH3 adsorbed on Lewis acid sites, while peaks at 1734, 3270, and 3350 cm−1 were attributed to NH4+ adsorbed on Brønsted acid sites. NH3 on the Lewis acid site and NH4+ on the Brønsted acid site decreased gradually with NO + O2 passage for 8 min, suggesting that both the coordinated NH3 molecules and NH4+ were involved in the NH3-SCR process of NOx [22]. After NO + O2 passage for 8 min, bridged nitrate strips (1638 cm−1), monodentate nitrite strips (1365 cm−1), bidentate nitrate strips (1550 cm−1) [16], and other nitrogen compounds (1215 and 1892 cm−1) began to appear one after the other, and nitrogen compounds gradually increased with exposure duration. Therefore, the related results suggested that Cu-ZSM-5 catalyst followed Langmuir–Hinshelwood mechanism.

In situ DRIFTS spectra of NO + O2 reacted with pre-adsorbed NH3 species (a) and NH3 reacted with pre-adsorbed NOx species (b) over 0.1Cu-ZSM-5 catalyst
The in situ DRIFTS spectra of 0.1Cu-ZSM-5 catalyst pre-adsorbed NO + O2 are shown in Fig. 3b. After the addition of NO + O2, nitrate strips (1628 cm−1), and other nitrogen compounds (1215 and 1892 cm−1) were detected for the 0.1Cu-ZSM-5 catalyst. The bands of nitrogen compounds on the catalyst converged to a straight line within 8 min after the addition of NH3. Liganded NH3 (1330 cm−1) adsorbed at the Lewis acid site appeared after the passage of NH3 for 2 min, and adsorbed NH3 and NH4+ species-associated bands (1180, 1606, and 3184 cm−1) could be detected after the passage of NH3 for 14 min. Therefore, the related results suggested that Cu-ZSM-5 catalyst followed Langmuir–Hinshelwood mechanism. According to the in situ DRIFTS results of 0.1Cu-ZSM-5 catalyst pre-adsorbed NO + O2 and NH3, the catalyst followed Langmuir–Hinshelwood mechanism during the SCR process.
CeO2-coated 0.1Cu-ZSM-5 catalysts
SCR performance
0.1Cu-ZSM-5 catalyst was selected to compare the denitrification performances with xCeO2–0.1Cu-ZSM-5, as shown in Fig. 4. The NO conversion of 10%CeO2–0.1Cu-ZSM-5, 20%CeO2–0.1Cu-ZSM-5, and 40%CeO2–0.1Cu-ZSM-5 catalysts decreased compared with 0.1Cu-ZSM-5, and the denitrification activity decreased significantly with the increased cerium loadings. That might be due to CeO2 coating onto the surface of 0.1Cu-ZSM-5, covering some active ingredients and reducing the percentage of active ingredient copper ions on the surfaces. However, 30%CeO2–0.1Cu-ZSM-5 had approximately the same catalytic activity as 0.1Cu-ZSM-5, with similar maximum NO conversion up to nearly 100%. Meanwhile, in the low-temperature region (below 200 ℃), the NO conversion of 30%CeO2–0.1Cu-ZSM-5 was higher than that of 0.1Cu-ZSM-5, and the NO conversion of 30%CeO2–0.1Cu-ZSM-5 decreased slightly after 320 ℃ compared with 0.1Cu-ZSM-5. By adding a small amount of cerium, the synergistic effect of Ce and Cu was not obvious, and Ce only showed the dilution of active sites, causing the decrease of SCR performances. Meanwhile, by adding an excessive amount of cerium, the decrease of exposure area played a dominant role in SCR performance rather than the synergistic effect between Cu and Ce, illustrating the decrease of catalytic activities. Moreover, when the addition of cerium was appropriate, the strong synergistic effect of Ce and Cu compensated for the capping effect of CeO2, causing an increase in the catalytic activity of 30%CeO2–0.1Cu-ZSM-5 [23].

