
Abstract
Nowadays, the issue of CO2 conversion becomes an urgent necessity for human civilization, which any ignorant will be caused irreparable damage to the future of human life. Thus, all researchers in the CO2 utilization field have been employed their facilities to overcome the CO2 issue. In the past decade, computational chemical modeling was mainly used to describe the observed results of a carried out reaction. Nowadays, regarding the rapid evolution of computational software and hardware, computational chemical sciences have been converted to powerful tools in the description of the obtained results, studying the mechanism of a reaction and designing the novel structures. The use of computational techniques in the investigation of CO2 transformation to value-added chemicals affords brilliant results that attract remarkable attention among scientists. Among various theoretical techniques, density functional theory (DFT) modeling is a powerful and efficient approach to explore the mechanisms of CO2 conversion and investigate novel catalysts for more efficient CO2 transformation. DFT-based approaches are valuable strategies to overcome the disadvantages of trial-and-error experimental processes such as tedious and time-/labor-consuming repetitions. In this chapter, a comprehensive discussion by the DFT calculations is represented about the investigation of the mechanism of the catalytic CO2 transformation into value-added materials such as CO, CH4, CH3OH, HCOOH, and heterocyclic compounds in the presence of heterogeneous, homogeneous, organo-based, and photo- and electro-catalysts. Moreover, the DFT-based design of novel catalytic systems, challenges, and opportunities in CO2 transformations is outlined.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
1 Carbon Dioxide Properties
CO2 is a colorless, odorless, and non-flammable gas that is heavier than air. Carbon dioxide is odorless gas at low concentrations, but at high concentrations, it has a potent acidic smell. Also, it is an inert material that does not react with many substances. Thus, its storage, liquefaction, solidification, and handling are very easy and safe. Table 1 shows the molecular properties of CO2 (Topham et al. 2000; Nakamura et al. 2015).
As a simple triatomic molecule, the carbon atom of CO2 is covalently double bonded to two oxygen atoms which consist of linear geometry, having short and equivalent C–O bonds (1.1602(8) Å) (Gershikov and Spiridonov 1983). This fashion of bonds causes a nonpolar molecule. The observed behaviors of CO2 in the solid, liquid, and gas phases are the outcomes of a molecular quadrupole that is due to the shape of molecule and electron distribution. Figure 1a illustrates Lewis structure of CO2, based upon valence shell electron pair repulsion theory (VSEPR), and its electrostatic potential (Fig. 1b), depicted on the electrostatic potential map (0.002 a.u.) at the PBE1PBE/aug-cc-pVTZ level of theory (Murphy et al. 2015). It can be concluded that the oxygen and carbon atoms are the centers of electrophilic and nucleophilic attacks, respectively. In other words, the carbon and oxygen atoms can behave as Lewis acid and base, respectively.

Lewis structure (a) and electrostatic potential map (0.002 a.u.) (b) of CO2 molecule (Murphy et al. 2015)
The molecular orbitals (MOs), which are the outcome of the molecular orbital calculations, are helpful tools to afford the basic information about the studied molecule (Nakamura et al. 2015). Figure 2 depicts the molecular orbital diagram and frontier orbitals (high occupied molecular orbital (HOMO), low unoccupied molecular orbital (LUMO)) of CO2. The illustrated MOs in this figure are not experimentally detectable as a physical object. However, they are used frequently in organic and organometallic chemistry as an efficient concept to justify and evaluate the molecule properties. From the orbital viewpoint, the MOs are considered as the most figurative images to explanation and characterization of the molecular behavior in a typical reaction (Nakamura et al. 2015; Luther Iii 2004).

Molecular orbital diagram and frontier orbitals (HOMO and LUMO) of CO2 (Luther Iii 2004)
It is notable that, both HOMO and LUMO energies are degenerate. Thus, the CO2 molecule has four degenerated electrons with similar energy levels, which can transfer in a typical reaction. Energy calculation reveals that the transformation of one of the four electrons diminishes the degeneracy, immediately. Therefore, the transformation of four electrons to or from the outside of CO2 does not take place, simultaneously. Moreover, CO2 possesses two degenerate orbitals including four unoccupied sites that give electron acceptation character for this molecule. An organic molecule having such properties is rare. Indeed, CO2 is neither a complex contains transition metal, nor an electron-rich solid specie. The degenerated frontier orbitals of CO2 are slightly similar to the topological characters of the d orbitals (two HOMOs) and f orbitals (two LUMOs) in transition metals and lanthanoids, respectively (Nakamura et al. 2015).
2 CO2 Transformation as an Undeniable Necessity
Carbon dioxide is an important component of the earth matrix, found in the core, crust, and also the atmosphere, considerably. According to the carbon cycle, soluble carbon dioxide in water can react with other components, be solidified in carbonated stones, and freely emitted into the atmosphere (Hofmann et al. 2009). For centuries, the exchange cycle had established CO2 equilibrium until the Industrial Revolution. However, due to global industrialization in the past years, the consumption of fossil fuels has intensified, which has been resulted in an irregular increase in the concentration of greenhouse gases in the atmosphere. Respiration procedure by living creatures on the earth and the sea causes CO2 emission into the atmosphere (Rafiee et al. 2018). Similar to this respiration, the decay process of the corpse of buried organisms by oxygen releases CO2 molecules. The produced CO2 by respiration or decomposition processes is absorbed by plants and transforms into carbohydrates, via the photosynthesis process, gradually. The consumed water and CO2 by algae, plants, and cyanobacteria via the photosynthesis process yield carbohydrates and oxygen as the main product and by-product, respectively. Both of these products are essential for other living creatures. By starting the Industrial Revolution, excessive fossil fuel extraction by human beings has disrupted the CO2 equilibrium in the environment. This extraction causes two forms of CO2 emission including released pure CO2 gas to the atmosphere, due to the extraction procedure, and the by-product of the combustion process (Artz et al. 2018).
Until the Svante Arrhenius report, there was no anxiety about the irregular emission of CO2. However, in the 1880s, he was the first investigator that anticipated global warming due to the libration of CO2 from the combustion of fossil fuels (Maslin 2008), which received numerous criticisms, firstly. However, after an investigation of variations of climate temperature by Guy Stewart Callendar, Arrhenius's theory was attracted numerous attention (Rafiee et al. 2018).
The outcomes of irregular fossil fuel consumption are increasing the global temperature beyond the normal value (Saxena et al. 2014), socking the coastlines due to the increment of water level in the sea, and occurring unpredictable floods that affect the global economics and ecosystem, catastrophically (Mukherjee et al. 2019). Six main greenhouse gases, namely carbon dioxide (CO2), methane (CH4), nitrous oxide (N2O), hydrofluorocarbons (HFCs), perfluorocarbons (PFCs), and sulfur hexafluoride (SF6) are specified by Kyoto Protocol as the agents which remarkably affect the environment quality (Zhang and Da 2015).
Among the greenhouse gases, CO2 is the most important gas due to the most contribution to global warming (Abeydeera et al. 2019). Because of the combustion of fossil fuel, among the global industries, about 60% of CO2 production belong to industries, such as cement production, iron and steel factories, manufacturing petrochemical materials, gas refiner mills, generators of electrical power, and transportation sector (Yaumi et al. 2017). For example, the contribution of electricity manufacture, agriculture, and forestry is evaluated by about 25% and 24% of the total CO2 emission, respectively (Yaumi et al. 2017). However, regarding inexpensive and abundant sources of coal, it is applied in thermal power plants, abundantly. Thus, the emitted CO2 is evaluated up to 2249 lbs/MWh (Spigarelli and Kawatra 2013). Figure 3 depicts the produced CO2 from different energy sections (Mukherjee et al. 2019).

