
Abstract
Catalytic performance of zirconyl hydrogen tellurate (ZrOHTe) and hafnium hydrogen tellurate (HfHTe) was studied for the first time. A process of butyl acetate synthesis was used as a test reaction. A number of physicochemical techniques (XRD, SEM, TGA/DSC and XPS) was applied for catalyst characterization. Using molecular electrostatic potential and net atomic charges, both the electronic structure and the chemical reactivity of ZrOHTe and HfHTe were considered. Kinetic (activation energy, pre-exponential factor) and thermodynamic (enthalpy, entropy, Gibbs free energy barrier) parameters were calculated. A plausible reaction mechanism for esterification was offered. Particles of a low crystallinity degree or small-sized crystallites were found to form ZrOHTe and HfHTe. The fragments in ZrOHTe/HfHTe are prismatic/layered in shape with an average size around 160 μm. XPS exposed that \({\text{HTeO}}_{4}^{-}\) managed the surface phenomena for ZrOHTe and HfHTe. A higher butyl acetate yield using ZrOHTe than HfHTe due to higher surface acidity of the former was generated.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The esterification process is one of the most widely used methods in industrial chemistry for obtaining valuable components for pharmaceutical, perfumery, food and chemical industries [1,2,3,4,5,6,7,8]. For example, to enhance the hydrophilization of ibuprofen, its direct esterification with sorbitol in the presence of porcine pancreas lipase type II catalyst was investigated [2]. In another paper, esterification reaction between sorbic acid and glycerol in a solvent-free system were performed with an immobilized lipase B catalyst [6]. It was noted that glycerol sorbate is a promising antimicrobial compound against Saccharomyces cerevisie. Experimental and kinetic study on isobutyl acetate production catalyzed by n-sulfopropyl-3-methylpyridinium trifluoromethanesulfonate ionic liquid ([HSO3-C1Pyr]+[CF3SO3]–) was reported in a paper of Shi et al. [8]. Although esterification is generally carried out as a reaction between a carboxylic acid and alcohol, processes of alcoholysis and acidolysis for esters production are noticed as well [9, 10].
Esterification is a highly reversible process and to shift the equilibrium in the direction of the reaction products it is realized in the presence of acid homogeneous or heterogeneous catalysts. Among these, strong protic acids are widely applied as homogeneous catalysts due to their low cost, accessibility and high reactivity [11]. However, along with the advantages of the mineral acids, there are also significant technical disadvantages such as: (i) required neutralization and washing from the product mixture, resulting in the formation of waste water and (ii) high catalyst consumption. Indicated drawbacks forced development of heterogeneous catalyst systems that can be easily separated from the reaction mixture and completely regenerated [12]. For instance, a heterogeneous catalyst based on silicotungstic acid has been synthesized for methanol-acetic acid esterification [13]. The highest catalytic activity was obtained as 84% with 20% catalyst at initial methanol-to-acetic acid ratio of 1:3, 80 °C for 27 h. In this regard, –SO3H modified carbon/cellulose composite was studied as an efficient heterogeneous catalyst for oleic acid methylation [14]. The authors reported for ester yields of 84%, 88% and 94% in the presence of 5, 10 and 20 wt% catalyst.
Recently, zirconium- and hafnium-based composites have been reported as promising heterogeneous catalytic systems for esterification [15,16,17,18,19,20,21,22,23,24,25,26,27]. Hence, biodiesel production using 7%Sr/ZrO2 catalyst has been investigated in a paper of Asif and co-workers [15]. As a result, ester yield of 75% at initial molar ratio = 1:14, temperature 70 °C and catalyst loading 1% was obtained. Sulfated zirconium oxide was applied as an efficient catalyst for methylation of lignin and palmitic acid [16, 21]. Condensation of ethyl pyruvate into diethyl 2-methyl-4-oxopent-2-enedioate and diethyl 2-methylene-4-oxopentanedioate (an itaconic acid ester analogue) with selectivity of 80% and 60% conversion were evaluated in the presence of Hf-containing BEA zeolites as catalysts [18].
Catalytic conversion of furfuryl alcohol and levulinic acid into alkyl levulinates using sulfonic acid-functionalized Hf-based MOFs was analyzed by Gupta et al. [19]. Xerogel catalysts based on mesoporous zirconia doped with telluric acid was investigated for acetic acid esterification with benzyl alcohol at alcohol-to-acid ratio of 10:1, and reaction temperature 80 °C [22]. After 4 h of reaction, ester yield of 30% was achieved. Speaking for benzyl alcohol, the latter was used for 4-phenylbutyric acid esterification in the presence of Zr(Hf)Cl4, Zr(Hf)Cl4(THF)2, Zr(Hf)O2, ZrOCl2⋅8H2O, HfOCl2, Zr(OH)4, Cp2HfCl2 salts as catalysts [23]. In this regard, ZrOCl2⋅8H2O was applied as an efficient, cheap and reusable catalyst for the esterification of acrylic acid with equimolar amounts of alcohols [25]. In another paper, heterogeneous acetylation of glycerol in the presence of Zr-modified hierarchical mordenite was represented [27].
Although a variety of papers focused on zirconium- and hafnium-based composites as catalysts for esterification, the development of highly effective, low-cost and stabile heterogeneous catalytic systemts is in progress. This matches well with requirements such as: (i) direct esterification of carboxylic acids with alcohols under solvent-free conditions, (ii) esters should be obtained in high yields using equimolar amounts of reactants, (iii) esterification should proceed at low temperature and (iv) the esters should be easy to isolate and purify.
Based on the literature review, the catalytic behavior of different type of hafnium and zirconium acidic compounds, namely zirconyl hydrogen tellurate (ZrO(HTeO4)2⋅4H2O) and hafnium hydrogen tellurate (Hf(HTeO4)4⋅8H2O) were explored in this paper for the first time. It should be pointed out here that Zr- and Hf-based hydrogen tellurates are noted previously, where reflux and precipitation methods for synthesis were used [28, 29]. However, a completely different preparation procedure for ZrO(HTeO4)2⋅4H2O and Hf(HTeO4)4⋅8H2O was applied in this work—hydrothermal synthesis. Catalytic performance of the samples obtained was studied in the reaction of butyl acetate synthesis.
Textural properties, thermal behavior and surface effects for ZrO(HTeO4)2⋅4H2O and Hf(HTeO4)4⋅8H2O samples were characterized in details via a number of physicochemical techniques such as X-ray diffraction (XRD), scanning electron microscopy (SEM), thermal gravimetric/differential scanning calorimetry (TGA/DSC) and X-ray photoelectron spectroscopy (XPS). To evaluate the electronic structure and chemical reactivity of the title compounds toward esterification, molecular electrostatic potential (MEP) surface and net atomic charges (NPA) were considered. In addition, essential kinetic (rate constant, activation energy, pre-exponential factor) and thermodynamic (enthalpy, entropy, Gibbs free energy barrier) parameters for butyl acetate synthesis were calculated. A plausible reaction mechanism was offered as well.
The results represented in this paper are expected to be valuable for design of new stable and active catalytic systems for acid-catalyzed processes.
Experimental section
Synthesis of ZrO(HTeO4)2⋅4H2O and Hf(HTeO4)4⋅8H2O
Preparation of ZrO(HTeO4)2⋅4H2O (abbreviated as ZrOHTe in this paper) was conducted via hydrothermal synthesis at 250 °C for 5 days in a stainless steel autoclave (50 ml), equipped with a polypropiolactone inner body. For that purpose, equimolar amounts of zirconyl chloride (ZrOCl2⋅8H2O, 0.80 g; 4.49 mmol) and telluric acid (Te(OH)6, 1.03 g; 4.49 mmol) in aqueous medium (15 g; 833.33 mmol) were used. After slow cooling to ambient temperature, a white solid phase was resulted (see supporting material, Fig. S1). The latter was washed with ethyl alcohol and dried at 80 °C overnight.
Synthesis of Hf(HTeO4)4⋅8H2O (denoted as HfHTe in this work) followed the above mentioned conditions, in general. However, 3 g (9.37 mmol) HfCl4, 4.3 g (18.74 mmol) Te(OH)6 and 25 ml (1388.88 mmol) H2O were applied in that case. HfHTe compound appeared as a yellow-colored crystalline phase (see supporting material, Fig. S1).
Samples characterization
XRD analysis was performed by a Bruker D8 Advance diffractometer using Cu Kα (λ = 0.15406 nm) radiation and a PW 2200 Bragg–Brentano θ/2θ goniometer. Diffraction peaks are recorded in the 2θ range from 10° to 90° with step size of 0.030 and step scan of 10.0s. Phase identification was carried out by comparison with database cards.
SEM images were generated by means of SEMoscope IEM11 instrument (10–30 kV, maximum resolution 5 nm), equipped with EDS detector.
TGA/DSC profiles of ZrOHTe or HfHTe were evaluated by means of a thermal gravimetric analyzer Setsys Evolution 2500 SETARAM. The experimental conditions are as follows: heating rate—10 °C/min, temperature range 30–1000 °C, argon flow—0.020 l/min, corundum pans. DSC measurements were carried at the same experimental conditions.
