
Abstract
In this paper, the conversion of oxygen atoms in Fischer–Tropsch synthesis of low carbon olefin (FTO) catalyzed by Co2C was investigated. Density functional theory (DFT) was used to investigate the surface reaction mechanism. The mechanism of H2O and CO2 formation was studied. Then the rate control steps for the generation of both were found. Research shows the key step in the formation of water is the hydrogenation of O to OH with an activation energy of 1.18 eV. The formation of COOH is the key step to the formation of CO2 with an activation energy of 1.78 eV. It is clear that the formation of H2O is kinetically dominant over that of CO2. The formation frequency of water and CO2 at different temperatures was investigated using the kinetic Monte Carlo (KMC) method, where the conversion frequency of H2O is about 1.6 times higher than that of CO2. The article indicates that O atoms are more readily converted to H2O than CO2 in the FTO process occurring over the Co2C catalyst.
Graphical abstract

Similar content being viewed by others

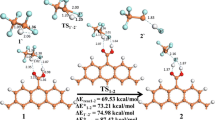
Avoid common mistakes on your manuscript.
Introduction
Fischer–Tropsch synthesis of low carbon olefins (FTO) refers to a green synthetic route for the direct synthesis of olefins from syngas via biomass [1,2,3,4]. The distribution of conventional FTO products follows the Anderson-Schulz-Flory (ASF) distribution (Fig. 1) [5], with a maximum selectivity of about 57 wt% for C2-4 olefins. Sun et al. [6] reported that Co2C catalysts have a tangential effect and can effectively promote the formation of low carbon olefins compared to conventional iron-based catalysts [7, 8]. Especially the activation energy barrier and total reaction energy of ethylene generation at specific exposed crystal planes (020) and (101) of Co2C are low and can effectively inhibit the formation of methane. At a reaction pressure of 523 K and 1 bar, the selectivity of the product hydrocarbons obtained from low carbon olefins was 60.8%, while the selectivity of CH4 was suppressed to 5%.

FTO product distribution. \(W_{n}\) is the molar fraction of \(C_{n}\), \(\alpha\) is the probability of chain growth and \(n\) is the number of carbon atoms [5]
Researchers have studied the mechanism of Fischer–Tropsch synthesis on Co2C to generate methane, methanol and ethylene [9], and the activation mechanism of CO [10], but it is worth mentioning that the current DFT studies on Fischer–Tropsch synthesis on Co2C to low-carbon olefins are limited to ethylene, and there are few studies on propylene, butene, and by-products including CO2, but it cannot be ignored that although in the previous studies C2-4 low carbon olefins selectivity reached 32.0%, but the selectivity of CO2 was still as high as 47.3%.
CO2 and another oxygenated byproduct, H2O, both of which are important destinations for O species in the FTO reaction and are often considered to remove O species from the system. When the FTO reaction feed ratio is a typical H2:CO = 2:1 [6], the molar fraction of oxygen atoms is 16.7% and the mass fraction is 50%, which is half of the reactant feed mass. For the target product low carbon olefins, oxygen-containing by-products are undoubtedly not needed, but the form in which O species leave the system is still a key issue. For C and H atoms, the competition for oxygen atoms is a competitive relationship, and when the selectivity of CO2 is as high as 30–50%, it accordingly means that 30–50% of oxygen leaves the system in the form of CO2, and this will disguisedly reduce olefin selectivity; On the contrary, if more oxygen leaves the system in the form of H2O, it can significantly reduce CO2 emissions and significantly improve the efficiency of carbon atom utilization, while the source of H element is wide, and the raw hydrogen can come from wind power, solar power, hydroelectric power, etc. [11].
Therefore, the article aims to investigate the mechanism of surface generation of water and carbon dioxide clearly and provide basic information to find the method of regulating FTO products in the future. The competition between CO2 and H2O for oxygen on the exposed crystalline surface of Co2C(020) has been systematically and comprehensively investigated using density flooding theory calculations, and the preferential generation of H2O species has been elucidated from the molecular scale. The kinetic Monte Carlo method (KMC) was further used to investigate the species coverage and the conversion frequency of the catalyst surface reaction.