Produced CO2 from different energy sections (Mukherjee et al. 2019)
In 2014, the International Panel on Climate Change (IPCC) declared that the released greenhouse gas via human activities is the main reason for global warming and the related climate change. Because of industrialization and climate variation, caused by human activities including emission of CO2, nitrous oxide (N2O), methane (CH4), and water vapor (H2O) into the atmosphere, human life is put in danger (Lee and Park 2015). Investigations show that 76% of the overall volume of greenhouse gases is CO2, as the consequence of the combustion of fossil fuels in various sections of the industry. Thus, CO2 emission has undeniable effects on global warming, environmental changes, and ecosystem conditions (Siqueira et al. 2017). As reported by world environmental organizations, carbon dioxide emissions in ambient air have been increased from 22.15 Gt in 1990 to 36.14 Gt in 2014. The beginning of the Industrial Revolution has become the main reason for the global warming phenomenon (Rashidi and Yusup 2016).
Therefore, to reach sustainable development, consciousness about increasing environmental problems and global climate change is necessary. Hence, scientific societies need to apply their potentials toward resolving new challenges, such as mitigation of climate change, conservation of the environment, and replacement of fossil fuels by renewable energies. The first investigation on carbon emission was reported in 1981, and afterward, carbon emission researches have been increased, gradually. Figure 4 depicts the gradual growth of research publications corresponding to the carbon emission starting from 1980 to June 2019. According to this plot, about 479 research articles were reported in 2018, which reveal the growing importance and concern about this issue among the scientific communities (Abeydeera et al. 2019).

Trend of research publications about carbon emission starting from 1980 to June 2019 (Abeydeera et al. 2019)
Various solutions, such as a decrease of irregular use of energy resources, changing sources of fossil fuels by renewable and sustainable energies, and utilization of CO2, have been considered to reduce CO2 emission and consumption of fossil fuels. All these solutions are considerable approaches to solving the greenhouse gas problem. However, most of the new sources of energy are less efficient than fossil fuels (mainly natural gas) and regarding the economic possibility viewpoint are not competitive. Based on reported studies, it is not possible to attain the target of 100% sustainable energy systems in a short or medium period. Hence, to provide energy, fossil fuel consumption will continue (OECD I 2016). Therefore, CO2 capture and utilization are approaches that can be regarded as a solution to global warming. But, after three decades of investigation, these approaches need greater maturity.
After the industrialization and the maturation of chemical industries, CO2 has been applied in numerous industries. The utilization techniques can be considered into two categories. First, the direct use of CO2 without any transformation to value-added compounds (Huang and Tan 2014). The manufacturing of carbonated drinks in beverage industries can be considered as the earliest and direct application of CO2. Also, enhanced oil recovery (EOR) techniques by CO2 injection remarkably improves the economic efficiency of oil extraction from the oil reservoir owning to high mutual solubility of supercritical CO2 and oil, as a hydrophobicity material (Dai et al. 2014). As well as, increasing the CO2 pressure leads to a substantial decrease in the viscosity of the CO2–oil mixture. Accordingly, CO2 injection can enhance production by about 15%. Moreover, it can also be used in enhanced gas recovery (EGR) and enhanced geothermal systems (EGS). Application of CO2 in the production of dry ice, fire extinguisher, supercritical solvent, and the refrigerant is the next direct CO2 utilization without any transformation. However, these direct usages for CO2 are limited in volume and have a small consequence on the overall CO2 concentration (Muradov 2014), as well as the CO2 molecules in all the mentioned applications stay pure and do not convert to another chemical. The left side of Fig. 5 depicts the physical utilization of CO2.

The second category of CO2 utilization is its transformation to value-added material and fuels (Aresta 2010). This approach has been attracted very attention among scientific communities. Hence, widespread attempts are accomplished for converting CO2 to C1 building block chemicals (Bertau et al. 2010; Keim and Offermanns 2010; Ola et al. 2013; Aresta and Dibenedetto 2007). In this approach, using the specific heat or pressure conditions, the CO2 molecules mostly decompose to the simple substances (such as pure carbon or CO) or in the presence of a catalyst react with other chemicals to produce value-added materials, such as hydrocarbons. Thus, the CO2 molecule is a constituent of a chemical product, in which the product cannot be synthesized without CO2. The right side of Fig. 5 shows the application of CO2 in the manufacture of value-added chemicals, as chemical utilization.
3 CO2 Activation
The activation of a molecule means elevation of the reactivity of the molecule. Therefore, it can be considered by making an obvious change in molecular properties in comparison with the properties of a stable ground state. Regarding the molecular structure, CO2 activation can be considered in three methods such as (1) bending of the O–C–O angle from linear equilibrium geometry. Notably, more bending causes a greater decrease in LUMO energy. (2) One or two C–O bond stretching and (3) CO2 polarization due to charge transfer to the molecule (Song et al. 2017; Álvarez et al. 2017).
CO2 is a stable compound with low reactivity, and its carbon atom is in the highest oxidization state, thermodynamically. Thus, overcoming thermodynamic barrier energy is an essential step in CO2 activation, which is an important challenge in its conversion to value-added chemicals. The greatest limitation in the industrial application of CO2, as a starting material, is its thermodynamic stability and/or kinetically inert in a typically favorable transformation. It can be concluded that a large energy source is essential to CO2 transformation (Sakakura et al. 2007). Generally, most of the usual reported methods in CO2 activation are limited to the use of highly reactive chemicals (i.e., energy-rich substrates, epoxides, and aziridines) and/or vigorous situations, such as high temperature and pressure, although side reactions are unavoidable. Thus, designing and using efficient catalyst systems have remarkable advantages such as effectually activate CO2 and/or the substrate, decrease the energy barrier of activation, modify the rate of the activation reaction, and decrease the production of by-products. It can be concluded that developing a convenient functional catalyst with an efficient active site to achieve high selectivity of the goal products is an important necessity in CO2 transformations. Therefore, impressive functional catalysts have key roles to get an efficient and selective procedure for CO2 transformation (Liu et al. 2015).
3.1 Methodologies of CO 2 Activation
The oxygen atom of CO2, as a Lewis base, has a nucleophilicity property, while the carbon atom, as Lewis acid, behaves as an electrophile (Song et al. 2017). Thus, as shown in Fig. 6, two general methods for CO2 activation can be considered. One method involves the catalysts having nucleophile characters such as superbase, N-heterocyclic carbine (NHC) (Kayaki et al. 2009), N-heterocyclic olefin (NHO) (Wang et al. 2013), tungstate, frustrated Lewis pair (FLP) (Bontemps 2016), and hydroxyl group-containing compounds (Yang et al. 2011) which involve with the carbon atom of CO2. In another method, the oxygen atom is affected by the species having Lewis acid orbital such as a transition metal, yielding metal–CO2 complex. In 1971, Aresta and coworkers reported the first almost planar metal–CO2 complex, as (PCy3)2Ni(CO2) in which Ni atom is linked to CO2 molecule and phosphine ligands (Aresta et al. 1975). This complex has two unequal C–O bonds (1.17 and 1.22 Å) and an O–C–O angle of 133° in the linked CO2. Afterward, various transition metals such as iron, ruthenium, and palladium, with different oxidation states, have been investigated in this issue (Paparo and Okuda 2017). Moreover, using high energy and active chemicals such as unsaturated chemicals (alkene, alkyne, etc.), three-membered rings (aziridines, epoxides), and organometallic structures is another key to successful CO2 conversion. Figure 7 depicts the energy profile of the CO2 reaction with high energy substrates, which leads to low energy chemicals (e.g., organic carbonates and carbamates) (Paparo and Okuda 2017).