NH3-SCR performances of xCeO2-Cu-ZSM-5 catalysts
XRD analysis
XRD results of xCeO2–0.1Cu-ZSM-5 catalysts are shown in Fig. S2. All catalysts showed typical peaks corresponding to ZSM-5 molecular sieves, but the intensity of the characteristic peaks became weaker, indicating that the original structure of the molecular sieves remained intact after the addition of cerium. Compared with 0.1Cu-ZSM-5 catalyst, weak CuO peaks were detected and no obvious CeO2 diffraction peaks could be found for xCeO2–0.1Cu-ZSM-5 catalysts, indicating that cerium oxides as an amorphous oxide were well dispersed on the surfaces.
Surface morphologies of xCeO2–0.1Cu-ZSM-5 catalysts
Surface morphologies of the xCeO2–0.1Cu-ZSM-5 catalysts were investigated by FESEM, as shown in Fig. S3. When 0.1Cu-ZSM-5 catalyst was doped with a small amount of Ce, the surfaces were covered with a layer of fine granular material, which was due to a small amount of CeO2 clustered into nanoparticles on the surfaces. As the amount of Ce loadings increased, the surfaces became rougher and rougher, which was attributed to the fact that CeO2 was well dispersed on the surfaces of catalyst. The coverage of CeO2 on the surfaces of the catalysts increased and the aggregation took place, when the amount of cerium loadings increased. Although the surfaces of the catalysts became rougher and rougher, the overall structures of the catalysts remained relatively intact.
NH3-TPD analysis
NH3-TPD results of 0.1Cu-ZSM-5 and xCeO2–0.1Cu-ZSM-5 catalysts are shown in the Fig. 5. Three NH3 desorption peaks were clearly observed for the 0.1Cu-ZSM-5 catalyst at 105, 205, and 310 °C, and the NH3 desorption peak areas are named A, B, and C in the Table S1. The peaks located at 105 °C and 200 °C were assigned to weakly acidic sites and attributed to NH3 bound to weak Brønsted acid. The peak at 310 °C was corresponded to moderately acidic sites (Lewis). According to the previous study, CuO appeared when the surfaces of catalysts were saturated with copper ions, and agglomerated CuO nanoparticles produced the Lewis acid sites [22]. When Ce was introduced, the xCeO2–0.1Cu-ZSM-5 catalysts formed new acidic sites at 420 °C and led to new NH3 desorption peaks. The area of the new peak was named D in the Table S1. The desorption peak of NH3 at around 420 °C indicated that CeO2 on the catalyst surface was effective in increasing the strong acid sites [24]. After the introduction of Ce, the number of weak acidic sites at 105 °C roughly trended toward decreasing, the number of weak acidic sites at 205 °C roughly trended toward increasing, and the number of medium acidic sites at 310 °C decreased with the increase in Ce loading. The introduction of Ce might be effective in reducing the formation of CuO, causing the decrease of the number of medium acid sites. However, the total area of the NH3 desorption peak of the 30%CeO2–0.1Cu-ZSM-5 catalyst was the smallest, indicating that the number of acidic sites did not directly determine the catalytic activity.