Computational models and methods
Density functional theory
Based on the characterization in the available literature and the related simulation results, Co2C (020) is the exposed crystalline surface of nanoprismatic cobalt carbide catalysts with high activity in FTO reactions [6]. Therefore, we developed a surface model of the Co2C (020) dense stack structure as shown in Fig. 2. A 4-layer p (2 × 3) supercell with a 15-vacuum layer in the z-direction, two layers (two Co layers and the corresponding C) at the bottom of fixation, and two layers at the top of relaxation is used to periodically model the Co2C(020) planar model. The adsorption sites are as follows: hollow, top, and bridge.

Top view of Co2C(020) catalyst and surface adsorption sites
This chapter investigates the mechanism of H2O and CO2 generation on Co2C (020) based on first-principles calculations of DFT, done using the VASP (Vienna ab initio Simulation Package) software package for ab initio quantum mechanics-molecular dynamics calculations developed by Hafner's group in Austria [12, 13]. The PAW method was used to describe the interactions between valence electrons and ions [14]. Since the system contains Co atoms with magnetic properties, the spin-polarization effect was considered. The Monkhorst–Pack grid was used to sample within the first Brillouin zone, setting k points to 2-2-1. The GGA-PBE generalization was used for structure optimization. The truncation energy was set to 400 eV, the energy convergence to 10−5 eV, and the force convergence to 0.05 eV/Å, according to the convergence test and related literature. The flat plate model (slab) was used to calculate the adsorption energy of the species involved in the FTO reaction (Eads, Eq. 1), determine the stable adsorption configuration, calculate the heat of reaction of the primitive reaction (ΔE, Eq. 2), and use the NEB method [15] to search for the lowest energy reaction path, the Dimer method [16] to optimize the transition state structure, the convergence criterion is reached when the transition state structural force constant is less than 0.05 eV/Å, then the activation energy barrier (Ea, Eqs. 2, 3) of the primitive reaction is calculated and the transition state frequency is verified; The transition state is correct when there is only one imaginary frequency in the transition state frequency calculation and the vibration is in the direction of the final state structure.
Here Eadsorbate/slab, Eadsorbate and Eslab denote the total energy when adsorbed on the surface, the energy of the adsorbed species and the energy of the empty surface.
Here EFS is the final state energy and EIS is the initial state energy. ΔE > 0, the reaction absorbs heat, and ΔE < 0, the reaction exerts heat.
The zero-point-energy (ZPE) correction of the total energy was obtained by:
Here \(h\) is the Planck constant, \(v_{i}\) is the vibrational frequency. In the following, the activation energies are all results after the zero-point energy correction without any special remarks. The specific frequency information used for the zero-point energy correction is given in Table S3.
Kinetics Monte Carlo
Monte Carlo method (MC) is a traditional computer simulation tool whose basic idea is based on the probabilistic model of physical phenomena and random sampling experiments based on the law of variation of random numbers to be sought, where the percentage of occurrence of an event will converge to the probability of occurrence of the event under a large number of experiments [17]. The kinetic Monte Carlo method (KMC), which combines the Monte Carlo method and the molecular dynamics method (MD), is to some extent a coarsening of MD, coarsening the focus from atoms to systems, coarsening the atomic trajectories to leaps in the system grouping, and correspondingly rising the time scale span to a scale of seconds [18].
The KMC method has an important role in studies on Fischer–Tropsch synthesis, Tian et al. [19] studied the effect of reaction conditions on the Fischer–Tropsch synthesis reaction of iron-based catalysts. The reaction conditions directly change the site distribution of H*, CO* and vacancies, which have an effect on the macroscopic properties of iron catalysts. Lin et al. [20] studied the Fischer–Tropsch synthesis on Pd(111) at different temperatures. The product generation was shown to be methanol only at 500 K. When the temperature was increased to 1000 K, the product had methanol, methane and water, but the main product was still methanol. In addition, this method has played an important role in the studies of carbon deposition by Fischer–Tropsch synthesis reaction on Co(0001) surface [21], CO dissociation on Fe(100) and Mn-modified Fe(100) surfaces [22, 23], low-carbon olefins by Fischer–Tropsch synthesis on CoFe(100) surface [24], and the role of MnO in CO activation and O removal on Co(0001) surface [25].