Various methods in CO2 activation (Song et al. 2017)

Energy profile of the CO2 reaction with high energy substrates (Song et al. 2017)
Besides the CO2 activation, there are still various scientific and practical difficulties in CO2 conversion. Other problems are CO2 collection from the atmosphere by proper technologies and economical aspects of the whole process of CO2 conversion using renewable resources of energy. Nevertheless, low reactivity and selectivity of the catalysts for the chemical transformations are the most considerable challenges, which are related to the high chemical durability of CO2. Thus, understanding the basic chemical procedure of the transformation and investigation of efficient, cost-effective, and eco-friendly catalysts are essential solvation for this challenge.
4 Theoretical Insight of CO2 Transformation
Despite the various investigations, the determination of the mechanism of a catalytic CO2 conversion is a challenging problem (Cheng et al. 2013). From the experimental viewpoint, the catalyst activity and complex structures, the proportion of reactant, catalyst, and substrate in the reaction, the circumstance of the interactions between the involved species and energy levels in the catalytic reactions make understanding the mechanism of CO2 conversion as a very difficult issue. Consequently, the mechanism investigation based on a trial-and-error attitude is a very tedious and time-consuming task that includes designing synthesis–structure–property correlations. This procedure is not so effective nor fully perfect and is very labor-consuming due to the various number of conditions to be studied. Therefore, low yield and negligible selectivity are two obstacles in the catalytic transformation of CO2, which can be improved remarkably by the logical design of catalysts, predicting key structure–activity relationships, and evaluation of specific structures (Wang et al. 2011; Centi and Perathoner 2009).
Experimental limitations give a unique opportunity to computational modeling. Due to current advances in theoretical methods such as novel methods, algorithms, and efficient computational tools, the accessibility of applicable software packages for electronic structure calculations, theoretical CO2 transformation and simulations can play a significant role in two overall main approaches including understanding catalytic reaction mechanisms and designing new efficient catalysts to CO2 transformation (Pople 1999; Kohn and Sham 1965; Kohn 1999; Ben-Nun and Martínez 2002; Bartlett and Musiał 2007). However, theoretical approaches can aid in a deep understanding of determining factors on the structures, binding energies, bond lengths, vibrational frequencies, intermolecular long-range interactions, dispersion forces, and reactivity of the catalysts, which are the main factors in the investigation of the reaction mechanism. Also, computational methods can be used as a predictive tool in the catalyst designing and the controlled synthesis of the target materials. Based on previous experience in the theoretical transformation of CO2, in this chapter, we focus on the mechanism investigation and catalyst designing based on computational approaches. It is noticeable that the experimental features of CO2 transformation are discussed in several reviews (Olah et al. 2009; Riduan and Zhang 2010; Mikkelsen et al. 2010; Ma et al. 2009; Hunt et al. 2010; Darensbourg 2010; Spinner et al. 2012; Cokoja et al. 2011; Yaashikaa et al. 2019; Schilling and Das 2018) and are not considered in this chapter. The chapter is divided into two sections. The first section deals with the theoretical aspects and possible reaction pathways of the catalytic CO2 conversion to value-added materials such as CO, CH4, HCOOH, CH3OH, and heterocyclic compounds. In the second section, theoretical modeling and design of novel catalysts are discussed, and successful examples, within challenges and opportunities, are provided in this approach.
4.1 The Theoretical Approach in CO 2 Conversion to Value-Added Chemicals
4.1.1 Carbon Monoxide
The water gas shift reaction (WGSR) is a traditional process, which is applied for the transformation of synthesis gas to needed hydrogen for ammonia manufacture in the fertilizer industry and petroleum refineries (Bs et al. 2010). Nowadays, the produced hydrogen of this reaction is used as fuel for power production and transportation. The primary application of the reaction back to 1888 (Bs et al. 2010) and its advantage refer to the Haber ammonia synthesis process and evolution of catalyst by Bosch and Wilde in 1912 (Bs et al. 2010). The developed catalyst is based on iron and chromium, which catalyzed the reaction at 400–500 °C and reduce the existing carbon monoxide. Nowadays, reverse water gas shift (RWGS) reaction (Eq. 1), as an endothermic reaction, is a well-known procedure in CO production from CO2, which is regarded as the elementary step for many other hydrogenation reactions, such as the Sabatier reaction (Liu et al. 2015; Wang et al. 2018a) and methanol synthesis (Hu et al. 2013). RWGS reaction is carried out in the gas phase and thermodynamically favorable at high temperatures.
The mechanisms of RWGS reaction can be classified into two overall classes, namely the redox mechanism and associative mechanism (Su et al. 2017). In the redox mechanism, a catalyst bed is responsible for the oxidation–reduction cycle (Bs et al. 2010), in which CO2 and H2 are firstly adsorbed on the active site of reducing metal or metal oxide. Equation 2 depicts steps of the reactions on an adsorbed species (star notation denotes a typical metal or metal oxide). At the end of the mechanism, the oxidized catalyst is reduced by H2, and the active sites are reproduce again (Saeidi et al. 2017; Yan et al. 2014).
The second mechanism named associative mechanism also named dissociative (Aresta et al. 2016; Chen et al. 2000) or formate (Cheng et al. 2013; Scibioh and Viswanathan 2018) mechanism. The critical step in this mechanism is described by an adsorption–desorption model, in which the adsorbed intermediate (carbonate, formate, carbonyl, etc.) is produced during the reaction between the absorbed species. Then, the produced product decomposes to form H2 and a monodentate carbonate (Podrojkova et al. 2020). On the other hand, the reaction of CO2* and dissociated H* produces bidentate formate intermediate by the adsorption of oxygen atoms on the metal surface. The reaction is followed by the formation of HCOOH via more hydrogenation which decomposes into HCO* and OH* and afterward into CO* and H*. In another pathway, CO2 is adsorbed on the surface of the carbon atom, followed by the hydrogenation of the oxygen atoms. Due to more hydrogenation, the produced COOH* decomposes into COH* and OH* forming CO* and H*. Also, COOH* intermediate can be directly decomposed into CO* and OH*. Finally, H2O is formed by the addition of OH* and H* intermediates. Equation 3 shows the steps of this mechanism, and Fig. 8 depicts two separate pathways for the associative mechanism.

Two separated pathways for the associative mechanism, a formation of HCOO intermediate and b formation of COOH intermediate (Podrojkova et al. 2020)
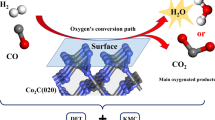
Cai and coworkers were employed the DFT method to investigate the performance of the MoP (0001) surface, in comparison with the Ni2P (0001) surface (Fig. 9) by the generalized gradient approximation (GGA) using the Perdew−Burke−Ernzerhof (PBE) functional. Higher adsorption energies and charge for MoP (0001) confirm that the intermediates are more stable on the MoP (0001) surface. However, both MoP and Ni2P are active in the RWGS reaction. Additionally, the obtained potential energy diagram (PED) (Fig. 10) in mechanistic study illustrates that in the case of MoP (0001) the direct path is more favorable, whereas the COOH-mediated mechanism is a better path for progressing the reaction in the presence of Ni2P (0001) as a catalyst (Guharoy et al. 2019).

Crystal structures of the applied MoP (0001) and Ni2P (0001) surfaces in RWGS reaction (Guharoy et al. 2019)

Potential energy diagram (PED) for the RWGS reaction on a MoP (0001) and b Ni2P (0001) (Guharoy et al. 2019)
In another study, the performance of the surfaces of Cu@Mo2C (001) and Cu4@Mo2C (001) (Fig. 11) was explored in the RWGS reaction by DFT calculations using the PBE functional (Jing et al. 2018).