NH3-TPD results for xCeO2–0.1Cu-ZSM-5 catalysts
H2-TPR analysis
H2-TPR results for 0.1Cu-ZSM-5 and xCeO2–0.1Cu-ZSM-5 catalysts are shown in Fig. 6. Three reduction peaks from low to high temperatures were corresponded to the reduction of free Cu2+ to Cu+, CuO to Cu0, and Cu+ to Cu0y. From the Table S2, the area of the reduction curve of the 0.1Cu-ZSM-5 catalyst was the largest. The area of the reduction curve decreased after the addition of Ce, illustrating that the coating of CeO2 reduced the proportion of copper ions on ZSM-5, which was consistent with the SCR performances and TPD results. Compared to other Ce-loaded catalysts, the reduction peak area of Cu2+ → Cu+ in 30%CeO2–0.1Cu-ZSM-5 was larger, and that of Cu+ → Cu0 was smaller. It has been reported that copper ions in close proximity to CeO2 are more susceptible to reduction by hydrogen [25]. It might be due to the fact that copper ions could form Ce–O–Cu with cerium oxide, changing the redox properties of catalysts [26]. Some copper ions could be bound with the vacancies of cerium oxides. The copper in the vacancies entering the cerium oxide might migrate out of the cerium oxide network and become highly dispersed copper ions on the surface, which could be easily reduced. Therefore, the reduction peak area of Cu2+ → Cu+ was increased for the catalyst loaded with 30% Ce. The reduction peak of Cu+ → Cu0 was shifted toward high temperature after the introduce of Ce, and it might be that the CeO2 coating affected the reaction of Cu+ → Cu0. And the reduction peak of Cu+ → Cu0 in 20%CeO2–0.1Cu-ZSM-5 was shifted the most toward high temperature, leading to its poor denitrification activity. The above showed that the addition of appropriate amount of Ce to Cu-ZSM-5 catalysts could improve the low-temperature reducibility of the catalysts. In addition, the synergistic effect of Ce and Cu could be maximized by suitable loading of Ce, and the synergistic effect between Ce and Cu was the most significant for the 30%CeO2–0.1Cu-ZSM-5 catalyst. The reduction peak area of CuO → Cu0 in 30%CeO2–0.1Cu-ZSM-5 catalyst was the smallest, suggesting that the loading of Ce inhibited the generation of CuO on the surfaces [27].

H2-TPR results for xCeO2–0.1Cu-ZSM-5 catalysts
XPS analysis
The valence states of the surface elements of the catalysts were detected by the XPS spectroscopy, as shown in Fig. S4. The electronic states of the O element in the 0.1Cu-ZSM-5 and xCeO2–0.1Cu-ZSM-5 catalysts are shown in Fig. S4a. The O1s peak of xCeO2–0.1Cu-ZSM-5 could be fitted to three peaks, such as the lattice oxygen Oα peak (529.6 eV), the chemisorbed oxygen Oβ peak (531.4 eV), and the hydroxyl oxygen or surface adsorbed water Oγ peak (532.3 eV) [28]. The peak with a binding energy of 529.6 eV belonged to the lattice oxygen in CeO2. In Table 1, except for 20%CeO2–0.1Cu-ZSM-5, the percentage of Oγ peak decreased and the percentage of Oα peak increased with the increase of Ce content in the other catalysts. Therefore, with the increase of the introduction of Ce, the thickness of the surface-coated CeO2 in Cu-ZSM-5 also increased. The XPS results of Cu in catalysts are shown in Fig. S4b. In Table 1, the copper content on the catalyst surface changed with the introduction of Ce. The highest percentage of Cu+ was found in 30%CeO2–0.1Cu-ZSM-5, enhancing the best catalytic activity.
The Ce3d XPS peak in Fig. S4c can be fitted to a map of the electronic states in eight 3d orbitals. Peaks at 883.2 eV, 885.9 eV, 890.0 eV, and 898.7 eV were corresponded to the Ce3d5/2 orbitals, while peaks at 900.9 eV, 903.6 eV, 908.0 eV, and 917.5 eV were consisted with the Ce3d3/2 orbitals. Peaks at 885.9 eV and 903.6 eV were related to Ce3+ species, while 883.2 eV, 890.0 eV, 898.7 eV, 900.9 eV, 908.0 eV, and 917.5 eV were ascribed to Ce4+ species. Therefore, there were mainly two valence states of Ce element. Ce3+ ↔ Ce4+ could achieve the role of oxygen storage and release. The electron transfer from Cu+ to Ce4+ induced the formation of surface oxygen vacancy on the CeO2, and Cu-redox (Cu+ ↔ Cu2+) and Ce-redox (Ce3+ ↔ Ce4+) cycles could activate NH3 and O2, causing the synergistic promotion of SCR performances [29,30,31]. Therefore, the highest percentage of Ce4+ (shown in Table 1) for the 30%CeO2–0.1Cu-ZSM-5 catalyst was beneficial to SCR reaction.