The Kinetix program is used for the KMC simulations, and the lattice gas-model is the most commonly used model to describe the surface reaction system. Based on the kinetic parameters provided by the DFT calculations, the system was calculated for a total of six temperatures from 423 to 673 K. The O reaction paths on the Co2C catalyst were further discussed in depth.
DFT study of oxygenated products
Activated dissociation of reactants
The FTO reaction is complex and the product distribution is complicated, including a series of reactions to produce alkanes, olefins and alcohols and aldehydes, water–gas conversion reaction, carbon monoxide disproportionation reaction and methanation reaction. The specific reaction equations are listed below.
Primary reaction for the formation of alkanes:
The main reaction to produce olefins:
Reactions to produce oxygenated compounds, alcohols and aldehydes:
Water–gas shift reaction(WGS):
Carbon monoxide disproportionation reaction (Boudouard):
Methanation reaction:
The FTO reaction needs to be activated by the adsorption activation of CO and H2 species, and their presence and form of action on the surface of the catalyst Co2C(020) are studied in order to reveal the reaction mechanism of the step. The mechanism of H2 adsorption activation in the feedstock is relatively simple. The H2 molecule is automatically dissociated at the bridge site after geometrical optimization to generate two H atoms, which constitute a stable adsorption state, and thus it is judged that the activated adsorption of H2 is actually a spontaneous dissociation adsorption process, and the H2 molecule can quickly and spontaneously dissociate into the adsorbed H state when it is close to the catalyst surface, and the H is preferentially adsorbed on the vacancy site with an adsorption energy of − 3.07 eV. Therefore The adsorbed H is considered to be the main form of H2 on Co2C(020), and the above process can be expressed by the reaction equation H2(g) + 2* → H* + H*.
CO activation dissociation
CO dissociation pathways are direct dissociation, H-assisted dissociation via CHO, and H-assisted dissociation via COH [26]. CO preferentially adsorbs at the vacant site with an adsorption energy of − 2.23 eV. Compared to H, the adsorption energy of CO is lower, so CO is more easily dissociated by activation. The dissociation of CO leads to O, which has a higher adsorption energy of − 6.77 eV and preferentially adsorbs at the vacant site. The strong adsorption property of O makes it difficult to convert and is one of the reasons for the selectivity of the target olefins. CHO and COH preferentially adsorb at the bridge and vacant sites, with activation energies of − 4.46 eV and − 4.96 eV, and their conversion usually encounters some difficulties due to the high adsorption energy of COH. The strong adsorption of COH on the catalyst surface may therefore also hinder the Fischer–Tropsch reaction. CH preferentially adsorbs at the vacant site with an adsorption energy of − 7.04 eV and can be stably adsorbed on the catalyst surface. OH has an adsorption energy of − 4.24 eV and preferentially adsorbs at the vacant site.
Figure 3 integrates the energy barrier curves for different CO activation energies, for CO direct dissociation. The direct dissociation of CO has only one step of the radical reaction. As seen in Fig. 4, in the initial state, the C atom of CO is located in the vacant position, the C–O bond length is gradually stretched and the CO direct dissociation reaction is activated. In the transition state, the C atom remains in the vacant position, while the O atom moves to the top position. The C and O atoms in the final state are each adsorbed in the vacant position. The activation energy barrier of this radical reaction is 2.30 eV and the heat absorption is 0.54 eV, which shows that the direct dissociation of CO is difficult to occur on Co2C(020).

Potential energy profiles for CO dissociation. Calculation Software: Vienna ab-initio Simulation Package (VASP), Models: Co2C(020) 4-layer p (2 × 3) supercell, Interaction: PAW, K points: 2-2-1, Structure optimization: GGA-PBE, Truncation energy: 400 eV, Energy convergence: 10–5 eV, Force convergence: 0.05 eV/Å
CO dissociation via CHO. The H-assisted dissociation path of CO via CHO consists of two radical reactions, hydrogenation on C of the adsorbed state CO to form CHO and then breaking the C-O bond to form CH and O. As shown in Fig. 4, in the reaction CO* + H* → CHO* + *, the initial state CO and H are adsorbed on the vacancy site, after which H moves toward the C atom and CO is deflected to the bridge site to form the transition state. The final state CHO adsorbs at the bridge site. The activation energy base of this radical reaction is 0.69 eV and the heat uptake is 0.46 eV, which is significantly lower and easier to occur compared to the direct dissociation of CO in CHO* + * → CH* + O*. The initial state CHO adsorbs at the bridge site, followed by the continued elongation of the C-O bond and the passage of the O atom through the top site to form the transition state. The final state CH and O are adsorbed in the vacancy site. The activation energy barrier of this radical reaction is 1.42 eV with an exotherm of 1.09 eV. More information is available in Table S1.