Optimized crystal structures of the Mo-terminated β-Mo2C (001) surface, Cu@Mo2C (001), and Cu4@Mo2C (001) surfaces, in which Mo, C, and Cu atoms have been detected by blue, gray, and orange color, respectively (Jing et al. 2018)
The results show that the doped β-Mo2C (001) surface by Cu needs lower energy for desorption of CO. Therefore, the introduction of non-noble metal copper is a useful technique in CO release. Also, mechanism studies illustrate that the reaction progress by the redox mechanism is the most preferred path for both Cu@Mo2C (001) and Cu4@Mo2C (001) surfaces. Also, regarding the rate-determining step (RDS) of the redox mechanism on the PEDs, corresponding to Cu@Mo2C (001) and Cu4@Mo2C (001) surfaces (Fig. 12), the latter surface is a better candidate for the proceeding of the RWGS reaction (Jing et al. 2018).

PED of the RWGS reaction in the presence of a Cu@Mo2C (001) and b Cu4@Mo2C (001) surfaces and corresponding energy values for RDS (Jing et al. 2018)
As mentioned in two previous examples, DFT calculations based on the plane-wave basis sets are a privileged approach to exploring the solid-state materials as catalysts in the RWGS reaction. However, DFT calculations by Gaussian-type orbitals are another method for the investigation of this reaction. Guo and coworkers investigated Cu12TM (TM = Co, Rh, Ir, Ni, Pd, Pt, Ag, Au) as the bimetallic metal catalysts in RWGS reaction, in which 6-31G(d,p) basis set and LANL2DZ pseudopotential were used to describe the H, C, O atoms and metal elements, respectively (Zhang and Guo 2018).
Figure 13 illustrates the optimized structure of Cu12TM and adsorbed reactants, intermediates, and products, in the RWGS reaction. Figure 14 depicts the PED of the reaction on the Cu13 and Cu12TM clusters. The results indicate the redox mechanism, in which CO2 is cleaved to CO* and O* is more favorable than both carboxyl and formate mechanisms. Also, as it is shown in PED, H2O formation is the RDS of the reaction (Zhang and Guo 2018).

Optimized structure of Cu12TM and adsorbed reactants, product, and intermediates in the RWGS reaction (Zhang and Guo 2018)

PED of the RWGS reaction on Cu13 and Cu12TM clusters, within the involved species in the different steps (Cao et al. 2016)
In other DFT calculations by the Gaussian-type orbitals, four mechanisms were considered to explore the thermodynamic and kinetic aspects of the RWGS reaction in the presence of the Rh–Mo6S8 cluster as a catalyst. Besides the general mechanisms in the RWGS reaction, the authors considered the possibility of formic acid formation for the possible paths of the reaction mechanism. Equation 4 illustrates the proposed steps of the formation and consumption of formic acid in the RWGS reaction (Cao et al. 2016).
Figure 15 depicts the optimized species in the reaction, and Fig. 16 illustrates the PEDs corresponding to (A) the redox mechanism, (B) the carboxyl mechanism, (C) the directly formic acid decomposition to CO, and (D) the formation of a CHO as the intermediate through the reaction. Based on the studied mechanisms, the reaction progress through the direct decomposition of HCOOH to CO and H2O (mechanism C) leads to a lower value of barrier energy in comparison with other mechanisms. Also, due to the high energy barrier of RDS (76.01 kcal mol−1), the decomposition of HCOOH to CHO* is unlikely to proceed (Cao et al. 2016).

Stable reactants, intermediates, and products in the catalyzed reaction by Rh–Mo6S8 cluster, in which Mo, S, Rh, H, O, and C atoms are colored by light blue, yellow, dark blue, white, red, and gray, respectively (Cao et al. 2016)

PED corresponding to a the redox mechanism, b the carboxyl mechanism, c the directly formic acid decomposition to CO, and d the formation of a CHO intermediate along with the reaction (Cao et al. 2016)
Despite the various studies about the RWGS reaction, the subjects, such as the effects of the metal catalysts and related mechanisms along the spontaneous, dynamic, and high-temperature reaction, need more investigation and clarification (Choi et al. 2017). It was revealed that to reach a satisfactory level of conversion, high temperature is required, thermodynamically. Therefore, remarkable attempts are performed to modify catalytic activity and selectivity for carrying out the RWGS reaction at lower temperatures (Yang et al. 2018).
4.1.2 Methane
CO2 reduction to methane (CO2 methanation or Sabatier reaction) shows numerous benefits among other chemical reaction because methane can be straightly driven into the natural gas pipelines, and it can be applied as fuel or starting material for the generation of other chemicals (Frontera et al. 2017). Also, CO2 conversion to methane is a simple reaction that proceeds under atmospheric pressure and room temperature. Regarding thermodynamic property, CO2 methanation remains the most advantageous transformation than other reactions, which form hydrocarbons or alcohols (Aziz et al. 2015). This reaction is exothermic, pressure-dependent, and thermodynamically favorable at low temperatures (Eq. 5). But, the full reduction of the carbon atom of CO2 with the highest oxidation state has a significant limitation, kinetically. Thus, to attain an acceptable rate and selectivity, the use of a catalyst is necessary.
Two general reaction mechanisms can be considered for CO2 methanation on the solid surfaces. The first route is passing through the CO intermediate via RWGS reaction followed by further reduction and CO conversion to CH4 (Wei and Jinlong 2011). However, the second mechanism is a direct reduction, in which the reaction of CO2 with H2 forms methane without CO formation as an intermediate. Figure 17 depicts (A) direct hydrogenation mechanism and (B) possible routes for the mechanism including the CO intermediate (Lapidus et al. 2007).

Two general mechanisms of CO2 methanation on the solid surfaces (Podrojkova et al. 2020)
Ren and colleagues report a DFT investigation about methanation of CO2 on the Ni(111) surfaces within the GGA and Perdew–Wang (PW91) functional (Ren et al. 2015). Figure 18 depicts the most stable configurations of the reactants and intermediates involved in the investigated mechanisms.

Involved species in the mechanism of CO2 methanation on Ni(111) surfaces, in which C, O, H, and Ni atoms are depicted by as the gray, red, white, and blue colors, respectively (Ren et al. 2015)
Three mechanisms were considered for this conversion. In the first path, HCOO species are formed by the reaction between CO2 and H. CH4 is produced due to the dissociation reaction of HCOO to CO and OH and hydrogenation of CO. In this path, dissociation of HCOO shows the highest energy barrier (306.8 kJ mol−1). In the second route, CO2 directly is split into CO and O on the Ni(111) surface, without any HCOO formation. Then, CO decomposition and hydrogenation of C atoms lead to CH4 molecules. The RDS for this mechanism is 237.4 kJ mol−1, which belongs to the decomposition of CO into C and O species. In the third route, CO2 is firstly hydrogenated to C(OH)2 species, which dissociate into CH2O and OH in the next step. Further hydrogenation leads to CH2O dissociation into CH2 species. RDS for this path belongs to the formation of C(OH)2 on the Ni(111) surface, corresponding to 292.3 kJ mol−1. Therefore, regarding all studied mechanisms, path 2 is the best candidate for reaction progress. Figure 19 depicts the PED diagrams for the studied pathways.

PED diagrams for the methanation of CO2 on the Ni(111) surfaces (Ren et al. 2015)
In other DFT calculations, CO2 methanation is studied on Ni4/t-ZrO2(101), Ni4/VO-t-ZrO2(101), and Ni4/H-t-ZrO2(101) surfaces within the GGA method. Geometry optimizations were obtained using the PBE functional and double numeric polarized (Han et al. 2017) basis set. This basis set is analogous in measure and feature to the 6-31G(d,p) basis set, as a Pople basis set. Figure 20 shows the optimized crystal structures of the studied surfaces.