In situ DRIFTS results of 30%CeO2–0.1Cu-ZSM-5 catalyst
The in situ DRIFTS spectra of pre-adsorbed NH3 over 30%CeO2–0.1Cu-ZSM-5 catalyst are shown in Fig. 7a. Peaks at 1180 cm−1 and 1330 cm−1 were corresponded to liganded NH3 adsorbed on Lewis acid sites, while peaks at 1397 cm−1, 3276 cm−1, and 3360 cm−1 were attributed to NH4+ adsorbed on Brønsted acid sites. After the passage of NO + O2 for 4 min, the NH3 on Lewis acid sites and the NH4+ on Brønsted acid sites gradually decreased until they disappeared. After the introduction of NO + O2 for 8 min, bridged nitrate strips (1638 cm−1), monodentate nitrite strips (1365 cm−1), and bidentate nitrate strips (1540 cm−1) were observed. With the introduction of Ce, adsorbed NH4+ and NH3 on the catalyst decreased more rapidly and more compounds of nitrogen were found, enhancing excellent denitrification activity of 30%CeO2–0.1Cu-ZSM-5 catalyst in the low temperature region. Meanwhile, according to the above results, the 30%CeO2–0.1Cu-ZSM-5 catalyst followed the Eley–Rideal mechanism.

In situ DRIFTS spectra of NO + O2 reacted with pre-adsorbed NH3 species (a) and NH3 reacted with pre-adsorbed NOx species (b) over 30%CeO2–0.1Cu-ZSM-5 catalyst
The in situ DRIFTS spectra of pre-adsorbed NO + O2 over 30%CeO2–0.1Cu-ZSM-5 catalyst is shown in Fig. 7b. Monodentate nitrite bands (1365 cm−1), bidentate nitrate bands (1570 cm−1), and other compounds of nitrogen (1215, 1675 cm−1) C were observed due to the introduce of NO + O2. The adsorbed NH3 and NH4+ species-related bands (1185, 1330, 3278 and 3358 cm−1) appeared successively after the NH3 passage for 14 min. Therefore, the 30%CeO2–0.1Cu-ZSM-5 catalyst followed the Langmuir–Hinshelwood mechanism based on the above results. According to the in situ DRIFTS results of 30%CeO2–0.1Cu-ZSM-5 catalyst pre-adsorbed NO + O2 and NH3, the catalyst followed Langmuir–Hinshelwood and Eley–Rideal mechanism during the SCR process.
Sulfur resistance
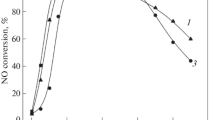
In practical applications, the sulfur resistance is the quite important criterion for catalysts. In order to investigated the effect of sulfur on the catalytic activity, sulfur resistance experiments were conducted over 0.1Cu-ZSM-5 and 30%CeO2–0.1Cu-ZSM-5 catalysts, as shown in Fig. 8. When the 0.1Cu-ZSM-5 catalyst was used for SCR reaction at 300 ℃ for 1 h, the NO conversion decreased from 99 to 95%. When the 30%CeO2–0.1Cu-ZSM-5 catalyst was used for SCR reaction at 300 ℃ for 1 h, the NO conversion decreased from 99 to 97%. It can be seen that the stability of 0.1Cu-ZSM-5 is not as good as that of 30%CeO2–0.1Cu-ZSM-5, indicating that the addition of Ce can improve the stability of the catalysts for a long time. It might be due to the fact that the presence of Ce could synergize with Cu to promote the rapid switching of Cu2+ ↔ Cu+, thus exhibiting excellent stability.

Sulfur resistance results of 0.1Cu-ZSM-5 and 30%CeO2–0.1Cu-ZSM-5 catalysts
When SO2 was introduced into the reaction system for a few hours, the NO conversion of the 0.1Cu-ZSM-5 catalyst decreased significantly from 95 to about 77%, and the NO conversion of the 30%CeO2–0.1Cu-ZSM-5 catalyst decreased insignificantly from 97 to about 93%. After cutting off SO2, the catalytic activities of both catalysts could not be restored to the initial level. Therefore, these observations illustrated that the sulfur resistance of 30%CeO2–0.1Cu-ZSM-5 catalyst was greatly improved compared to 0.1Cu-ZSM-5 catalyst due to the loading of Ce.