CO dissociation via COH. As shown in Fig. 4, the reaction mechanism of this pathway is to first hydrogenate the O of the adsorbed state CO to form a COH intermediate, and then the C-O bond is broken to form C and OH. CO* + H* → COH* + * in which the initial state CO and H are adsorbed on the vacant site, after which H moves through the bridge site toward the O atom and CO is deflected toward the top site to form the transition state. The final state COH adsorbs to the vacancy. The activation energy base of this radical reaction is 1.49 eV with a heat uptake of 0.60 eV. This reaction has a relatively high activation energy and occurs with difficulty. COH* + * → C* + OH* reaction The initial state COH adsorbs at the vacancy site, followed by continued elongation of the C-O bond and OH passing through the top site to form the transition state. The final states C and OH are adsorbed in vacancies. The activation energy barrier of this radical reaction is 1.34 eV with an exotherm of 0.82 eV.
Generation of H2O
H2O is generated from the dissociated O and H species via the OHx (x = 0–1) stepwise hydrogenation mechanism and the OH disproportionation mechanism. The stepwise hydrogenation mechanism consists of a total of two steps of radical reactions. In the O* + H* → OH* + * reaction, O, H and OH are adsorbed on the vacancy site. The activation energy base of this reaction is 1.18 eV and the heat of reaction is 0.39 eV. In the OH* + H* → H2O* + * reaction, the initial state OH and H are adsorbed at the vacant site. H and OH move to the top site simultaneously and form a transition state when H moves to the bridge site. The final state H2O adsorbs at the top site. The activation energy base of the reaction is 1.30 eV and the heat uptake is 1.18 eV.
The OH disproportionation mechanism has two steps of radical reaction, the first step of hydrogenation has been discussed, OH* + OH* → H2O* + O* reaction in the initial state of the two OH adsorbed in the vacant position, after the O–H bond of OH-1 breaks, H moves to OH-2 and forms a transition state in the bridge position, the final state O and H2O adsorbed in the vacant and top positions. The activation energy base of the reaction is 0.64 eV and the heat uptake is 0.44 eV.
Generation of CO2
The generation of CO2 in the FTO process is usually considered as a water gas change reaction, CO + H2O → CO2 + H2, unlike the aforementioned H2 generation, there is a CO direct oxidation mechanism and a carboxylation mechanism as shown in Fig. 5 [26, 27].

Geometries of elementary reactions and reaction path involved in CO activation dissociation unit on Co2C(020). Structural optimization with Material studio, NEB method to search for the lowest energy reaction path, Dimer method to optimize the transition state structure
The CO direct oxidation mechanism consists of a one-step radical reaction, i.e., CO* + O* → CO2* + *. The conformation of this reaction is shown in supplementary information. The initial state CO and O are adsorbed at the vacancy site after which O moves toward CO and forms a transition state at the bridge site. Finally, CO2 is adsorbed at the bridge site. The activation energy base of this reaction is 1.58 eV with a heat uptake of 1.35 eV.
The carboxyl pathway contains two steps of radical reactions, in CO* + OH* → COOH* + *, the initial state CO and OH are adsorbed at the vacancy site and both move towards each other and form a transition state at the bridge site. The final state COOH is adsorbed in the vacant site. In the second step COOH* + * → CO2* + H*, the initial state COOH adsorbs at the vacant position, and then the O–H bond breaks and H moves to the bridge position to form the transition state. Eventually, CO2 and H are adsorbed in the bridge and vacant positions. The activation energy base of this reaction is 0.24 eV with an exotherm of 1.05 eV.
In addition, the forward activation and reverse activation energy barriers of CO hydrogen-assisted dissociation over Fe catalysts are listed in Table 1. In agreement with the literature, the H-assisted dissociation of CO via CHO is the dominant dissociation path on both Fe (110) and Co (0001) surfaces, and on the other hand, the OH disproportionation path is also more dominant for water generation [28].