Optimized crystal structures of the Ni4/t-ZrO2(101), Ni4/VO-t-ZrO2(101), and Ni4/H-t-ZrO2(101) surfaces (Han et al. 2017)
Based on the obtained results, the reaction passes through the CO intermediate for all surfaces, and direct reduction of CO2 to methane without any formation of CO intermediate was not observed. On the other hand, methanol production, based on the steps of Eq. 6, is a competitive reaction with methane formation. In the case of Ni4/t-ZrO2(101) catalyst, the difference between the highest barrier energies of CH4 and CH3OH formation is 261.8 kJ mol−1 and 197.9 kJ mol−1, respectively. Therefore, CH3OH formation reduces the productivity and selectivity of the CO hydrogenation into the CH4 product. In the case of Ni4/VO-t-ZrO2(101) catalyst, the highest barrier energies for the production of CH4 and CH3OH from CO are 157.8 kJ mol−1 and 202 kJ mol−1, respectively, which specify that the formation of CH4 is more favorable, thermodynamically and kinetically. However, regarding the highest barrier energies for the conversion of CO to CH4 (246.9 kJ mol−1) and CH3OH (274 kJ mol−1) for Ni4/H-t-ZrO2(101) catalyst, it can be concluded that the hydroxyl groups and oxygen vacancies on Ni4/t-ZrO2 surfaces are useful to the development of CH4. Moreover, the selectivity of CO conversion to methane is enhanced due to the oxygen vacancies. Figure 21 depicts PED diagrams of the reaction for the investigated surfaces (Han et al. 2017).

PED of the reaction for hydrogenation of CO to CH4 on Ni4/t-ZrO2(101), Ni4/VO-t-ZrO2(101), and Ni4/H-t-ZrO2(101) surfaces (Han et al. 2017)
Applied catalysts in CO2 methanation are not limited to heterogeneous surfaces, and Wei and coworkers reported a DFT investigation by the cationic Ir-pincer complex ((POCOP)Ir(H)(acetone)+ (POCOP = 2,6-bis(dibutylphosphinito)phenyl)) as the catalyst and silane as the reducing agent. The investigation of a four-step mechanism was performed by the hybrid meta exchange–correlation M06 functional, in which the effective core potentials (ECPs) of Hay and Wadt with double-ζ valence basis sets (LanL2DZ) were applied to study the Ir metal atom. Also, these basis sets have been improved by polarization functions (ζf = 0.938). The 6-311G(d, p) basis set was applied to describe other atoms. Figure 22 depicts the overall reaction and the studied mechanism for the reaction (Fang et al. 2018).

Overall CO2 methanation reaction by the Ir-pincer complex and studied four-step mechanism for the reaction (Fang et al. 2018)
The reaction progresses by sequential reduction of CO2 molecule to silylformate, bis(silyl)acetal, methoxysilane, and methane as the final product, respectively. In stage 1, the hydrosilylation of CO2 to silylformate was investigated via three routes. In the first step of path A, the cationic Ir-pincer complex activates CO2, accomplished by the Ir–CO2 moiety activation by a silane molecule. Bridged hydrogen between Ir and Si atoms is the result of Path B, which is started by the interaction between the cationic Ir-pincer complex and the silane molecule. This path is followed by the activation of a CO2 molecule by the silane iridium adduct. CO2 molecule inserts into the iridium-hydride bond of the cationic Ir-pincer complex in path C. Then, the produced iridium formate reacts with a free silane to produce silylformate (HCOOSiMe3). Figure 23 depicts the involved species and corresponding relative energies in stage 1.

Schematic representation of the involved species and corresponding relative energies in stage 1 silylformate (HCOOSiMe3) formation via pathways A and B (Fang et al. 2018)
Similar to step 1, three pathways can be considered for the conversion of silylformate to bis(silyl)acetal (H2C(OSiMe3)2) or formaldehyde (H2C=O) in step 2. Path A proceeds through the breaking of the next silane Si–H bond and C=O bond reduction of silylformate, path B is related to the ionic SN2 outer-sphere mechanism, accomplished by the silylformate nucleophilically attacking the 1-silane iridium complex, and pathway C corresponds to silylformate insertion into the Ir–H bond. The obtained results show that path B (Fig. 24) passes through lower barrier energy than A and C paths. The calculated RDS energy for this step is about 12.2 kcal mol−1. In the third step, bis(silyl) acetal is converted to methoxysilane, in which the obtained RDS energy for this step is 16.4 kcal mol−1. Finally, in the fourth step, by overcoming to activation energy about 22.9 kcal mol−1, methoxysilane is reduced to methane (Fig. 25). Also, it is notable that formaldehyde production by the cationic Ir-pincer complex is not probable.

PED of the silylformate conversion to bis(silyl)acetal or formaldehyde in step II via path B (Fang et al. 2018)

PED of the bis(silyl)acetal conversion to methoxysilane and methane within steps 3 and 4 through the ionic outer-sphere mechanistic routes (Fang et al. 2018)
Electrochemically CO2 reduction reaction (CO2RR) to fuels and organic feedstock is another approach toward CO2 utilization. CO2RR can be scaled to facilitate the large-scale storage of chemical products (Wang et al. 2011; Gattrell et al. 2007; Costentin et al. 2013). Since the products of CO2RR are extracted from petrochemical sources, their manufacture via CO2RR could decrease the global demand for fossil fuels. Lihui and coworkers reported the CO2 reduction into CH4 on the Cu (111) via electrochemical reaction (Ou et al. 2019). Figure 26 depicts the crystal structure of Cu (111) in water as the studied solvent in this reaction.

Arrangement of solvent molecule on Cu(111): a side view; b top view (Ou et al. 2019)
The DFT investigation was accomplished using the GGA method and the PBE exchange–correlation functional. Also, ultrasoft pseudopotential was applied to study the nuclei and core electrons, and the Kohn–Sam equations are solved by a plane-wave basis set. Two overall mechanisms including (a) CH2O and (b) CHOH pathways in the presence of the simulated low overpotential have been considered for the reaction. Common intermediates for the first and second mechanisms are CHO and CH2, respectively (Fig. 27). The results showed that both considered mechanisms may happen in a parallel way in the presence of the simulated low overpotential. Also, the formation of CO is the potential-limiting step, which is according to the observed experimental results. Based on the studied mechanisms, methanol formation is a side reaction, which can interrupt the methane formation; however, in the simulated low overpotential, methanol formation on Cu(111) is prohibited, kinetically. Figure 28 depicts the obtained PED of CO2 reduction into CH4 and CH3OH on Cu(111) in the presence of the considered low overpotential: (a) CH2O pathway; (b) CHOH pathway.

Considered CO2 reduction mechanisms on Cu(111): a CH2O pathway; b CHOH pathway (RHE = reversible hydrogen electrode) (Ou et al. 2019)