X-ray photoelectron spectroscopy (XPS) measurements were conducted utilizing the ESCALAB MkII electron spectrometer under a vacuum pressure of 5 × 10–9 mbar, employing an Al Kα X-ray source with an energy of 1486.6 eV. The hemispherical analyzer utilized a pass energy of 20 eV and 6 mm slit widths. The instrumental resolution, determined by the full width at half maximum (FWHM) of the Ag3d5/2 photoelectron peak, was 1 eV. The energy scale was adjusted using the Te3d5/2 peak maximum at 577.1 eV to correct for electrostatic charging effects.
The processing of the acquired spectra involved the subtraction of X-ray satellites and a Shirley-type background [30]. Peak positions and areas were assessed through symmetrical Gaussian–Lorentzian curve fitting. The accuracy of the binding energy (BE) values was within ± 0.1 eV. To determine the relative concentrations of different chemical species, the peak areas were normalized to their photoionization cross-sections, as calculated by Scofield [31].
Batch esterification
Butyl acetate synthesis was carried out at different reaction temperatures (60 °C, 70 °C, 80 °C) in the presence of ZrOHTe or HfHTe (0.42 g, 10 wt% with respect to the acetic acid mass), using equimolar amounts of acetic acid (0.0646 mol, 4.2 g) and butyl alcohol (0.0646 mol, 4.8 g). Ester yield was analyzed by at fixed intervals of time (10 min) using GC 7890A apparatus, equipped with FID detector and a HP-INNOWAX capillary column). Helium at a flow rate of 0.0015 l/min was used as a carrier gas. Both, the ster yield and the rate constant of esterification were investigated as a function of the catalyst content (0.21 g–0.50 g, 5 wt%–12 wt% with respect to CH3COOH mass) in this work as well.
Kinetic and thermodynamic calculations
The studied reaction in this paper is interaction between equimolar amounts of reactants and Eq. 1 was used to interpret the experimental kinetic data.
Here, \({\text{C}}_{\text{i}}\) (\(i{ } \equiv {\text{ A, Y, B, Z}}\)) corresponds to the acetic acid concentration (mol/l) (A), butanol (Y), butyl acetate (B) and water (Z) at given reaction time (t, min). The forward and backward constants were abbreviated as \({\text{k}}_{1}\) and \({\text{k}}_{2}\), respectively.
The equilibrium substrate conversion (\({\text{X}}_{{\text{A}}_{\text{e}}}\)) was determined as follows:
Here KC denotes the equilibrium constant. When the equilibrium constant at different temperatures was estimated, enthalpy (\(\Delta {\text{H}}_{r}^{^\circ }\), kJ/mol) and entropy \(\Delta {\text{S}}_{r}^{^\circ }\),J/mol/K) of butyl acetate production were calculated using Eq. 3.
To value the forward rate constant, a nonlinear least-squares method was applied, Eq. (4). SRS abbreviates the minimum of the sum of residual squares between measured and predicted ester yields (x).
The activation energy (\({\text{E}}_{\text{A}}\), kJ/mol) and pre-exponential factor (A, l/mol/min) were determined via the Arrhenius law:
Enthalpy (\(\Delta {\text{H}}^{ \ne }\), kJ/mol) and entropy (\(\Delta {\text{S}}^{ \ne }\), J/mol/K) of activation for the title reaction were calculated as follows:
Here, \({\text{k}}_{\text{B}}\) is Bolzmann constant (1.38 × 10–23 J/K) and h is Planck constant (6.63 × 10–34 Js).
Theoretical section
Computational details
The quantum estimations were completed by means of Gaussian 16 software at B3LYP/6–311 + G(d,p) and LANL2DZ for Hf, Zr and Te [32,33,34]. The calculated frequencies were adjusted by empiric scaling factor of 0.9679 [35]. No fanciful upsides of the wave numbers for the ground states were noticed, which shows that the optimized calculations are situated at the minimum points of the potential energy surfaces. A fanciful recurrence for the change state structure showed that a seat point was found. The programming Gauss View 6 was used for results illustration [36].
Molecular electrostatic potential surface
Electrophilic and nucleophilic reactivitys of ZrOHTe and HfHTe samples was investigated via MEP surface [37]. On the other hand, the optimal conditions for appropriate reactants orientation can be determined by MEP as well. In the field of MEP surface analysis: (i) red color corresponds to areas of high electron density (strong negative potential), (ii) light red-to-yellow color denotes regions of low negative potential, (iii) green-to-white color abbreviates molecular electrostatic potential close to zero, (iv) light blue color expresses low positive potential and (v) dark blue colored areas are for significant positive potential (less evident electron density).
Results and discussion
Phase identification and morphology
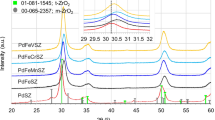
To investigate the composition of ZrOHTe and HfHTe samples, XRD analysis was used (Fig. 1). For comparison, X-ray patterns of the raw materials (telluric acid, zirconyl chloride and hafnium chloride) are also applied. Morphology of ZrOHTe and HfHTe was investigated by means of SEM method (see supporting material, Fig. S2).

XRD profiles for telluric acid (TA), hafnium chloride (HfCl), zirconyl chloride (ZrOCl), HfHTe and ZrOHTe samples
The results exposed that the XRD profile of telluric acid contained distinct peaks at diffraction angles (2θ) of 22.6°, 32.2° and 37.9° (ICSD 15-922-91). On the other hand, the diffractogram of the ZrOCl sample is characterized by intense peaks localized at 2θ = 21.9°, 22.6°, 24.2°, 25.9°, 28.5° and 29.5° (ICSD 00-032-1498). Analysis of the X-ray pattern of HfCl revealed 2θ = 21.2°, 27.3°, 28.6°, 29.2°, 31.2°, 34.6°, 35.2° and 39.5° (ICSD 17-247-62). Broad diffraction peaks positioned at 25.1°, 26.9°, 32.5°, 34.1° and 36.9° were found to be present in the X-ray diffractogram of ZrOHTe sample. Obviously, the latter does not contain peaks related to TA and ZrOCl, indicating the formation of a new phase. This was confirmed by peaks at 25.1°, 34.1° and 36.9° due to hexavalent tellurium (Te6+) in the form of TeO3 phase (ICSD 00-043-1048). On the other hand, patterns at 2θ = 26.9° and 32.5° related to a phase of zirconium tellurate (ZrTe3O8, ICSD 00-015-0692) were noticed as well.
The XRD profile of HfHTe was found to follow that of ZrOHTe, containing broad diffraction peaks at 2θ = 25.1°, 27.3°, 29.2°, 34.6°, 37.1°, 39.5°, 44.6° and 47.1°. Among these, the peaks located at 25.1°, 34.2° and 37.1° are identical to the patterns already established in the diffractogram of ZrOHTe and express a phase of tellurium oxide (TeO3, ICSD 00-043-1048). The peaks at 2θ = 27.3°, 29.2°, 39.5°, 44.6° and 47.1° characterize the hafnium tellurate phase (HfTe3O8, ICSD 01-070-2441). It should be noted that peaks at 27.3°, 29.2°, 34.2° and 44.6° can be found in both the diffractorgams of HfHTe and pure hafnium chloride. In other wods, the presence of a low-crystalline hafnium chloride phase in the composition of HfHTe sample should not be excluded.
A detailed analysis found that peaks assigned to phases of zirconyl hydrogentellurate (ZrO(HTeO4)2⋅4H2O) and hafnium hydrogentellurate (Hf(HTeO4)4⋅8H2O) are not observed in the diffractograms of ZrOHTe and HfHTe. Two possible reasons have been proposed to explain these effects. The first was related to significantly small crystallites of ZrO(HTeO4)2⋅4H2O and Hf(HTeO4)4⋅8H2O, which cannot be registred by XRD method. The second reason implies ZrO(HTeO4)2⋅4H2O and Hf(HTeO4)4⋅8H2O particles of a low crystallinity degree (highly amorphous state). The latter is supported to some extent by the broad diffraction peaks, characterizing ZrOHTe and HfHTe samples. Similar results were observed for amorphous XZrTe2.5H and XZrTe10H [22]. The authors reported for a broad diffraction peak between 20° and 30° which denotes XRD profiles of XZrTe2.5H and XZrTe10H.
To study the morphology of ZrOHTe and HfHTe, scanning electron microscopy was used (Fig. 2). The results revealed that the synthesized hydrogen tellurates are composed of particles with a dense structure. It was found that the fragments in zirconyl hydrogen tellurate structure are prismatic in shape with an average size of 160 μm. Similar to ZrOHTe, an identical average size (157 μm) was found for the particles constituting HfHTe. In contrast to zirconyl hydrogen tellurate, a layered structure denotes the fragments in the HfHTe composition.

TGA (left) and DSC (right) profiles for HfHTe and ZrOHTe samples
Thermal behavior
The thermal properties of ZrOHTe and HfHTe samples were studied in the range from 30 °C to 1000 °C (Fig. 2). To investigate the thermal effects during decomposition, differential scanning calorimetry was used. Detailed thermal decomposition mechanisms for ZrOHTe and HfHTe samples are represented in Table S1 (see supporting material).