Discussion of DFT results
The potential energy curves of different CO activation dissociation paths on the surface of Co2C(020) were plotted on the basis of the kinetic parameters of the designed primitive reactions with different CO activation dissociation paths, as shown in Fig. 4.
It is clear that the activation energy barrier for direct CO dissociation is the highest, while the activation energy barriers for the primitive reactions in the two H-assisted dissociation paths are relatively low. Among them, the activation energy barrier of CHO decomposition is higher than that of the CHO generation reaction, which is the rapid control step of the CHO-assisted dissociation path; the generation of COH in the COH path is the rapid control step.
In the potential energy curve, the difference between the highest value of transition state energy in each path and the energy at the beginning of the path is called the effective energy barrier, which can be used to determine the ease of occurrence of different paths. The effective energy barriers of the three paths are 2.30 eV for direct dissociation, 1.88 eV for H-assisted dissociation via CHO, and 2.04 eV for H-assisted dissociation via COH, so the H-assisted dissociation via CHO is the preferred activation dissociation path for CO.
In addition, the CO2 generation path potential energy curves were plotted in Fig. 5 on the basis of kinetic parameters for different CO2 generation path primitive reactions.

Potential energy profiles for CO2 formation. Calculation Software: Vienna ab-initio Simulation Package (VASP), Models: Co2C(020) 4-layer p (2 × 3) supercell, Interaction: PAW, K points: 2-2-1, Structure optimization: GGA-PBE, Truncation energy: 400 eV, Energy convergence: 10–5 eV, Force convergence: 0.05 eV/Å
Among the two H2O generation mechanisms, the effective energy barrier of the reaction path of the stepwise hydrogenation mechanism is 1.91 eV and that of the disproportionation mechanism is 1.18 eV, and it is obvious that the OH disproportionation mechanism is the more occupied mechanism in H2O generation. Further comparison, both of these two mechanisms are two-step reactions, the first step reaction O* + OH* → OH* is the same, the second step reaction, OHx (x = 0–1) stepwise hydrogenation mechanism second step reaction OH* + H* → H2O* activation energy barrier is 1.30 eV, OH disproportionation mechanism second part reaction OH* + OH* → H2O* + O* activation energy barrier is 0.64 eV, again The disproportionation mechanism also prevails. In addition, among the reaction paths corresponding to the disproportionation mechanism, O* + OH* → OH* has the highest activation energy and is the rate control step of the preferential H2O generation path.
The effective energy barrier of the direct CO oxidation mechanism is 2.45 eV, and that of the carboxyl mechanism is 2.38 eV, which is not much different between them. Considering the potential energy curve and the effective energy barrier together, it can be seen that among the different CO2 generation pathways on Co2C(020), the carboxyl mechanism is more advantageous, and the rate control step is CO* + OH* → COOH* + * with an activation energy barrier of 1.78 eV.
KMC study of oxygenated products
KMC model
Based on the DFT study in the previous section for the reaction paths of oxygen atoms during Fischer–Tropsch synthesis, the primitive processes and the corresponding forward activation energy barriers Ea,f, reverse activation energy barriers Ea,r, forward pointing front factors υ0,f and reverse pointing front factors υ0,r are listed in Table S2, and the reaction paths are shown in Fig. 6.

Generation pathways of H2O and CO2
The rate constant K for the surface reaction is calculated using the Arrhenius formula:
Here \(v_{0}\) is the finger front factor, \(E_{a} \) is the activation energy barrier, \(R\) is the ideal gas constant, and \(T\) is the temperature (K). The finger front factor is calculated using the following equation:
Here \(v_{i}^{IS}\) is the vibrational frequency of the reaction initial state, \(v_{i}^{TS}\) is the vibrational frequency of the reaction transition state (excluding imaginary frequencies, and n is the atomic number of the reactants (or products) [29,30,31].
Discussion of KMC results
The reaction network of KMC simulation was constructed, including adsorption and desorption of reactants, product desorption, surface forward and reverse reactions and surface diffusion, which involved the main species CO, H, O and OH, and focused on the diffusion effect of these four species.
In KMC simulations, the degree of activity of each primitive step in the reaction system is judged by the number of times it occurs in the KMC simulation results, which can also be called the reaction frequency.