SO2 can react with NH3 and H2O to form sulfate substances ((NH4)2SO4, NH4HSO4, and (NH4)2SO3). SO2 can also react with the active ingredient Cu2+ to form CuSO4, which has a large particle size and clogs up the active sites on the surfaces of the catalyst [32, 33]. Meanwhile, the above effects can decrease the catalytic activities. The decomposition temperature of the generated CuSO4 is above 550 ℃, and they cannot be restored to its initial level. According to the previous studies, Ce species could restrain the deactivation of the catalyst by SO2 [34]. Firstly, CeO2 reacts with SO2 to produce Ce(SO4)2 and Ce2(SO4)3, increasing the number of surface hydroxyls and providing more Brønsted acid sites for the catalysts [28]. The unstable cerium sulphate inhibits the production of ammonium sulfate and CuSO4 on the surfaces of catalysts. Secondly, CeO2 can reduce the thermal stability of sulfates on the surfaces and promote the decomposition of toxic substances [35], and appropriate amount of Ce3/4+ ions and reaction conditions can realize the dynamic equilibrium between sulfates formation and decomposition to achieve a good SO2 resistance [36]. Therefore, it can be determined that the introduction of Ce not only improves the thermal stability but also enhances the sulfur resistance for 30%CeO2–0.1Cu-ZSM-5 catalyst.
Conclusion
In this study, the introduction of Cu significantly enhanced the denitrification performance of the catalyst, illustrating a wider temperature window. The 0.1Cu-ZSM-5 catalyst could obtain the NO conversion above 90% in the temperature range of 200–400 ℃. 30%CeO2–0.1Cu-ZSM-5 catalyst was prepared by coating CeO2 on the surfaces. The denitrification performance of 30%CeO2–0.1Cu-ZSM-5 catalyst was better than that of 0.1Cu-ZSM-5. According to the related characterizations, Ce-coated catalysts illustrated excellent SCR performances at low temperature due to the synergistic effect of Ce and Cu. The redox properties of the catalysts and the dispersion of Cu species on the catalyst surface were improved by the synergistic effect. Related experiments showed that the 30%CeO2–0.1Cu-ZSM-5 catalyst exhibited excellent thermal stability and sulfur resistance.
Data availability
The data are available from the corresponding author on reasonable request.
References
Anenberg S, Schwartz J, Shindell D, Amann M, Faluvegi G, Klimont Z, Maenhout G, Pozzoli L, Dingenen R, Vignati E, Emberson L, Muller N, West J, Williams M, Demkine V, Hicks W, Kuylenstierna J, Raes F, Ramanathan V (2012) Global air quality and health co-benefits of mitigating near-term climate change through methane and black carbon emission controls. Environ Health Persp 120:831–839
Xie X, Peng J, Zhao S, Wang L, Ge R, Wu S, Mai Y, Zeng K, Sun Z (2022) DeNOx characteristics of commercial SCR catalyst regenerated on–line by dry ice blasting in a coal-fired power plant. Ind Eng Chem Res 61(38):14382–14392
Ren Q, Zhang G, Huang X, Tang Z, Zhang J (2023) Revealing the deactivation mechanism of Ca poisoning on V-W-Ti catalysts in cement kilns. Fuel 351:128999
Lietti L, Nova I, Ramis G, Dall’Acqua L, Busca G, Giamello E, Forzatti P, Bregani F (1999) Characterization and reactivity of V2O5-MoO3-TiO2 De-NOx SCR Catalysts. J Catal 187:419–435
Maqbool M, Pullur A, Ha H (2014) Novel sulfation effect on low–temperature activity enhancement of CeO2-added Sb-V2O5/TiO2 catalyst for NH3-SCR. Appl Catal B-Environ 152–153:28–37