The results demonstrated that the thermogram of ZrOHTe contains four main stages of mass loss: 30 °C–190 °C, 200 °C–660 °C, 670 °C–750 °C and 770 °C–960 °C. A detailed analysis found that the second temperature interval can be considered as a set of three additional areas of mass loss, namely 200 °C–360 °C, 365 °C–535 °C and 540 °C–660 °C. The mass loss in the first temperature interval is associated with complete dehydroxylation of the ZrO(HTeO4)2⋅4H2O sample, resulting in the formation of ZrO(HTeO4)2 phase. A broad endothermic peak with a maximum at 100 °C characterized this stage. According to the decomposition mechanism, a good correlation between the experimentally measured mass loss (\({\text{m}}_{\text{exp}}\) = 11.91%) and the calculated one (\({\text{m}}_{\text{calc}}\) = 12.76%) was found. The resulting ZrO(HTeO4)2 phase undergoes additional mass loss (\({\text{m}}_{\text{exp}}\) = 5.99%; \({\text{m}}_{\text{calc}}\) = 5.28%) within 200 °C–360 °C, where release of 0.5 H2O and 0.5O2 generates Zr(TeO3)2. The evolution of oxygen as a decomposition product was the reason for the appearance of an exothermic peak at 360 °C in the heat flow profile of the ZrOHTe sample. The thermal decomposition of ZrOHTe in the temperature range from 360 to 750 °C includes successive stages of oxygen release (\({\text{m}}_{\text{exp}}\) = 15.05%; \({\text{m}}_{\text{calc}}\) = 14.62%) with formation of a phase, containing mixed-valent tellurium oxides—Zr(TeO)(TeO2).
The significantly larger amount of generated oxygen (2O2) in the interval 360 °C–750 °C compared to the oxygen amount (0.5O2) released between 200 °C and 360 °C was marked via appearance of a more pronounced exothermic peak (maximum at 650 °C) in the DSC profile of ZrOHTe. Increasing the temperature above 750 °C produces a non-stoichiometric phase of Zr(Te1.5O2) due to release of TeO2 (\({\text{m}}_{\text{exp}}\) = 20.44%; \({\text{m}}_{\text{calc}}\) = 20.23%). Since the experimentally measured thermal stability of ZrOHTe was conducted in the range within 30 °C–1000 °C, additional calculations for its probable thermal behavior at temperatures above 1000 °C were made. Hence, the phase of Zr(Te1.5O2) can undergo two different stages of thermal decomposition. The first one releases 0.5TeO2 and a phase of ZrTeO was formed. A mass loss amount of 25.36% was calculated in this case. The second stage expressed formation of a new non-stoichiometric phase of ZrTeO0.5 (\({\text{m}}_{\text{calc}}\) = 27.91%) with due to 0.5TeO3 release (Table S1).
The thermogram of HfHTe indicated that the sample is less thermally stable than ZrOHTe at temperatures up to 400 °C, while an opposite trend was observed between 400 and 1000 °C. The analysis discovered that the thermal decomposition of HfHTe involves five distinct mass loss stages: 30 °C–190 °C, 200 °C–535 °C, 540 °C–640 °C, 650 °C–770 °C, and 780 °C–990 °C. Similar to ZrOHTe, the first stage is associated with an endothermic dehydroxylation process (\({\text{m}}_{\text{exp}}\) = 14.34%; \({\text{m}}_{\text{calc}}\) = 13.18%), leading to Hf(HTeO4)4 formation. Comparing the mass loss at this stage for the two samples (ZrOHTe and HfHTe), \({\text{m}}_{\text{exp}}\) for HfHTe (14.34%) is greater than \({\text{m}}_{\text{exp}}\) for ZrOHTe (11.91%). This effect was related to the presence of a larger number of hydroxyl groups in HfHTe compared to the OH– groups in ZrOHTe structure. The thermal behavior of HfHTe in the range from 200 to 535 °C includes the release of additional molecules of water and oxygen, resulting in the formation of Hf(TeO3)4 (\({\text{m}}_{\text{exp}}\) = 6.91%; \({\text{m}}_{\text{calc}}\) = 7.17%). At temperatures up to 770 °C, Hf(TeO3)4 undergoes successive stages of decomposition with release of additional amounts of oxygen (\({\text{m}}_{\text{exp}}\) = 2.74%; \({\text{m}}_{\text{calc}}\) = 2.73%), where Hf (TeO3)4 was transformed into a non-stoichiometric hafnium phase, Hf(TeO3)3(TeO1.5).
The formation of a phase of Hf(TeO3)3(TeO1.5) was confirmed by two distinct exothermic peaks (maxima at 667 °C and 762 °C) in the DSC profile of HfHTe. In the region between 780 and 990 °C, significant mass loss of 15.16% (\({\text{m}}_{\text{calc}}\) = 15.09%) due to release of TeO2 and formation of Hf(TeO)3(Te0.5O3.5) was observed. Similar to Zr(Te1.5O2), several individual stages for thermal degradation of Hf(TeO)3(Te0.5O3.5) at temperatures above 1000 °C were proposed: (i) remove of 1.5TeO3 with \({\text{m}}_{\text{calc}}\) = 36.13% or (ii) release of 0.5TeO at \({\text{m}}_{\text{calc}}\) of 9.85% (Table S1).
In summary, the thermal decomposition of the studied materials (ZrOHTe and HfHTe) starts at temperatures above 90 °C, which means that under the chosen temperature (80 °C) of conducting the catalytic experiment, these are stable systems.
Electronic structure
To know the electron density distribution in ZrOHTe and HfHTe, their electronic structures were studied by means of MEP surface analysis (Fig. 3) and net atomic charges (NPA, Table 1).

Molecular electrostatic potential surfaces for ZrOHTe (left) and HfHTe (right): O-atoms are in red, Te-atoms are in yellow, Zr(Hf)-atoms are in light-blue and H-atoms are in white
NPA analysis exposed that O6 atom (– 1.148) localizes the highest negative charge in ZrOHTe, followed by O5 (– 1.128), O14 (– 1.113), O12 (– 1.092), O4 (– 1.087), O7 (– 1.021) and O13 (– 1.015). In addition, a significant negative potential (in the range from – 0.910 to – 0.994) was also registered for the remaining oxygen atoms (O1, O11, O15, O16 and O22) in the zirconyl hydrogentellurate molecule as well. This suggests that O6 is the highest nucleophilic center in ZrOHTe sample and determines its electrophilic reactivity. However, the molecular electrostatic potential surface for ZrOHTe discovered that the O6 atom is located in a light yellow-colored zone with a significantly reduced negative potential. In other words, the negative charge on O6 atom appears to be highly compensated, most probably a consequence of interaction with the positively charged Zr23 (+ 1.744) and Te25 (+ 3.145). A similar trend was also found for the oxygen atoms localized around Zr23 and Te24.
On the other hand, however, the rest of the oxygen atoms (O7, O13 and O14) attached to Te25 are positioned in an yellow-colored area with a clear negative potential, more pronounced in the case of O13 atom. The latter suggests that O13 atom is the site of the highest nucleophilicity in ZrOHTe and defines its reactivity towards electrophilic reactions. At first sight, this contradicts the registered pure atomic charges, since among the atoms O7, O13 and O14, the last one is characterized by the highest negative potential (– 1.113), followed by O7 (– 1.021). Considering the existence of O7–H21 bond in the ZrOH–Te structure, it can be assumed that the negative charge localized on O7 is significantly compensated by the positive charge on H21 (+ 0.524). Based on the latter, the O7 atom should be excluded as a variant defining the electrophilic reactivity of the ZrOHTe sample. The less pronounced negative charge around the O14 atom (placed in an area with a slightly saturated yellow color) can be assigned to an intramolecular electrostatic interaction (O14···H10) with the participation of a coordinated water molecule around the Zr23 atom.
Analyzing the atoms in ZrOHTe with a localized positive charge, the highest one was observed for zirconium and tellurium ones—Te25 (+ 3.145), Te24 (+ 3.139) and Zr23 (+ 1.744). A significantly lower positive potential (from + 0.495 to + 0.524) was observed around the hydrogen atoms in the ZrOHTe structure. Thus, it was assumed that the nucleophilic reactivity of ZrOHTe depends to the greatest extent on tellurium atoms and to a lesser extent on zirconium. However, the molecular electrostatic potential surface showed that Te24 and Zr23 are located in a region with a neutral charge, which means that their positive potential is completely compensated by the negative one on the surrounding oxygen atoms. Moreover, Te25 is present in the structure of the studied compound as an atom located in an are with a light yellow color, i.e. in an area of weak negative potential. As a result, it was found that the nucleophilic reactivity of ZrOHTe is determined by the positive charge on the hydrogen atoms.