Obtained PEDs of CO2 reduction into CH4 and CH3OH by Cu(111) in the presence of the considered low overpotential: a CH2O pathway; b CHOH pathway (Ou et al. 2019)
4.1.3 Methanol
The vision of liquid sunshine was applied by Shih and coworkers that mentioned that solar energy is an enormous resource to make renewable alcohol fuels, such as methanol (Shih et al. 2018). Also, regarding obtaining a sustainable energy source, the “Methanol Economy” suggested by George A. Olah has been widely accepted among the scientific community (Olah et al. 2018).
Nowadays, the main starting material for the industrial production of methanol is mostly from syngas (a mixture of CO and H2 molecules). Syngas is mainly produced by coal and natural gas, as fossil resources, due to the gasification of coal and the reforming of natural gas by steam (Zhong et al. 2020). To improving the kinetic aspect and to achieve a desired stoichiometry for the reaction, a few amounts of CO2 (about 2–8%) are added to the CO/H2 mixture. Equation 7 shows the stoichiometric ratio for methanol synthesis from CO and CO2. It can be concluded that in comparison with syngas (CO), methanol synthesis from CO2 needs an excessive value of hydrogen. Because an excess amount of H2 is required to eliminate one oxygen atom from CO2 via the production of water as a by-product. Also, the thermodynamic aspect of methanol formation from CO2 is not favorable, similar to CO. Thus, methanol production from CO2 leads to a lower yield than that of the syngas reaction (Mikkelsen et al. 2010; Macquarrie 2005).
The lower temperature and higher pressure for the reaction favor the methanol production from CO2, thermodynamically. Accordingly, an increase in reaction temperature is helpful for CO2 activation and methanol formation, subsequently. Also, other by-products are formed. However, the formation of undesired by-products, such as CO, hydrocarbons, and higher alcohols, causes the use of a highly selective catalyst to become an undeniable necessity. Regarding the various investigation, methanol synthesis over an active surface catalyst such as Cu-based can proceed through three different reversible reaction paths including (1) formate production as intermediate via CO2 reaction with surface atomic H, (2) the formation of carboxyl intermediate through RWGS mechanism, (3) the formation of *C(OH)2 as the produced intermediate by CO2 hydrogenation (Dang et al. 2019; Saeidi et al. 2014; Wu et al. 2017). The progress of the hydrogenation for the mentioned three mechanisms leads to the formation of formyl (H2CO*), methoxy (H3CO*), and methanol (CH3OH), respectively. Mechanism 1 passes through the chemisorbed formate, which can be produced from the CO2 reaction with dissociated surface hydrogen. Afterward, the hydrogenation of surface-bound formate causes dioxomethylene formation that H2CO* is formed by the elimination of hydroxyl as H2O. The hydrogenation of H2CO* intermediate can be continued to methoxy and methanol formation. Mechanism 2 proceeds through the CO* formation by the elimination of hydroxyl from hydrocarboxyl. The sequential hydrogenation of the formed HCO intermediate leads to formyl and methanol. However, in mechanism 3, the hydrogenation of hydrocarboxyl leads to COOH* intermediate formation, which is converted to *COH and hydroxymethylene by continuous hydrogenation. Figure 29 depicts three mechanisms for CO2 reduction to methanol by a catalytic surface (Wu et al. 2017; Qiu et al. 2016; Liu et al. 2019).

Three possible mechanisms in CO2 reduction to methanol on the catalytic surface: a formate, b RWGS, and c hydrocarboxyl mechanisms (Wu et al. 2019)
Yang and coworkers reported a theoretical investigation on the CO2 hydrogenation to methanol on the PdIn(310) surface (Fig. 30) (Wu et al. 2019). The calculations were accomplished within the projector-augmented wave (PAW) method. Also, considering the long-range dispersion force, the Bayesian error estimation functional with van der Waals correlation (BEEF-vdW) exchange−correlation functional has been applied, which is widely used to study surface catalysis reactions by the plane-wave basis set. In the reported study, three overall mechanisms of CO2 conversion were investigated that are divided into four distinguishing paths corresponding to I, II, III, IV in Fig. 31. The results showed that the reaction mechanism of methanol formation on the clean PdIn(310) proceeds through the COOH intermediate and CO stepwise hydrogenation. Also, based on the applied microkinetic analysis, it was illustrated that Pd atom is the active catalytic site in comparison with In atom.

Optimized crystal structure of PdIn(310), in which Pd and In atoms are depicted in blue and brown colors, respectively. (Left) top and (right) side views (Wu et al. 2019)

Three overall mechanisms of CO2 conversion on the PdIn(310) surface (Wu et al. 2019)
However, on this basis of DFT calculations and microkinetic analysis, it was illustrated that the differential adsorption energies of formate at the Pd or the In site are close to each other when two formates are preadsorbed at the In step-bridge site. Moreover, in the presence of formate on the surface, the favorable path of methanol formation changes from the COOH/CO path to HCOO/HCOOH. Figure 32 depicts the obtained PED diagram for the studied paths (Wu et al. 2019).

Obtained PEDs of the studied paths for methanol synthesis in the Pd and In sites over PdIn(310) with two preadsorbed formates at the In step-bridge site (Wu et al. 2019)
In another DFT calculation, the consequence of the size of the Cu cluster on the reduction of CO2 to methanol was studied by using PAW potentials and the PBE functional. Figure 33 depicts the optimized structures of the adsorbed species on the crystal structure of Cu clusters having different sizes and corresponding adsorption energies. The studied mechanism reveals direct CO2 decomposition to CO and O rather than hydrogenation of CO2 to COOH because it has been reported that the former mechanism has lower barrier energy than the latter one on the Cu (111) and Cu (211) (Zhang et al. 2018a). Figure 34 illustrates a schematic presentation of the mechanism of CO2 reduction to methanol on the Cu clusters.

Adsorbed key intermediates on the Cun (n = 13, 15, 19, 55, and 79) clusters and corresponding adsorption energies (Zhang et al. 2018a)

Schematic representation of the reduction mechanism of CO2 transformation to methanol on the Cu clusters (Zhang et al. 2018a)
The DFT results showed that all barriers of the reaction, involved in the CO2 transformation to methanol, represent a linear correlation with the adsorption energies of CO and O. Also, based on the results of the microkinetics simulations, it can be concluded that Cu19 clusters are appropriate candidates for CO2 reduction. Moreover, particle measure of the Cu alters the adsorption energies of the involved intermediates, which can be associated with the position of the d-band of the Cu clusters. Thus, the upshift of the d-band center has remarkable effects on the strengths of the bonding interaction between the metal and intermediates, which has a substantial influence on the CO2 reduction activity. Figure 35 depicts the PED of the CO2 reduction to methanol in the presence of different size of Cu clusters and involved species in each step.

PED of the CO2 reduction to methanol for different sizes of the Cu clusters (Zhang et al. 2018a)
The electrochemical approach based on the Cu surface is another method in CO2 hydrogenation to methanol. The next example is DFT calculation based on the PAW method to understand and comparison of the catalytic activity and mechanism investigation of CO2 reduction on the Cu85 nanocluster and Cu (111) surface. Also, the exchange–correlation interaction is treated by the generalized gradient approximation of Perdew–Burke–Ernzerhof (GGA-PBE) (Rawat et al. 2017).
Equation 8 shows electrochemical CO2 reduction in which two-electron reduction leads to H2, CO, and HCOOH formations. However, four- (4e−) and six-electron (6e−) reduction leads to CH2O and CH3OH formation, respectively. The authors describe two overall electrochemically C–O bond dissociation, as direct and indirect. The direct C–O bond dissociation (*CO2 → *CO + *O) improves the four- and six-electron reduction processes, kinetically. On the other hand, indirect C–O bond dissociation by hydrogenation (*CO2 + *H → *COOH) and then COOH dissociation (*COOH → *CO + *OH) are more favorable than direct C–O bond dissociation. Also, the direct C–O bond dissociation has a remarkable effect on the product selectivity.
The obtained results show that Cu85 nanocluster is more active than the Cu(111) surface in the reduction reaction. Also, the reduction of *CO to *CHO/*COH is the RDS of the hydrogenation reaction, whose barrier energy for Cu85 nanocluster is lower than that of Cu(111) surface. However, the formation of *CHO is more favorable than *COH, followed by CH3OH formation. The next obtained result is a lower required overpotential of Cu85 nanocluster for progressing the potential-limiting step of the reaction. Indeed, the reduction of *CO to *CHO is a potential-limiting step on both Cu(111) and Cu85 surfaces. The overpotential of this step for Cu(111) is 0.71 eV, which is the reaction Gibbs energy for the development of *CO to *CHO. While the reaction Gibbs energy for this reaction on Cu85 nanocluster is 0.53 eV. Figure 36 depicts the PED of the reduction reaction on the Cu85 nanocluster and their dependence on the applied electrode potentials (Rawat et al. 2017).