Fang D, He F, Mei D, Zhang Z, Xie J, Hu H (2014) Thermodynamic calculation for the activity and mechanism of Mn/TiO2 catalyst doped transition metals for SCR at low temperature. Catal Commun 52:45–48
Thirupathi B, Smirniotis P (2011) Co-doping a metal (Cr, Fe Co, Ni, Cu, Zn, Ce, and Zr) on Mn/TiO2 catalyst and its effect on the selective reduction of NO with NH3 at low-temperatures. Appl Catal B-Environ 110:195–206
Han L, Cai S, Gao M, Hasegawa J, Wang P, Zhang J, Shi L, Zhang D (2019) Selective catalytic reduction of NOx with NH3 by using novel catalysts: state of the art and future prospects. Chemical Rev 119(19):10916–10976
Yao X, Chen L, Kong T, Ding S, Luo Q, Yang F (2017) Support effect of the supported ceria–based catalysts during NH3-SCR reaction. Chinese J Catal 38(38):1423–1430
Pan W, He J, Huang G, Zhang W, Fang D (2023) Research progress of the selective catalytic reduction with NH3 over ZSM-5 zeolite catalysts for NOx removal. Catalysts 13(10):1381
Yang J, Li Z, Yang C, Ma Y, Li Y, Zhang Q, Song K, Cui J (2022) Significant promoting effect of La doping on the wide temperature NH3-SCR performance of Ce and Cu modified ZSM-5 catalysts. J Solid State Chem 305:122700
Brandenberger S, Krocher O, Tissler A, Althoff R (2010) The determination of the activities of different iron species in Fe-ZSM-5 for SCR of NO by NH3. Appl Catal B-Environ 95(3–4):348–357
Vennestrom P, Janssens T, Kustov A, Grill M, Puig-Molina A, Lundegaard L, Tiruvalam R, Concepcion P, Corma A (2014) Influence of lattice stability on hydrothermal deactivation of Cu-ZSM-5 and Cu-IM-5 zeolites for selective catalytic reduction of NOx by NH3. J Catal 309:477–490
Ma Y, Li Z, Zhao N, Teng Y (2021) One-pot synthesis of Cu-Ce co-doped SAPO-5/34 hybrid crystal structure catalysts for NH3-SCR reaction with SO2 resistance. J Rare Earths 39(10):1217–1223
Masoumifard N, Guillet-Nicolas R, Kleitz F (2018) Synthesis of engineered zeolitic materials: from classical zeolites to hierarchical core–shell materials. Adv Mater 30(16):1704439
Liu J, Du Y, Liu J, Zhao Z, Cheng K, Chen Y, Zhang X (2017) Design of MoFe/Beta@CeO2 catalysts with a core–shell structure and their catalytic performances for the selective catalytic reduction of NO with NH3. Appl Catal B-Environ 203:704–714
Chen L, Wang X, Cong Q, Ma H, Li S, Li W (2019) Design of a hierarchical Fe-ZSM-5@CeO2 catalyst and the enhanced performances for the selective catalytic reduction of NO with NH3. Chem Eng J 369:957–967
Fang D, He F, Xie J, Xue L (2020) Calibration of binding energy positions with C1s for XPS results. J Wuhan Univ Technol-Mat Sci Edit 35:711–718
Chen X, Wang X, Fang D (2020) A review on C1s XPS-spectra for some kinds of carbon materials. Fuller Nanotub Car N 28:1048–1058
Xie J, Ye Y, Li Q, Kang T, Hou S, Jin Q, He F, Fang D (2023) Denitrification performance and sulfur resistance mechanism of Sm-Mn catalyst for low temperature NH3-SCR. Front Chem Sci Eng 17(5):617–633
Fang D, Qi K, Li F, He F, Xie J (2022) Excellent sulfur tolerance performance over Fe-SO4/TiO2 catalysts for NH3-SCR: Influence of sulfation and Fe-based sulfates. J Environ Chem Eng 10(1):107038
Lai S, Meng D, Zhan W, Guo Y, Zhang Z, Lu G (2015) The promotional role of Ce in Cu/ZSM-5 and in situ surface reaction for selective catalytic reduction of NOx with NH3. RSC Adv 5(110):90235–90244