A detailed analysis found that the positive charge localized on the hydrogen atoms present in ZrOHTe decreases in the order: H10 (+ 0.527) > H21 (+ 0.524) ≈ H20 (+ 0.523) > H2 (+ 0.520) ≈ H17 (+ 0.519) > H18 (+ 0.513) > H19 (+ 0.507) > H26 (+ 0.495). Thus, it was suggested that the nucleophilic reactivity of ZrOHTe depends on H10, H20 and H21 atoms. However, MEP surface for ZrOHTe displayed that the these hydrogen atoms populate in areas with an electrostatic potential close to zero, i.e. their positive charge is completely compensated by the negative one localized on the oxygen atoms connected to them. On the other hand, Fig. 3 clearly shows that the H26 atom is placed in an area of the lowest electron density (the highest positive potential). To explain the observed effect, the total negative charges of the oxygen atoms around the tellurium ones were taken into account. For example, the total negative potential generated by O7, O13 and O14 atoms is greater than the positive charge around Te25 atom. In other words, a region of weak negative potential characterizes this part of the ZrOHTe molecule. However, analyzing the total negative potential of the oxygen atoms around Te24, practically identical and opposite to the positive charge on the tellurium atom was found. This means that the negative charge on the O11 atom affects the positive potential localized on H26 atom in a low extent. As a result, H26 was defined as the site responsible for the nucleophilic reactivity of the ZrOHTe sample.
Examining the electronic structure of the HfHTe sample, the highest positive charge (blue-colored area) was registered on the hydrogen atoms H33 (+ 0.469) and H44 (+ 0.472) in the water molecules coordinated around Hf35 (Fig. 4). Considering that the difference in electron density of H33 and H44 is practically absent, the mentioned hydrogen atoms were defined as equivalent electrophilic centers determining the nucleophilic reactivity of HfHTe. Comparing the charges for H33 and H44 in HfHTe sample with that of H26 in ZrOHTe sample, a notably higher positive potential (+ 0.495) on H26 in comparison with H33 and H44 was found. On the other hand, the positive charges located on Te19 (+ 3.188), Te20 (+ 3.198), Te22 (+ 3.188) and Te25 (+ 3.215) atoms in HfHTe were established to be more pronounced than the positive charges around Te24 (+ 3.139), Te25 (+ 3.145) in ZrOHTe, i.e. more reduced telluric atoms in HfHTe with respect to those in ZrOHTe are observed. This effect could be explained with the presence of intramolecular hydrogen bonds (O29···H21, O32···H28, O47···H46 and O42···H41) between water molecules and hydrogen tellurate anions in HfHTe. For comparison, such type of interactions is missing for ZrOHTe. The mentioned hydrogen bonds imply a decreased positive potential on the hydrogen atoms H21, H28, H46 and H41. As a result, strong electron density distribution from Te19, Te20, Te22 and Te25 toward O9, O11, O45·and O40 occurred. Based on the latter, a more pronounced positive potential characterizes telluric atoms in HfHTe than ZrOHTe.

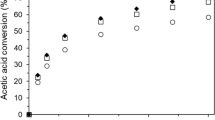
Effect of the catalyst nature on butyl acetate yield (left) and comparison between measured and predicted butyl acetate yield (right). Reaction conditions: initial reactants molar ratio of 1.0, reaction temperature 80 °C and 0.42 g (10 wt%) catalyst. Fit lines are in accordance with Eq. 4
Electron core levels description
It is well known that the surface phenomena play a crucial role in the catalytic performance of a given heterogeneous catalytic system. For that purpose, C1s, O1s, Zr3d, Te3d5/2 and Hf4f electron core levels were described (see supporting material, Figs. S3 and S4). XPS atomic concentrations (atom%) for C1s, O1s, Zr3d, Te3d5/2 and Hf4f are depicted in Table 2. To evaluate the surface effects in the synthesized catalysts more accurately, electron core levels for the raw materials (telluric acid, zirconyl chloride and hafnium chloride) were defined first. Although the samples studied in this paper do not contain carbon as a structural element, adventitious carbon is usually detected during XPS analysis and it should be considered.
Results exhibited that the C1s line of telluric acid (TA) contains two peaks at binding energy (BE) values of 284.3 eV and 288.2 eV. Taking into account that higher BEs (284.8 eV–285.0 eV) are typicall for sp3 bulk bonded carbon (C–C), the fragment at 284.3 eV was related to carbon in the vicinity of a slightly electropositive atom—most probably tellurium [38]. The peak at 288.2 eV was assigned to O–C=O groups due to the presence of Te–O and Te=O bonds in the TA structure [39]. Analysing the C1s spectrum of zirconyl chloride (ZrOCl), peaks located at BE of 285.6 eV and 289.6 eV were registered. While the first reflection was connected with C–O fragment, the peak at 289.6 eV was interpreted as a carbon atom in the form of Cl(O)–C=O groups [40]. Similar to ZrOCl, sp3 bulk bonded carbon in the atmosphere of oxygen was detected in the C1s spectrum of hafnium chloride (HfCl) due to a peak positioned at 285.3 eV. However, a fragment with a BE value of 283.2 eV was observed in HfCl sample as well. It was denoted as carbon attached to the hafnium atom (Hf–C interaction) in HfCl. Hafnium carbide investigated via XPS and discovered a peak with BE of 282.0 eV due to Hf–C bond formation [41]. Obviously, the fragment in this paper possesses notably higher BE (283.2 eV) in comparison with those observed by Rodenbücher et al. [41]. This can be explained by the presence of chlorine in HfCl, which decreases the electron density amount on the carbon atom. Being less shielded, this carbon atom appreared at higher BE value. It was found that the components in C1s spectrum for ZrOHTe sample are very similar to that for bare TA and ZrOCl since peaks at BEs of 284.5 eV (related to C–C in the reductive atmosphere of tellurium) and 285.4 eV (denoted as carbon in the neighborhood of oxygen, C–O) were detected (Fig. S3). However, a peak with notably lower BE (287.8 eV) than these in the C1s lines for TA (288.2 eV) and especially for ZrOCl (289.6 eV) was registered as well. It could be correlated with C=O surface moieties in ZrOHTe [42].
Unlike ZrOHTe, C1s line of HfHTe sample expressed components at 282.1 eV and 285.0 eV (Fig. S4). The first of these completely matched with the BE value (282.0 eV) for hafnium carbide, while the peak at 285.0 eV denotes C–C/C–H fragments in areas with electrostatic potential close to zero. Based on the MEP surface analysis for HfHTe (Fig. 4), regions with electrostatic potential close (white colored) were observed around the hydrogen atoms in hydrogen tellurate anions and coordinated water molecules). Comparing the C1s spectrum of ZrOHTe and HfHTe, it appears that hydrogen tellurate anions (\({\text{HTeO}}_{4}^{-}\)) have an active role in the surface properties of the studied samples. The presence of a different type of oxygen-containing fragments on ZrOHTe surface suggests that the oxygen atoms in \({\text{HTeO}}_{4}^{-}\) and Zr=O are mainly involved. For comparison, influence of the Hf and Te atoms on the surface adsorption capacity of HfHTe was primarily advocated. Based on these, a notably more pronounced surface carbon concentration for ZrOHTe (48.02 atom%) than HfHTe (24.89 atom%) was revealed (Table 2).
An evidence that \({\text{HTeO}}_{4}^{-}\), Zr and Hf structures are involved in the surface behavior of HfHTe and ZrOHTe was gained when Zr3d, Hf4f and Te3d5/2 electron core levels were analyzed. Prior to that, Zr3d, Hf4f and Te3d5/2 lines for ZrOCl, HfCl and TA were analized first. The Zr3d spectrum of ZrOCl is composed of a doublet of peaks (with spin–orbit splitting of 2.4 eV) located at 184.0 eV for Zr3d5/2 and 186.4 eV for Zr3d3/2 features. Based on literature data, BE for Zr3d5/2 was established to be: (i) notably higher than that observed for ZrO2 (182.4 eV) and (ii) identical to that displayed for ZrCl4 (184.0 eV) [22, 43]. Hence the binding energies of 184.0 eV and 186.4 eV in ZrOCl can be assigned to Zr4+ oxidation state due to O=Zr–(Cl)2 bonds. A doublet of peaks (with spin–orbit splitting of 1.7 eV) related to Hf4f7/2 (18.3 eV) and Hf4f5/2 (20.0 eV) was observed in the Hf4f line of HfCl as well. The BEs detected in this work were comparable with those for HfCl4, where characteristic peaks of Hf4+ ions (Hf–Cl interactions) at 20.3 eV (Hf4f5/2) and 18.6 eV (Hf4f7/2) were detected [44]. Except patterns at 18.3 eV and 20.0 eV, Hf4f for HfCl revealed a component of low intensity with BE of 16.8 eV, assigned to Hf–O bonds (probably due to adsorbed water) [45]. Evaluating the Te3d5/2 line of telluric acid, a single peak with BE of 577.1 eV due to Te6+ ions was detected [46].