When there are mutually reversible pairs of primitive reactions in the reaction system, the difference in their reaction frequencies can be regarded as the degree of activity of the reaction paths in which the pair of primitive reactions are carried out in the forward or reverse direction, which is called the net conversion frequency. The simulation results were analyzed using steady-state data, and the simulation time was long enough to reach a steady state, mainly for the species coverage and the net conversion frequency of the primitive processes.
The surface coverage of the four species CO, H, O and OH was analyzed and the results are shown in Table 2, where the variation of the surface coverage of CO and OH species was observed in focus and the results are shown in Figs. 7 and 8.

Surface coverage of the reacting species on the catalyst surface at different temperatures. Calculation software: material studio, modules: kinetix, configuration size: 128 \(\times\) 128, lattice type: hexagonal, symmetry: p 6 m m, lattice dimensions: 4.370 × 4.370, lattice angle: 60.0, simulation time: 0.01 s, simulation temperature: a 423 K b 473 K c 523 K d 573 K e 623 K f 673 K

Left: variation of surface coverage of CO and OH with temperature; right: transient states on simulation adsorption sites of Co2C
CO as the feed to the reaction has a high coverage on the surface at the beginning of the reaction, and the dissociation of CO is the first step of the Fischer–Tropsch reaction, so it can be seen in Fig. 8 that the surface coverage of CO is as high as 28%, while the coverage of O and OH species generated after the dissociation of CO is much lower. For the two speed-controlled steps OH + OH → H2O + O and CO + OH → COOH in the generation pathways of H2O and CO2, the reaction rates are affected by the low coverage of OH species, and the apparent activation energy of the H2O-generating primitive reaction is known to be lower than that of the CO2-generating primitive reaction in the DFT analysis. Also as listed in Table 3, conversion frequency of H2O is on average 1.62 times higher than CO2.
The conversion frequencies of the reactions related to the formation of H2O and CO2 at different reaction temperatures are given in Table 3, from which the Fig. 9 is plotted. It can be clearly seen that the conversion frequencies of the systems all increase with increasing temperature, reaction rate increases with increasing temperature. There is a strong similarity in the formation frequencies of water and carbon dioxide with increasing temperature form Table 3. The rate of increase of water production with temperature is slightly higher than that of carbon dioxide (in the temperature interval from 524 to 624 K). It can be seen from Fig. 9 that for both pathways of CO activation dissociation, the H-assisted dissociation pathway via COH is the most active at 573 K, and this result is consistent with the results of DFT simulations; in the reactions related to H2O generation, O* + H* → OH* + * conversion frequency is larger and OH species are easily generated, while the two conversion pathways of OH* are significantly different, and the OH disproportionation mechanism corresponding to OH* + OH* → H2O* + O* reaction is more active than OH* + H* → H2O* + *.

Net conversion frequency(per active site·per second) of each elementary reaction at 573 K, represents the number of times the primitive reaction occurs
Therefore, H2O generation is more inclined to OH disproportionation. In the CO2 generation reaction, the net conversion frequency value corresponding to the carboxyl mechanism is close to that of the CO direct oxidation mechanism, so both mechanisms generate CO2 in the reaction system studied here, with the carboxyl mechanism slightly dominating.
Conclusion
In this paper, density functional theory (DFT) and kinetic Monte Carlo KMC method were used to study the conversion path of oxygen atoms in the FTO process over Co2C catalyst. Conversion frequency was employed to evaluate the formation path of the two major oxygen-containing products H2O and CO2. The main conclusions are as follows:
-
(1)
The H-assisted path of CO through CHO is the dominant path, among three studied paths of CO in vivo dissociation on the surface of Co2C (020) with an effective energy barrier of 1.88 eV.
-
(2)
The disproportionation mechanism of OH in the H2O formation pathway is dominant, the rate-controlling step is O* + H* → OH* + * with an effective energy barrier of 1.18 eV; The carboxyl mechanism is more dominant in the formation of CO2, the rate control step is CO* + OH* → COOH* + * with an effective energy barrier of 1.78 eV. In contrast the formation of H2O is easier than that of CO2.
-
(3)
CO species is the most adsorbed species on the surface, with a surface coverage of up to 28% at 573 K according to KMC results.