Zhang L, Tian F, Li H, Meng J, Liu Q, Guo X, Qiu Y, Zhang J, Li C (2023) Ce (III)-modulation over non-enzymatic Pt/CeO2/GO biosensor with outstanding sensitivity and stability for lactic acid detection. J Rare Earths 41(9):1437–1447
Dou B, Lv G, Wang C, Hao Q, Hui K (2015) Cerium doped copper/ZSM-5 catalysts used for the selective catalytic reduction of nitrogen oxide with ammonia. Chem Eng J 270:549–556
Wang W, Yu W, Du P, Xu H, Jin Z, Si R, Ma C, Shi S, Jia C, Yan C (2017) Crystal plane effect of ceria on supported copper oxide cluster catalyst for CO oxidation: Importance of metal–support interaction. ACS Catal 7(2):1313–1329
Marco P, Samir B, Tahrizi A, Nunzio R, Raffaele P, Debora F (2017) Cerium-copper oxides prepared by solution combustion synthesis for total oxidation reactions: from powder catalysts to structured reactors. Appl Catal B-Environ 205:455–468
Mai H, Sun L, Zhang Y, Si R, Feng W, Zhang H, Liu H, Yan C (2005) Shape–selective synthesis and oxygen storage behavior of ceria nanopolyhedra, nanorods, and nanocubes. J Phys Chem B 109(51):24380–24385
Di Z, Wang H, Zhang R, Chen H, Wei Y, Jia J (2022) ZSM-5 core–shell structured catalyst for enhancing low-temperature NH3-SCR efficiency and poisoning resistance. Appl Catal A-Gen 630:118438
Bin F, Wei X, Li B, San Hui K (2015) Self-sustained combustion of carbon monoxide promoted by the Cu-Ce/ZSM-5 catalyst in CO/O2/N2 atmosphere. Appl Catal B-Environ 162:282–288
Li Y, Song W, Liu J, Zhao Z, Gao M, Wei Y, Deng J (2017) The protection of CeO2 thin film on Cu-SAPO-18 catalyst for highly stable catalytic NH3-SCR performance. Chem Eng J 330:926–935
Wang F, Li S, You R, Han Z, Yuan W, Zhu B, Gao Y, Yang H, Wang Y (2023) Unraveling the synergy between MnOx and CeO2 in MnOx-CeO2 SCR catalysts based on experimental and DFT studies. Appl Surf Sci 638:15812
Wang H, Xu X, Hao J, Ning P, Zhang Q (2022) Unravelling the phosphorus–induced effect on NH3-SCR catalytic performance, hydrothermal stability and SO2 resistance of Cu/SAPO-34. Appl Catal A-Gen 646:118888
Zhang L, Wang D, Liu Y, Kamasamudram K, Li J, Epling W (2014) SO2 poisoning impact on the NH3-SCR reaction over a commercial Cu-SAPO-34 SCR catalyst. Appl Catal B-Environ 156:371–377
Li B, Ren Z, Ma Z, Huang X, Liu F, Zhang X, Yang H (2016) Selective catalytic reduction of NO by NH3 over CuO-CeO2 in the presence of SO2. Catal Sci Technol 6:1719–1725
Jin R, Liu Y, Wang Y, Cen W, Wu Z, Wang H, Weng X (2014) The role of cerium in the improved SO2 tolerance for NO reduction with NH3 over Mn-Ce/TiO2 catalyst at low temperature. Appl Catal B-Environ 148:582–588
Ma Z, Sheng L, Wang X, Yuan W, Chen S, Xue W, Han G, Zhang Z, Yang H, Lu Y, Wang Y (2019) Oxide catalysts with ultrastrong resistance to SO2 deactivation for removing nitric oxide at low temperature. Adv Mater 31:1903719
Acknowledgements
This work was financially supported by National Natural Science Foundation of China (52102367).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Fang, D., Sheng, F., Hou, S. et al. Effect of surface coating CeO2 on the SO2 resistance of Cu-ZSM-5 with highly selective catalytic reduction activity. Reac Kinet Mech Cat 137, 2215–2230 (2024). https://doi.org/10.1007/s11144-024-02667-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11144-024-02667-1