Results showed that synthesis of ZrOHTe and HfHTe decreses the BE of Zr3d5/2 (from 184.0 eV to 183.2 eV) and Hf4f7/2 (from 18.3 eV to 17.7 eV) with 0.6 eV–0.8 eV. In other words, zirconium and hafnium in ZrOHTe and HfHTe seem more reduced than the same ones in bare materials (ZrOCl and HfCl). The registered BEs of 183.2 eV and 17.7 eV were widely attributed to metal–oxygen interactions, namely Zr–O and Hf–O [47, 48]. Knowing that Zr and Hf possess identical electronegativity, the uniform BE shift to lower values for Zr3d5/2 and Hf4f7/2 proposes a similar electron density amount around Zr and Hf. It agrees well with the MEP surface and NPA analysis where charges of 1.744 and 1.887 were calculated for Zr23 and Hf35, respectively. To clarify the reduced BE for Zr3d5/2 and Hf4f7/2, different reasons have been announced: (i) a diminished number of chemical bonds around Zr and Hf atoms due to surface exposure and/or (ii) interaction between Zr/Hf and hydrogen tellurate anions. Studying the first reason, a more pronounced surface Zr and Hf concentrations for ZrOHTe and HfHTe samples in comparison with those for bare ZrOCl and HfCl was expected. However, surface Zr (7.47 atom%) and Hf (6.81 atom%) concentrations for ZrOHTe and HfHTe are found to be notably lower than the ones for ZrOCl (17.17 atom%) and HfCl (11.33 atom%) (Table 2). Thus, it was stated that formation of chemical interactions in the form of Zr–O–Te and Hf–O–Te bonds were responsible for the reduced BE for Zr3d5/2 and Hf4f7/2. The latter was conformed when Te3d5/2 electron levels for ZrOHTe and HfHTe were investigated. In details, peaks at 577.3 eV (ZrOHTe) and 577.1 eV (HfHTe) were recorded. Obviously, these are identical to the observed one (assigned to Te6+ ions in TeO3 or Te(OH)6) in the Te3d5/2 line of pure TA. Observed phenomena supported XRD data (Fig. 1), where phases of TeO3, ZrTe3O8 and HfTe3O8 have been noted. Except a pattern at 577.1 eV in the Te3d5/2 electron level of HfHTe, a component at lower BE (575.4 eV) was also identified. It indicates that tellurium in a more reduced oxidation state (lower than Te6+) is present in HfHTe. Considering that the BE value of 575.1 eV is between these reported for Te0 (elemental Te, 573.0 eV) and Te4+ (TeO2, 576.1 eV), exact assignment of the low-intensive component in Te3d5/2 line for HfHTe proved to be difficult [22, 46]. Nevertheless, several reasons were proposed: (i) tellurium in a close contact with an electropositive atom (such as hafnium) and/or (ii) suboxidic species of Te present at the surface resulting from a lower coordination number for Te. Possible explanation for the reasons offered above was tried to be given using the intramolecular hydrogen bonds (due to O29···H21, O32···H28, O47···H46 and O42···H41) between water molecules and \({\text{HTeO}}_{4}^{-}\), being discovered in the electronic structure of HfHTe (Fig. 3 and Table 1). It was assumed that these interactions induce additional charge transfers (Te → O) in \({\text{HTeO}}_{4}^{-}\) anions, leading to the formation of low-oxidized tellurium fragments. However, the less evident intensity of the peak at 575.4 eV with respect to that located at 577.1 eV (Te6+ ions) indicates that the additionally encouraged charge transfers in \({\text{HTeO}}_{4}^{-}\) (observed for the HfHTe sample only) are sparsely populated. This is probably due to the electrostatic nature (weak interaction) of the O29···H21, O32···H28, O47···H46 and O42···H41 bonds.
Comparing the surface tellurium concentration for the title compounds, a notably higher value for HfHTe (19.61 atom%) than ZrOHTe (10.22 atom%) was distinguished (Table 2). This was related to the higher number of \({\text{HTeO}}_{4}^{-}\) anions around hafnium atom with respect to these attached to zirconium. In light of this, more prounced surface oxygen concentration for HfHTe (48.69 atom%) in comparison with ZrOHTe (34.29 atom%) was detected as well. Since the surface oxygen concentration for all of the samples is higher than the surface tellurium one, it was assumed that the oxygen atoms in \({\text{HTeO}}_{4}^{-}\) anions are exposed in a greater extent than tellurium atoms. From one side, these observations correlated well with the C1s data for ZrOHTe, where functional groups such as C=O and O–C are present. On the other hand, higher surface oxygen concentration for HfHTe could be related to an impoved number of unaffected oxygen atoms in \({\text{HTeO}}_{4}^{-}\) anions and coordinated water molecules (being revealed by C1s, Zr3d5/2, Hf4f7/2 and Te3d5/2 electron core levels). More noticeable surface oxygen concentration than tellurium might be an indication for perpendicular arrangement of oxygen and tellurium atoms on the surface, where the OH– groups in Te–OH are mainly exposed.
To define the surface oxygen species for ZrOHTe and HfHTe more accurately, O1s core levels were considered. Prior to that, O1s spectra for bare TA, ZrOCl and HfCl were described in details (Figs. S3 and S4). It was found that O1s electron level for TA contains peaks located at 529.3 eV, 531.3 eV and 532.5 eV. The O1s component located on the lowest BE is attributed to stoichiometric oxygen in the oxide main matrix [49]. The peak at medium BE (531.3 eV) expresses surface oxygen like Te–OH interactions in TA, while the fragment positioned at BE of 532.5 eV characterizes weakly adsorbed water [50, 51]. Similar to TA sample, peaks positioned at 531.4 eV and 532.7 eV have been detected in the O1s peak of HfCl. Considering that HfCl sample does not contain structural water: (i) the first component was assirgned to adsorbed hydroxyl groups in the form of Hf–OH bonds and (ii) the pattern at BE of 532.7 eV denotes oxygen in the vicinity of the chlorine atom in HfCl structure. Analysisng the O1s spectrum of ZrOCl, peaks located at BEs of 530.1 eV, 531.7 eV, 533.1 eV and 534.8 eV were registered. While the first fragment abbreviates the O2− ions in ZrOCl due to Zr=O interaction, the second one is assigned to the hydroxyl groups (OH−) of Zr [52, 53]. The pattern with BE of 533.1 eV could be attributed to surface C–O moieties (already revealed as a peak at 285.6 eV in the C1s spectrum of ZrOCl) and/or OH− groups in the environment of the chlorine atom [54]. The peak at the highest BE (534.8 eV) in the O1s spectrum of ZrOCl was related to weakly bonded oxygen-containing species, most probably adsorbed water. The deconvolution of the O1s peak for HfHTe sample exposed components with BEs of 529.6 eV, 531.2 eV and 533.0 eV, which are identical to those for pure telluric acid. However, the peaks at 529.6 eV, 531.2 eV for HfHTe are notably more intensive than the ones for TA due to higher surface concentration of \({\text{HTeO}}_{4}^{-}\) anions, supporting the C1s and Te3d5/2 lines description.
Unlike HfHTe, a component around 529.0 eV was not detected in the O1s spectrum of ZrOHTe. In addition, a lower surface oxygen concentration for ZrOHTe than pure telluric acid and HfHTe was observed. These effects are in accordance with a significantly less noticed surface concentration of \({\text{HTeO}}_{4}^{-}\) for ZrOHTe in comparison with that for HfHTe, already established by means of Te3d5/2 spectra. The identical BE values for the hydroxyl groups in ZrOHTe (531.1 eV) and HfHTe (531.1 eV) corresponded well to the thermal properties for the title compounds (Fig. 3), where complete dexydroxylation in similar temperature regions (up to 200 °C) was detected. In this regard, a higher surface oxygen concentration for HfHTe than ZrOHTe implies more evident mass loss for HfHTe with respect to ZrOHTe. Indeed, values of 14.34% and 11.91% were registered as \({\text{m}}_{\text{exp}}\) in the temperature area up to 200 °C for HfHTe with respect to ZrOHTe. However, more pronounced thermal stability of HfHTe than ZrOHTe at elevated temperatures could be connected with higher surface population of \({\text{HTeO}}_{4}^{-}\) for HfHTe with respect to ZrOHTe, clearly observed from the Te3d5/2 electron levels.
Catalytic performance
The catalytic behavior of the synthesized hydrogen tellurates was studied at an initial mole ratio of reactants of 1.0 and reaction temperature of 80 °C. The results are presented in Fig. 4 and Table 3. In addition, a correlation between the experimentally measured and calculated butyl acetate yields is depicted in Fig. 4 as well.
The results demonstrated that the esterification of acetic acid with 1-butanol in the absence of catalyst (blank reaction) generates a butyl acetate yield of 4.91% after 90 min reaction time. Hence, it was claimed that ester production in the absence of catalyst practically does not occur. However, the addition of 0.42 g (10 wt%) HfHTe and ZrOHTe increased the butyl acetate yield significantly up to 16.51% and 47.44%, respectively. Confirmation for the improved butyl acetate yield in the presence of HfHTe and ZrOHTe with respect to the non-catalyzed reaction was obtained by means of the calculated forward rate constant: 0.53 × 10–4 l.l/mol × min (blank reaction), 4.87 × 10–4 .l/mol × min(HfHTe catalyst) and 14.00 × 10–4 .l/mol × min(ZrOHTe catalyst). A good linear correlation between experimentally measured and calculated butyl acetate yields was found (Fig. 4). The latter means that Eq. 4 can describe adequately the kinetics of acetic acid esterification with 1-butanol.