PED of the reduction reaction on the Cu85 nanocluster and the difference between the applied electrode potentials (Rawat et al. 2017)
Nowadays, the photocatalytic CO2 conversion to methanol has been attracted specific attention among the scientific community (Wang et al. 2011; Bensaid et al. 2012; Centi et al. 2013) because, by using sustainable solar energy, methanol can be formed without any emission of greenhouse gases. Recently, a defect-laden indium oxide, In2O3-x(OH)y, with a rod-like nanocrystal superstructure was reported, which is a photocatalyst in the reduction of CO2 to methanol with 50% selectivity under the atmospheric pressure. This report is a suitable investigation for the formation of a low-pressure solar methanol process using CO2 and renewable H2 sources (Wang et al. 2018b).
To investigate the mechanism based on the DFT calculation, all involved species were optimized by the PBE exchange–correlation functional, together with the Rappe–Rabe–Kaxiras–Joannopoulos (RRKJ) ultrasoft pseudopotentials. Structural analysis shows that the surface of In2O3-x(OH)y possesses hydroxide groups that coordinate to unsaturated indium positions. These positions behave as a Lewis base and a Lewis acid, respectively, which form a surface-frustrated Lewis pairs (SFLP) that plays a key role in catalytic hydrogenation of CO2 to CO and CH3OH. Also, compared with the ground state, Lewis acidity and Lewis basicity of the SFLPs increase the excited state leading to a remarkable facility of the photochemical CO2 transformation to methanol. Figure 37 shows the investigated mechanism of CO2 reduction on the In2O3-x(OH)y. As shown in this figure, CO2 reduction to CO is a side reaction developed by CO2 conversion to methanol. However, the results illustrate that methanol selectivity increases from 40% for nanocrystals to >50% for nanocrystal superstructure of In2O3-x(OH)y, which shows that the nanocrystal superstructures enhance both yield and selectivity toward the methanol production. Figure 38 depicts the PED diagram of the CO2 reduction to methanol over In2O3-x(OH)y (Wang et al. 2018b).

Investigated mechanism of CO2 reduction on the In2O3-x(OH)y surface (Wang et al. 2018b)

PED of the CO2 reduction to methanol over the In2O3-x(OH)y, in which gray, red, green blue atoms corresponds to In, O, C, and H, respectively (Wang et al. 2018b)
The application of organic molecules, as a catalyst in CO2 transformation, has remarkable properties, such as cost-effectiveness, metal-free status, and sustainability (Fiorani et al. 2015). The investigations show that, in comparison with the organo-based catalysts, the activation of the substrates by metal-based catalysts, via the coordination of the functional group to metal, is more effective. But, organocatalysts have some privileges, such as non-toxicity, stability along with the reaction, and resistance toward moisture and air. Thus, they can be considered safe catalysts (Fiorani et al. 2015). Moreover, in some cases, they can be readily provided from renewable chemicals. Based on DFT studies, our group investigated the performances of the proton sponges in CO2 reduction to methanol through the B3LYP/6–311 + + G (d,p) level of theory. Thermodynamic and kinetic aspects of the CO2 reduction to methanol were investigated in the presence of six proton sponges, as the organocatalysts, and borane molecule as the reducing reagent. Figure 39 illustrates the investigated proton sponges and the overall reduction of CO2 to methoxyborane (MeOBO)3, as the final product of the reaction, which is decomposed to methanol and boric acid by the reaction with water (Sabet-Sarvestani et al. 2018).

Overall reduction of CO2 within the studied proton sponges (Sabet-Sarvestani et al. 2018)
Two different cycles can be considered for the catalytic reaction, and the performance of considered proton sponges was studied in gas and solvent phases. BH3OCOH is the output of cycle 1, which can be considered as the starting material of cycle 2. Based on the kinetic studies, the development of boronium–borohydride ion pair (step 2) is the key step of the reaction in cycle 1. Also, the activation energy of this step is affected by the steric effects of the linked methyl groups to the proton sponges. Structural analysis of the proton sponges reveals that dihedral angles (θ) between two aromatic rings of the proton sponges have a key factor in the ΔG≠ value of step 2. Figure 40 illustrates the involved species of cycle 1 (Sabet-Sarvestani et al. 2018).

Involved species of cycle 1 in CO2 reduction in the presence of PS1 as a typical proton sponge (Sabet-Sarvestani et al. 2018)
In the next part of the study, the probable pathways of the (MeOBO)3 formation from BH3OCOH were investigated. Three paths that are nominated as green, blue, and red routes were considered for this conversion (Fig. 41). BH3OCOH is the starting material for all these paths. Figure 41 depicts the involved species in red (above), green and blue paths (down) in cycle 2.

Involved species in red (above), green, and blue pathways (down) of cycle 2 (Sabet-Sarvestani et al. 2018)
Based on the PED of cycle 2 (Fig. 42), the red route is not an appropriate path for the (MeOBO)3 formation, thermodynamically and kinetically. Also, in comparison with the green path, the blue route is not a probable path for the reaction, kinetically. Therefore, the green route is the most probable pathway for cycle 2, thermodynamically and kinetically.

PED of cycle 2 corresponding to the green, blue, and red paths (Sabet-Sarvestani et al. 2018)
4.1.4 Formic Acid
Due to the wide application of formic acid as a preservative, insecticide, and industrial material, the development of the efficient approaches of synthesis has been considered. The formic acid fuel cell is the next usage of this compound to provide electricity (Wang et al. 2018c). Moreover, properties such as non-toxicity, biodegradability, being liquid at the ambient conditions, easiness of storage and transportation, relatively high hydrogen capacity (4.4 wt%), and sustainability cause it to become one of the most proper materials for hydrogen storage. In the gas phase, direct CO2 hydrogenation to formic acid is endergonic; however, in the aqueous phase or the presence of an inorganic base, such as ammonia, the reaction becomes exergonic and feasible (Eq. 9) (Wang et al. 2018c). The use of an inorganic base, as a catalyst for the reaction, leads to formate production, which by adding a strong acid is converted to formic acid. Thus, the inorganic base changes the reaction equilibrium toward higher selectivity to formic acid. Other approaches, such as the application of buffers, basic ionic liquids, and solvents addition (like DMSO as coordinating agent), are alternative methods for the production of pure formic acid, without the use of amine or other strong bases.
In 1976, the first study of the CO2 hydrogenation to formic acid was reported by Inoue and et al that applied the complexes of Ru, Rh, and Ir and triphenylphosphine (PPh3). This report did not attract significant regard until the 1990s, in which the attention to the CO2 transformation to formate was revived (Inoue et al. 1976). Based on the recent investigations, two different paths can be considered in CO2 hydrogenation to formic acid on a catalytic surface. These pathways depend on the different adsorption mode of the CO2 and generated formate. As shown in Fig. 43, in paths A and B, monodentate formate and bidentate formate are generated, respectively, in which formic acid is produced after hydrogenation of these intermediates (Peng et al. 2012; Chiang et al. 2018; Zhang et al. 2018b; Filonenko et al. 2016).

Two different paths for CO2 reduction to formic acid on a catalytic surface (Podrojkova et al. 2020)
A DFT investigation based on the M06-L density functional is reported to study the efficacy of the copper atoms implanted in the plane of graphene (Cu/dG) in catalytic hydrogenation of CO2 to formic acid. For this study, the Stuttgart–Dresden effective core potential and 6-31G(d,p) basis set are considered for copper and normal atom, respectively. Cu atom on the Cu/dG surface is an active site for the adsorption and heterolytic cleavage of the hydrogen molecule. Figure 44 depicts the optimized structure of (Cu/dG), atomic natural bond orbital (NBO) charges, and bond lengthen of Cu bond with the nearby atoms(Sirijaraensre and Limtrakul 2016).