-
(4)
The formation frequency of H2O is higher than that of CO2, and the formation frequency increases with the increase of temperature. Also the growth rate of water production is slightly higher than that of CO2.
-
(5)
Based on calculated results of whole pathway of the O atoms FTO from the feedstock CO to the products H2O and CO2, conclusions can be drawl that the H2O generation reaction gain advantages in competition of the scramble for O atoms.
Data availability
Materials described in the manuscript includes all relevant raw data and are available to any researcher who read this manuscript.
References
Amghizar I, Vandewalle LA, Van Geem KM, Marin GB (2017) New trends in olefin production. Engineering 3(2):171–178. https://doi.org/10.1016/J.ENG.2017.02.006
Tong XG, Zhang GY, Wang ZM, Wen ZX, Tian ZJ, Wang HJ, Ma F, Wu YP (2018) Distribution and potential of global oil and gas resources. Pet Explor Dev 45(4):779–789. https://doi.org/10.1016/S1876-3804(18)30081-8
Fischer FATH (1926) The synthesis of petroleum at atmospheric pressures from gasification products of coal. Brennstoff-Chemie 7:97–104
Yu F, Lin TJ, An YL, Gong K, Wang XX, Sun YH, Zhong LS (2022) Recent advances in Co2C-based nanocatalysts for direct production of olefins from syngas conversion. Chem Commun 58(70):9712–9727. https://doi.org/10.1039/d2cc03048a
Torres Galvis HM, de Jong KP (2013) Catalysts for production of lower olefins from synthesisgas: a review. ACS Catal 9(3):2130–2149
Zhong LS, Yu F, An YL, Zhao YH, Sun YH, Li ZJ, Lin TJ, Lin YJ, Qi XZ, Dai YY, Gu L, Hu JS, Jin SF, Shen Q, Wang H (2016) Cobalt carbide nanoprisms for direct production of lower olefins from syngas. Nature 538(7623):84. https://doi.org/10.1038/nature19786
Li ZJ, Yu DM, Yang LY, Cen J, Xiao K, Yao N, Li XN (2021) Formation mechanism of the Co2C nanoprisms studied with the CoCe system in the Fischer-Tropsch to Olefin reaction. ACS Catal 11(5):2746–2753. https://doi.org/10.1021/acscatal.0c04504
Zhai P, Li YW, Wang M, Liu JJ, Cao Z, Zhang J, Xu Y, Liu XW, Li YW, Zhu QJ, Xiao DQ, Wen XD, Ma D (2021) Development of direct conversion of syngas to unsaturated hydrocarbons based on Fischer–Tropsch route. Chem 7(11):3027–3051. https://doi.org/10.1016/j.chempr.2021.08.019
Zaffran J, Yang B (2021) Theoretical insights into the formation mechanism of methane, ethylene and methanol in Fischer-Tropsch synthesis at Co2C surfaces. ChemCatChem 13(11):2674–2682. https://doi.org/10.1002/cctc.202100216
Chen PPLJXL (2019) Carbon monoxide activation on cobalt carbide for Fischer-Tropsch synthesis from first-principles theory. ACS Catal 9(9):8093–8103
Sahrin NT, Khoo KS, Lim JW, Shamsuddin R, Ardo FM, Rawindran H, Hassan M, Kiatkittipong W, Abdelfattah EA, Da Oh W, Cheng CK (2022) Current perspectives, future challenges and key technologies of biohydrogen production for building a carbon-neutral future: A review. Bioresour Technol. https://doi.org/10.1016/j.biortech.2022.128088
Kresse GHJ (1993) Ab initio molecular dynamics for open-shell transition metals. Phys Rev B 17(48):13115
Kresse GFJ (1996) Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys Rev B 16(54):11169
Maintz S, Deringer VL, Tchougreeff AL, Dronskowski R (2013) Analytic projection from plane-wave and PAW wavefunctions and application to chemical-bonding analysis in solids. J Comput Chem 34(29):2557–2567. https://doi.org/10.1002/jcc.23424
Henkelman G, Jonsson H (2000) Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points. J Chem Phys 113(22):9978–9985. https://doi.org/10.1063/1.1323224