Taking into account the generally accepted statement that the esterification reaction proceeds through formation of an active carbonyl complex due to transfer of a proton from the catalyst to the carbonyl oxygen atom in the carboxylic acid, the enhanced ester yields in the presence of HfHTe or ZrOHTe in comparison with blank reaction were explained [55]. However, to describe more favored butyl acetate synthesis using ZrOHTe catalyst (47.44%) instead of HfHTe one (16.51%), electron core levels (XPS analysis) in a combination with the electronic structure (MEP surface and NPA charges) for the title compounds were evaluated. In details, C1s, Te3d5/2 and O1s lines showed that \({\text{HTeO}}_{4}^{-}\) anions manage the surface effects in ZrOHTe and HfHTe due to OH– groups population (in the form of Te–OH bonds and coordinated water molecules). Based on the electronic structure, the acidity of H26 atom (expressed as T24–O11–H26 fragment) in ZrOHTe sample was referred as stronger than that for H33 and H44 atoms (denoted as coordinated water molecules around T25–O40–H41 and T20–O11–H28 moieties) in HfHTe. Hence, superior catalytic behavior of ZrOHTe in comparison with HfHTe was related to a more prominent positive potential around H26 atom than H33 and H44 atoms, facilitating formation of a greater number of active carbonyl complexes (protonated carboxylic acid) between the catalyst and substrate.
Since the ZrOHTe samples was defined as a more active catalytic system for butyl acetate production, it was preferred for studying important thermodynamic parameters such as equilibrium constant, enthalpy and entropy. In addition, effect of the reaction temperature and catalyst content on the ester yield and rate constant of esterification were determined as well.
Thermodynamic calculations
The acetic acid esterification with 1-butanol in the presence of a ZrOHTe catalyst was conducted at equimolar amounts of reactants, without continuous product (water and/or ester) release during the reaction progress. The latter shows that the chemical equilibrium has a strong influence on the butyl acetate yield and the rate constant of esterification [56]. Thus, the equilibrium constant (KC) of the process needs to be determined. For this purpose, Eq. 2 was applied.
The results showed that the equilibrium constant value increased with increasing the reaction temperature: 5.239 (60 °C), 6.218 (70 °C) and 7.295 (80 °C). This was an indication that the esterification reaction of acetic acid with 1-butanol is an endothermic process. Knowing the KC values at different temperatures, the enthalpy (\(\Delta {\text{H}}_{r}^{^\circ }\) = 16.23 kJ/mol) and entropy (\(\Delta {\text{S}}_{r}^{^\circ }\) = 62.45 J/mol×K) for butyl acetate synthesis in the presence of ZrOHTe catalyst were calculated using Eq. (3). The latter was graphically represented in Fig. S5 (see supporting material). The positive \(\Delta {\text{H}}_{r}^{^\circ }\) value was an another indication that elevated temperatures favor the butyl acetate synthesis.
A practically identical \(\Delta {\text{H}}_{r}^{^\circ }\) (16.61 kJ/mol) was obtained using the standard heats of formation (Eq. (7)) of acetic acid (– 474.1 kJ/mol), 1-butanol (– 328.4 kJ/mol), butyl acetate (–533.5 kJ/mol) and water (– 285.6 kJ/mol) [57]. When \(\Delta {\text{H}}_{r}^{^\circ }\) and \(\Delta {\text{S}}_{r}^{^\circ }\) values were determined, Gibbs energy (\(\Delta {\text{G}}_{r}^{^\circ }\) = \(\Delta {\text{H}}_{r}^{^\circ }\)−T(\(\Delta {\text{S}}_{r}^{^\circ }\)) at 60 °C, 70 °C and 80 °C was found: – 4.567 kJ/mol, – 5.191 kJ/mol and – 5.816 kJ/mol. The same \(\Delta {\text{G}}_{r}^{^\circ }\) = values have been reached when KC was used (\(\Delta {\text{G}}_{r}^{^\circ }\) = – RTlnKC): –4 .581 kJ/mol (60 °C), – 5.211 kJ/mol (70 °C) and – 5.832 kJ/mol (80 °C).
Obviously, the Gibbs energy decreases with increasing the reaction temperature, which is consistent with: (i) the endothermic nature of the butyl acetate production process established above and (ii) the well-known postulate that the progress of a given reaction requires minimum \(\Delta {\text{G}}_{r}^{^\circ }\) values [58].
Influence of the reaction temperature
The influence of temperature on the butyl acetate production and the rate constant of esterification was investigated at an initial reactants mole ratio of 1.0 and 0.42 g (10 wt%) content of ZrOHTe catalyst. The results are presented in Fig. 5. Long-term testing of the catalyst at different temepratures is represented in Fig. S6.

Effect of the reaction temperature on butyl acetate yield (left) and comparison between measured and predicted butyl acetate yield (right). Reaction conditions: initial reactants molar ratio of 1.0 and 0.42 g (10 wt%) ZrOHTe catalyst. Fit lines are in accordance with Eq. 4
It was established that the acetic acid esterification of with 1-butanol at temperature of 60 °C leads to a butyl acetate amount of 10.83% at a forward rate constant of 2.15 × 10–4 l/mol × min (Table 3). Increasing the reaction temperature up to 70 °C and 80 °C led to significantly higher butyl acetate yields—14.84% and 47.44%, respectively. An identical tendency in the case of the forward rate constants at 70 °C (3.98 × 10–4 l/mol × min) and 80 °C (14.00 × 10–4 l/mol × min) was registered. Similar to the results presented in Fig. 4, a good linear regression between experimentally measured and calculated ester yields was found. The intensified esterification process at elevated reaction temperature was attributed to improved kinetic energy of the reactants, allowing them to pass an energy barrier known as activation energy (EA) [59]. The latter was determined for the forward (\({\text{E}}_{{\text{A}}_{\text{f}}}\)) and backward (\({\text{E}}_{{\text{A}}_{\text{b}}}\)) reactions in the process of butyl acetate synthesis by menas of Eq. 5, presented graphically in Fig. 6. The intercept from the y-axis allows to calculate the pre-exponential factor for the forward (\({\text{A}}_{\text{f}}\)) and backward (\({\text{A}}_{\text{b}}\)) reactions.

Arrhenius plot for finding the activation energy and pre-exponential factor (left) and Eyring plot for determine the entropy and enthalpy of activation (right) for butyl acetate synthesis. Reaction conditions: 0.42 g (10 wt%) ZrOHTe catalyst, reactants molar ratio of 1.0, reaction time 90 min
The results showed that values of 90.92 kJ/mol and 74.67 kJ/mol characterize \({\text{E}}_{{\text{A}}_{\text{f}}}\) and\({\text{E}}_{{\text{A}}_{\text{b}}}\), respectively. In addition, a noticeably higher value for \({\text{A}}_{\text{f}}\) (3536.54 × 107 l/mol × min) compared to that generated for \({\text{A}}_{\text{b}}\) (1.91 × 107 l/mol × min) was obtained. The higher activation energy of the forward reaction with respect to that of the reverse one indicates the need to supply energy (expressed as increased reaction temperature) to shift the equilibrium in the reaction products direction, in accordance with the expression \({\text{E}}_{{{\text{A}}_{{\text{f}}} }} {\text{ = E}}_{{{\text{A}}_{{\text{b}}} }} { + }\Delta {\text{H}}_{r}^{^\circ }\) The latter shows \(\Delta {\text{H}}_{r}^{^\circ }\) of 16.25 kJ/mol, i.e.which practically identical to those determined by Eq. 3 (16.23 kJ/mol) and Eq. 7 (16.61 kJ/mol). Baring in mind that the pre-exponential factor denotes the number of effective impacts (impacts leading to the formation of a reaction product) between reacting molecules, \({\text{A}}_{f} { } \gg {\text{A}}_{b}\) corresponds to the calculated equilibrium constant values and the positive temperature dependence for the butyl acetate production.
Analyzing the long-term catalyst activity in the reaction of acetic acid esterification with butanol (Fig. S6), a continuous increase in the ester yield is seen without decreasing the of the catalyst performance. In details, butyl acetate yields of 69.57% (60 °C), 71.38% (70 °C) and 72.92% (80 °C) were registered after 210 min time of reaction. After that no changes in the ester yields were observed. The latter means that the chemical equilibrium (expressed by \({\text{X}}_{{\text{A}}_{\text{e}}}\)) has been reached (Table 3).
It was mentioned above that butyl acetate synthesis proceeds via formation of an active complex between the carbonyl oxygen in acetic acid and an acidic hydrogen atom in the ZrOHTe catalyst. Based on the latter, enthalpy and entropy of activation for the forward (\(\Delta {\text{H}}_{f}^{ \ne }\) and \(\Delta {\text{S}}_{f}^{ \ne }\)) and backward (\(\Delta {\text{H}}_{{\text{b}}}^{ \ne }\) and \(\Delta {\text{S}}_{{\text{b}}}^{ \ne }\)) reactions were calculated, Eq. 6. Thus, values of 88.07 kJ/mol \(\Delta {\text{H}}_{f}^{ \ne }\) and –52.44 J/mol×K (\(\Delta {\text{S}}_{f}^{ \ne }\))were found. As expected, significantly lower \(\Delta {\text{H}}_{{\text{b}}}^{ \ne }\) (71.83 kJ/mol) and \(\Delta {\text{S}}_{{\text{b}}}^{ \ne }\) (–114.89 J/ mol × K) were determined for the backward reaction.