Henkelman G, Jonsson H (1999) A dimer method for finding saddle points on high dimensional potential surfaces using only first derivatives. J Chem Phys 111(15):7010–7022. https://doi.org/10.1063/1.480097
Metropolis NUS (1949) The monte carlo method. J Am Stat Assoc 247(44):335–341
Voter AF (2007) Introduction to the kinetic monte carlo method. In: Sickafus KE, Kotomin EA, Uberuaga BP (eds) Radiation effects in solids. Springer Netherlands, Dordrecht, pp 1–23
Tian L, Huo CF, Cao DB, Yang Y, Xu J, Wu BS, Xiang HW, Xu YY, Li YW (2010) Effects of reaction conditions on iron-catalyzed Fischer-Tropsch synthesis: A kinetic Monte Carlo study. J Mol Struct-Theochem 941(1–3):30–35. https://doi.org/10.1016/j.theochem.2009.10.032
Lin S, Ma JY, Ye XX, Xie DQ, Guo H (2013) CO Hydrogenation on Pd(111): competition between Fischer-Tropsch and oxygenate synthesis pathways. J Phys Chem C 117(28):14667–14676. https://doi.org/10.1021/jp404509v
Jansen A, Agrawal R, Spanu L (2016) Thermodynamics and kinetics of carbon deposits on cobalt: a combined density functional theory and kinetic Monte Carlo study. Phys Chem Chem Phys 18(41):28515–28523. https://doi.org/10.1039/c6cp04719j
Amaya-Roncancio S, Arroyo-Gomez JJ, Linares DH, Sapag K (2020) Direct versus hydrogen-assisted dissociation of CO on iron surfaces: Kinetic Monte Carlo and microkinetic modeling. J Mol Struct. https://doi.org/10.1016/j.molstruc.2019.127188
Huang HY, Yu YZ, Zhang MH (2020) CO dissociation mechanism on Mn-Doped Fe(100) Surface: a computational investigation. Catal Lett 150(6):1618–1627. https://doi.org/10.1007/s10562-019-03066-1
Yu YZ, Zhang J, Lei H, Zhang MH (2020) Carbon chain growth reaction of synthesis of lower olefins from syngas on Fe–Co catalyst. Appl Surf Sci. https://doi.org/10.1016/j.apsusc.2019.144211
Zhang MH, Chi SC, Huang HY, Yu YZ (2021) Mechanism insight into MnO for CO activation and O removal processes on Co(0001) surface: A DFT and kMC study. Appl Surf Sci. https://doi.org/10.1016/j.apsusc.2021.150854
Zhang MH, Yu HP, Yu YZ, Wang LT (2022) Key roles of formyl insertion mechanism and C-O scission of oxygenates on cobalt carbide in syngas Conversion: a detailed reaction network analysis. J Catal 413:455–466. https://doi.org/10.1016/j.jcat.2022.07.005
Zhang MH, Yu HP, Sun YZ, Yu YZ, Chen YF, Wang LT (2022) Theoretical and experimental insights into CO2 formation on Co2C catalysts in syngas conversion to value-added chemicals. Appl Surf Sci. https://doi.org/10.1016/j.apsusc.2022.154379
Manuel O, Rahul N, Anand UN, Akio I (2010) CO activation pathways and the mechanism of Fischer–Tropsch synthesis. J Catal 272(2):287–297
Henkelman G (2017) Atomistic simulations of activated processes in materials. Ann Rev Mater Res. https://doi.org/10.1146/annurev-matsci-071312-121616
Karim W, Spreafico C, Kleibert A, Gobrecht J, VandeVondele J, Ekinci Y, van Bokhoven JA (2017) Catalyst support effects on hydrogen spillover. Nature 541(7635):68. https://doi.org/10.1038/nature20782
Huai L-y STWH (2016) NO reduction by H2 on the Rh(111) and Rh(221) Surfaces: a mechanistic and kinetic study. J Phys Chem C 10(120):5410–5419
Acknowledgements
This research was supported by the National Natural Science Foundation of China (Grant No. 21978199)
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Geng, Z., Xiao, C., Gao, J. et al. Theoretical research of the main conversion path of oxygen atom on Co2C catalysts in the Fischer–Tropsch synthesis process. Reac Kinet Mech Cat 136, 1915–1932 (2023). https://doi.org/10.1007/s11144-023-02436-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11144-023-02436-6