A detailed analysis discovered that the differences \(\Delta {\text{H}}_{f}^{ \ne }\)–\(\Delta {\text{H}}_{{\text{b}}}^{ \ne }\) and \(\Delta {\text{S}}_{f}^{ \ne }\)–\(\Delta {\text{S}}_{{\text{b}}}^{ \ne }\) have values of 16.24 kJ/mol and 62.45 J/mol×K. These completely coincide with the above noted enthalpy and entropy for the process of butyl acetate synthesis. Knowing \(\Delta {\text{H}}_{f}^{ \ne }\), \(\Delta {\text{H}}_{{\text{b}}}^{ \ne }\), \(\Delta {\text{S}}_{f}^{ \ne }\) and \(\Delta {\text{S}}_{{\text{b}}}^{ \ne }\), Gibbs energy values for the forward (\(\Delta {\text{G}}_{f}^{ \ne }\) = \(\Delta {\text{H}}_{f}^{ \ne }\)–T \(\Delta {\text{S}}_{f}^{ \ne }\)) and backward (\(\Delta {\text{G}}_{f}^{ \ne }\) = \(\Delta {\text{H}}_{{\text{b}}}^{ \ne }\)–T \(\Delta {\text{S}}_{{\text{b}}}^{ \ne }\)) reactions were calculated for 60 °C, 70 °C and 80 °C. Hence, \(\Delta {\text{G}}_{f}^{ \ne }\) was determined as 105.53 kJ/mol (60 °C), 106.06 kJ/mol (70 °C) and 106.58 kJ/mol (80 °C). On the other hand, higher values were registered in the case of \(\Delta {\text{G}}_{b}^{ \ne }\), namely 10.12 kJ/mol (60 °C), 111.27 kJ/mol (70 °C) and 112.42 kJ/mol (80 °C). The positively valued \(\Delta {\text{G}}_{f}^{ \ne }\) and \(\Delta {\text{G}}_{b}^{ \ne }\) are reasonable, since energy for cleavage of existing chemical bonds in the reacting molecules is required. On the other hand, \(\Delta {\text{G}}_{f}^{ \ne }\) > \(\Delta {\text{G}}_{b}^{ \ne }\) means that the forward reaction (ester generation) is energetically more favored than the opposite one, i.e. it proceeds predominantly at a higher extent. The latter completely coincides with the statement that the reaction of butyl acetate synthesis is an endothermic process.
Influence of the catalyst loading
It was already mentioned that an essential step in the mechanism of the esterification process is the formation of an active complex due to the interaction between the catalyst and the carboxylic acid. At the beginning of the process, when the concentration of the substrate is significant, a large number of active complexes are formed and the reaction proceeds at a high speed. However, during the reaction progresses the carboxylic acid amount diminishes and the catalyst plays a key role to maintain a sufficiently high reaction rate (high ester yields).
The effect of the catalyst (ZrOHTe) content on the of butyl acetate yield and the rate constant of esterification was investigated at an initial reactants molar ratio of 1.0 and 80 °C. The results are presented in Fig. 7. Taking into account that butyl acetate synthesis in the absence of a catalyst practically does not occur (Fig. 4, Table 3), the non-catalytic reaction will not be considered here. The results showed that the introduction of small amounts 0.21 g (5 wt%) of ZrOHTe catalyst affected the esterification process significantly, expressed by more than two times higher butyl acetate yield (from 4.91% to 13.82%). The latter was also confirmed by the calculated forward rate constants: 0.53 × 10–4 l/mol × min (blank reaction) and 2.84 × 10–4 l/mol × min (0.21 g ZrOHTe catalyst). Increasing the ZrOHTe amount up to 0.29 g (7 wt%) practically did not change the ester yield (15.43%) and rate constant of esterification (3.22 × 10–4 l/mol × min).

Effect of the catalyst loading on butyl acetate yield (left) and comparison between measured and predicted butyl acetate yield (right). Reaction conditions: initial reactants molar ratio of 1.0, reaction temperature 80 °C and ZrOHTe catalyst. Fit lines are in accordance with Eq. 4
However, usage of 0.42 g (10 wt%) catalyst, led to a noticeable difference in the rate of esterification of acetic acid with 1-butanol, expressed as butyl acetate yield of 47.44% and forward rate constant value equal to 14.00 × 10–4 l/mol × min. This was attributed to the formation of a significantly higher number of active complexes in the presence of 0.42 g catalyst compared to those formed in the process involving 0.21 wt% and 0.29 g ZrOHTe. Similar to the effects observed for the butyl acetate synthesis in the presence of 0.21 wt% and 0.29 g ZrOHTe, the ester yield (48.61%) and the rate constant of esterification (15.03 × 10–4 l/mol × min) remained practically unchanged after introduction of 0.50 g (12 wt%) catalyst. Hence, 0.42 g of ZrOHTe was claimed as an optimal catalyst amount required for maximum butyl acetate yield under the selected reaction conditions. A good linear correlation between measured and predicted butyl acetate yields was illustrated on Fig. 7.
Reaction mechanism
The mechanism of butyl acetate production represented in the present work (Scheme 1) was entirely based on both the surface phenomena (investigated via XPS method) and the electronic structure (expressed by surface molecular electrostatic potential and net atomic charges) for ZrOHTe catalyst. Briefly, XPS revealed a high surface concentration of hydrogen tellurate anions, which generated an exposed acidic hydrogen atom (H26). The latter was recognized as the most electrophilic site in ZrOHTe, determining its reactivity towards nucleophilic attacks. On the other hand, the carbonyl atom in the acetic acid structure is the center with the highest electron density, responsible for its reactivity toward electrophilic addition. Considering that the coordinated water molecules are not directly involved in the reaction mechanism, they are not included in the catalyst structure presented in Scheme 1. Based on it, the carbonyl oxygen is protonated by the acidic hydrogen atom in ZrOHTe, activating it toward a nucleophilic attack from the ethanol (step I). The alcohol executes a nucleophilic attack on the carbonyl. A lone pair of electrons from the oxygen atom of the alcohol forms a bond with the carbonyl carbon, breaking its pi bond with the other oxygen. The π-bond electrons move up to the oxygen and neutralize its positive charge. This results in an oxonium ion (step II). A proton transfer occurs from the oxonium ion to the OH– group, giving rise to an activated complex (step III). This can be divided into two further steps where the alcohol first deprotonates the oxonium ion, giving a tetrahedral intermediate after which the hydroxyl group accepts the proton from the alcohol. Then, the 1,2 elimination of water occurs, giving the protonated ester. A lone pair of oxygen forms a π-bond with the carbon, thereby expelling the water (step IV). The remaining positively charged oxygen is deprotonated, giving the required ester as a product and initial catalyst form (step V).

Plausible reaction mechanism for butyl acetate synthesis in the presence of ZrOHTe
Conclusions
Zirconyl hydrogen tellurate (ZrOHTeO4)2⋅4H2O, ZrOHTe) and hafnium hydrogen tellurate (Hf(HTeO4)4⋅8H2O, HfHTe) were explored as acidic catalytic systems (butyl acetate synthesis was used as a test reaction) for the first time. It was found that ZrOHTe and HfHTe are amorphous in structure with particle size around 160 μm. TGA/DSC measurements showed that HfHTe is less thermally stable than ZrOHTe at temperatures up to 400 °C, while an opposite trend was observed between 400 °C and 1000 °C. A highly acidic hydrogen atom (H26) in the form of \({\text{HTeO}}_{4}^{-}\) fragments dictated the nucleophilic reactivity of ZrOHTe sample. For comparison, hydrogen atoms (H33 and H44) with a lower positive potential were establiched as the most electrophilic sites in HfHTe. These results were in accordance with XPS analysis, where \({\text{HTeO}}_{4}^{-}\) were recognized as the most populated surface moieties. Higher butyl acetate yield generated in the presence of ZrOHTe than HfHTe was directly related to the more evident H26 acidity in comparison with that of H33 and H44 atoms. Formation of an intramolecural hydrogen bond ([Te–O–H26]cat···[O=C]acet) between ZrOHTe catalyst ([Te–O–H26]cat fragment) and carbonyl oxygen in acetic acid ([O=C]acet moiety) was crucial for the mechanism of butyl acetate synthesis. Values of 16.24 kJ/mol, 88.07 kJ/mol, 90.92 kJ/mol, 3.537 × 1010 l/mol × min, 62.45 J/mol × K, – 52.44 J/mol × K, – 4.657 kJ/mol and 106.58 kJ/mol were calculated as \(\Delta {\text{H}}_{r}^{^\circ }\), \(\Delta {\text{H}}^{ \ne }\), EA, A, \(\Delta {\text{S}}_{r}^{^\circ }\), \(\Delta {\text{S}}^{ \ne }\), \(\Delta {\text{G}}_{r}^{^\circ }\) and \(\Delta {\text{G}}^{ \ne }\) .
Data availability
The datasets generated and analyzed during the current study are available from the corresponding author.
References
Kar S, Sanderson H, Roy K, Benfenati E, Leszczynski J (2022) Chem Rev 122(3):3637–3710
Zappaterra F, Rodriguez MEM, Summa D, Semeraro B, Costa S, Tamburini E (2021) Int J Mol Sci 22(6):3066
Lee J-W, Trinh CT (2020) Curr Opin Biotechnol 61:168–180
Khan Z, Javed F, Shamair Z, Hafeez A, Fazal T, Aslam A, Zimmerman WB, Rehman F (2021) J Ind Eng Chem 103:80–101
Ragavan KV, Hernandez-Hernandez O, Martinez MM, Gutiérrez TJ (2022) Trends Food Sci Technol 119:45–56
Zappaterra F, Summa D, Semeraro B, Buzzi R, Trapella C, Ladero M, Costa S, Tamburini E (2020) Fermentation 6(4):96
Shan J, Wang Q, Hao H, Guo H (2023) Ind Eng Chem Res 62(42):7135–17147
Shi M, Huang K, He R, Jiang Y, Zou Y, Xu J, Tong Z (2023) Chinese J Chem Eng 63:21–30
Mukdsi MCA, Maillard M-B, Medina RB, Thierry A (2018) LWT 89:38–43
Kárpáti L, Szarka G, Hartman M, Vargha V (2018) Period Polytech Chem Eng 62(3):336–344
Marchetti JM, Errazu AF (2008) Biomass Bioenerg 32(9):892–895
Sirsam R, Hansora D, Usmani GA (2016) J Inst Eng India Ser E97:167–181
Şimşek V, Şahin S (2019) J Porous Mater 26:1657–1665
Santos EM, de Carvalho Teixeira AP, da Silva FG, Cibaka TE, Araújo MH, Oliveira WXC, Medeiros F, Brasil AN, de Oliveira LS, Lago RM (2015) Fuel 150:408–414
Asif M, Javed F, Younas M, Gillani MA, Zimmerman WB, Rehman F (2024) Fuel 358:130125
Wu Z, Zhang J, Pan Q, Li X, Zhang Y, Wang F (2018) RSC Adv 8:12344
Kaur K, Jain P, Sobti A, Toor AP (2016) Green Process Synth 5:93–100
Wang Y, Lewis JD, Román-Leshkov Y (2016) ACS Catal 6:2739–2744
Gupta SSR, Kantam ML (2019) Catal Commun 124:62–66
Oohashi Y, Fukumoto K, Mukaiyama T (2005) Chem Lett 34(2):190–191
Wang S, Pu J, Wu J, Liu H, Xu H, Li X, Wang H (2020) ACS Omega 5:30139–30147
Ksila W, Younes MK, Ghorbel A, Rives A (2021) J Sol-Gel Sci Technol 99:376–390
Ishihara K, Nakayama M, Ohara S, Yamamoto H (2002) Tetrahedron 58:8179–8188
Popova M, Lazarova H, Szegedi A, Mihályi MR, Rangus M, Likozar B, Dasireddy VDBC (2017) J Serb Chem Soc 82:1–12
Sun H-B, Hua R, Yin Y (2006) Molecules 11(4):263–271
Popova M, Shestakova P, Lazarova H, Dimitrov M, Kovacheva D, Szegedi A, Mali G, Dasireddy VDBC, Likozar B, Wilde N, Gläser R (2018) Appl Catal A: Gen 560:119–131
Popova M, Lazarova H, Kalvachev Y, Todorova T, Szegedi A, Shestakova P, Mali G, Dasireddy VDBC, Likozar B (2017) Catal Commun 100:10–14
Rahman MK, Huq AMS (1970) J Chromatog A 53(2):613–616
Shakshooki SK, El-Akari FA, El-Hamroni SM, Idris RH, El-Aouzi AM (2016) J Pharm Appl Chem 2(3):22–27
Végh J (2006) J Electron Spectrosc Relat Phenom 151(3):159–164
Scofield JH (1976) J Electron Spectrosc Relat Phenom 8(2):129–137
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Petersson GA, Nakatsuji H, Li X, Carica-to M, Marenich AV, Bloino J, Janesko BG, Gomperts R, Mennucci B, Hratchian HP, Ortiz JV, Izmaylov AF, Sonnenberg JL, Williams-Young D, Ding F, Lipparini F, Egidi F, Goings J, Peng B, Petrone A, Henderson T, Ranasinghe D, Zakrzewski VG, Gao J, Rega N, Zheng G, Liang W, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Throssell K, Montgomery JA Jr, Peralta JE, Ogliaro F, Bearpark MJ, Heyd JJ, Brothers EN, Kudin KN, Staroverov VN, Keith TA, Kobayashi R, Normand J, Raghavachari K, Rendell AP, Burant JC, Iyengar SS, Tomasi J, Cossi M, Millam JM, Klene M, Adamo C, Cammi R, Ochterski JW, Martin RL, Morokuma K, Farkas O, Foresman JB, Fox DJ (2016) Gaussian 16, Revision A.03. Gaussian Inc., Wallingford
Becke AD (1993) J Chem Phys 98:5648–5652
Hay PJ, Wadt WR (1985) J Chem Phys 82:270–283
Andersson MP, Uvdal P (2005) J Phys Chem A 109:2937–2941
Gauss View, Version 6, R. Dennington, Keith TA, Millam JM (2016) Semichem Inc., Shawnee Mis-sion, KS
Hasanzade Z, Raissi H (2018) J Mol Liq 255:269–278
Dolgov A, Lopaev D, Lee CJ, Zoethout E, Medvedev V, Yakushev O, Bijkerk F (2015) Appl Surf Sci 353:708–713
Sadri R, Hosseini M, Kazi SN, Bagheri S, Zubir N, Solangi KH, Zaharinie T, Badarudin A (2017) J Coll Interf Sci 504:115–123
Foelske-Schmitz A, Weingarth D, Kötz R (2011) Surf Sci 605:1979–1985
Rodenbücher C, Hildebrandt E, Szot K, Sharath SU, Kurian J, Komissinskiy P, Breuer U, Waser R, Alff L (2016) Appl Phys Lett 108(25):252903
Kim M-H, Yun S, Park HS, Han JT, Kim K-B, Roh KC (2015) J Mater Chem A3:2564–2567
Roy PC, Jeong HS, Doh WH, Kim CM (2013) Bull Korean Chem Soc 34(4):1221
Ismail M, Zhao Y, Yu XB, Dou SX (2012) EEST Part A: Energ Sci Res Special Issue 107–122.
Gaobo X, Qiuxia X (2009) J Semicond 30(2):1–5
Chen X, Yang Q, Chu B, An H (2015) Cheng Y RSC Adv 5:91295–91301
Hong S, Park SP, Kim Y-G, Kang BH, Na JW, Kim HJ (2017) Sci Rep 7:16265
Jayaram V, Reddy KPJ (2016) Adv Mater Lett 7(11):100–150
Iqbal MZ, Carleschi E, Doyle BP, Kriek RJ (2019) ACS Appl Energy Mater 2:8125–8137
Chen S, Luo Y, Liang J, Pei Y (2022) J Electron Mater 51:6297–6304
Kriek RJ, Iqbal MZ, Doyle BP, Carleschi E (2019) ACS Appl Energy Mater 2:4205–4214
Robledo-Peralta A, García-Quiñonez LV, Rodríguez-Beltrán RI, Reynoso-Cuevas L (2022) Polymers 14:1575
Teeparthi SR, Awin EW, Kumar R (2018) Sci Report 8:5541
Dey SK, Wang CG, Tang D, Kim MJ, Carpenter RW, Werkhoven C, Shero E (2003) J Appl Phys 93:4144–4157
Zhou D, Chen X, Wei X, Tang L, Liang J, Wang S, Wang L (2021) Ind Crops Prod 166:113486
Skoda-Földes R (2014) Molecules 19(7):8840–8844
Menikpura SNM, Basnayake BFA (2009) Renew Energ 34(6):1587–1594
Chen LQ (2019) MRS Bull 44:520–523
Radhakrishnan R, Thiripuranthagan S, Devarajan A, Kumaravel S, Erusappan E, Kannan K (2017) Appl Catal A Gen 545:33–43
Acknowledgements
1. Research equipment of the Distributed Research Infrastructure INFRAMAT, part of the Bulgarian National Roadmap for Research Infrastructures, supported by the Bulgarian Ministry of Education and Science was used in this investigation. 2. Scanning electron microscopy measurements have been conducted through the Project BG05M2OP001-1.002-0019: „Clean technologies for sustainable environment—water, waste, energy for circular economy“ (Clean & Circle) funded by the Operational programme “Science and Education for Smart Growth” 2014-2020, and co-financed by the EU.
Author information
Authors and Affiliations
Contributions
Ivaylo Tankov—Manuscript writing, results interpretation, Georgi Rusev—Formal analysis, Velyana Georgieva—TGA measurements, Hristo Kolev—XPS analysis, Rumyana Yankova—theoretical calculations, Svetlana Genieva—formal analysis.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
This declaration is “not applicable”.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Tankov, I., Rusev, G., Yankova, R. et al. Zirconyl and hafnium hydrogen tellurates as catalysts for esterification. Reac Kinet Mech Cat 137, 2105–2131 (2024). https://doi.org/10.1007/s11144-024-02655-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11144-024-02655-5