Optimized structures and atomic NBO charges of applied Cu/dG surface in CO2 hydrogenation to formic acid (Sirijaraensre and Limtrakul 2016)
Two different paths have been considered for CO2 hydrogenation. In the initial path, CO2 is reduced via the bimolecular adsorption, without H2 activation. As depicted in Fig. 45, this path passes through the barrier energy of 34.6 kcal mol−1 for the first hydrogenation of CO2, which produces an unsteady H-Cu-COOH intermediate. However, in the first step of the second path, the H2 molecule splits into coordinated hydride and proton on the Cu atom. The calculated barrier energy for this step is 19.7 kcal mol−1. The H2-activated Cu/dG can make easy the CO2 reduction to the formate kinds.

PED and optimized involved species for path 1 of CO2 hydrogenation to formic acid (Sirijaraensre and Limtrakul 2016)
In the second step of path 2, CO2 is hydrogenated by H2-activated Cu/dG, yielding a bidentate complex of formate on the Cu, which is more stable than the monodentate HCOO– complex. After the formation of formate intermediate (INT2b), two routes are possible for path 2. The first route includes the protonation of INT2b through the hydrogenated site of graphene to an oxygen atom of the HCOO–moiety, and the second route is the further reduction by the second hydrogen molecule. The obtained results show that hydrogenation of INT2b through the adsorbed hydrogen on the graphene has lower barrier energy than another hydrogen molecule. Also, as depicted in the PED (Fig. 46), the obtained energy of the RDS of path 2 (19.7 kcal mol−1) is lower than the first one (Sirijaraensre and Limtrakul 2016).

PED and optimized species for path 2 of the CO2 hydrogenation to formic acid (Sirijaraensre and Limtrakul 2016)
In another report based on the DFT calculation, CO2 activation and reduction to formic acid on the hybrid metal−organic frameworks (MOFs), namely Mo-Cu-BTC and W-Cu-BTC, has been investigated in which the Cu-BTC is formed from copper carboxylate dimers, [Cu2(COO)4 − (H2O)2], and organic linker, benzene-1,3,5-tricarboxylate (BTC). The metallic property of the bond of M-Cu dimer provides the unsaturated metal site which is distinguished from the metallic site of the pure M-M-BTC (Fig. 47) (Dong et al. 2018), in which a unit cell of Cu-BTC consisting of 48 coppers, 192 oxygens, 288 carbons, and 96 hydrogens is chosen for the calculation by exchange–correlation functional, the GGA-PW91 method, and double numeric plus polarization (DNP) basis set.

Structure of the periodic model (a) and cluster model (b) of Cu-BTC. Orange, red, gray, and white colors indicate copper, oxygen, carbon, and hydrogen atoms, respectively (Dong et al. 2018)
Two potential reaction paths were considered for the CO2 reduction to formic acid, called formate (HCOO*) and carboxyl (COOH*) mechanisms. Each path can be accomplished by two hydrogenation modes, and CO2 is hydrogenated either by H2 molecule (mode 1) or by activated atomic hydrogen (H*) linked to the metallic site (mode 2). Regarding the CO2 hydrogenation through the H2 molecule, the calculated energy barrier on the W-Cu-BTC via carboxyl path (1.27 eV) is lower than that of formate (1.48 eV), while on the Mo-Cu-BTC, the energy barriers in both paths are nearly equal. Thus, Mo-Cu acts as a relatively ideal catalytic system in hydrogenation via H2 molecule mode. However, in the case of hydrogenation through H*, there is a remarkable reduction of the activation energy between CO2 and H* in the formate (HCOO*) path for two bimetallic MOFs. The energy barrier of the carboxyl (COOH*) path stays almost similar. Also, the calculated energy barrier for CO2 reduction along the HCOO* path is 0.30 eV. Figure 48 shows the PED of Mo-Cu-BTC and W-Cu-BTC for formate (HCOO*) and carboxyl (COOH*) paths (Dong et al. 2018).

PED of the CO2 hydrogenation, in which a, b, c, and d are used for hydrogenation over W site by mode 1; over W site by mode 2; c over Mo site by mode 1 and d over Mo site by mode 2, respectively. (Black and red lines indicate HCOO* and COOH*mechanisms, respectively (Dong et al. 2018)
In an electrochemical study, In–Zn bimetallic nanocrystals were evaluated as the catalysts for CO2 conversion to formic acid. Electro-reduction of CO2 to formic acid formation by using several transition metals is an easy and highly efficient method (CO2 + 2H+ + 2e → HCOOH; E = −0.608 V versus normal hydrogen electrode (NHE) at pH = 7). However, the hydrogen evolution reaction (HER, 2H+ + 2e → H2) is a competitive reaction that distributes the high efficiency of the CO2 to HCOOH conversion. A composition ratio of In: Zn = 0.05, like Zn0.95In0.05, shows the highest catalysts selectivity toward HCOOH. In this research, a DFT calculation based on the PAW method with the PBE basis set was employed. Also, the effect of attractive van der Waals (vdW) interaction was considered by applying Grimme’s D3 correction (PBE-D3). Based on the obtained results, the energy of formate intermediate (*OCHO), (produced by CO2 + H+ + e → *OCHO) is a key factor in Zn0.95In0.05 selectivity. The relative Gibbs energy diagram of the CO2 hydrogenation to formic acid over Zn (002), Zn (101), In (110), In (101), (112) facets (Fig. 49), and Zn0.95In0.05 was calculated, in which deposited four-atom In monolayer on a three-layer (5 \(\times\) 5) Zn (002) surface is used to model the Zn0.95In0.05 nanocrystals (Fig. 50) (Kwon et al. 2019).

a ΔG diagrams for CO2 hydrogenation to HCOOH over In (110), In (101), In (112), Zn (002), and Zn (101) surfaces. b Optimized structures of the reaction intermediates (*OCHO) on each surface in the top (c-axis) and side (a-axis) views. Zn: bluish-purple, In: brown, C: gray, O: red, and H: white (Kwon et al. 2019)

Relative Gibbs energy diagrams for the CO2 hydrogenation to HCOOH over Zn (002), In (110), and Zn0.95In0.05 surfaces in which Zn, In, C, O, and H atoms are indicated by bluish-purple, brown, gray, red, and white colors (Kwon et al. 2019)
Based on ΔG values, the strong adsorption strength of *OCHO acts as a limiting factor in HCOOH formation. Based on these diagrams, the energy of *OCHO intermediate on the Zn0.95In0.05 surface is higher than those of other studied surfaces, and therefore, the adsorbed *OCHO easily releases. These results reveal that the Zn–In interface makes the HCOOH production energy-favorable, which causes a higher reaction rate of Zn0.95In0.05 (Kwon et al. 2019).
CO2 reduction by photocatalysts is the next approach toward formic acid production. Zhao and coworkers reported a theoretical study, in which the facet-dependent photocatalytic activity of TiO2 (101) and (001) was investigated in photocatalytic CO2 reduction to formic acid. All calculations were carried out using the GGA-PBE exchange–correlation potential, and the PAW method was applied to consider the effect of core electrons. Two TiO2 facets including (101) and (001) in this investigation are depicted in Fig. 51 (Ma et al. 2016).

Optimized structure of TiO2 (101) and (001) (Ma et al. 2016)
By analogy between the obtained results for the surface and the bulk of TiO2 (101) and (001), it can be deduced that the excited electron tends to stay on the surface than the bulk. Also, the barriers for transferring the excited electron through the bulk and among the surface are similar for both facets. Moreover, the energy of Ti3+ that appeared on the (001) facets is lower than that on the (101) facets, and thus, (101) facets possess a higher conduction band minimum (CBM). Figure 52 shows the PED of CO2 reduction to formic acid over both facets of TiO2 (101) and (001). The energy barrier of CO2 reduction to HCOOH over (101) facets is lower than that of the (001). A higher CBM generates stronger reducing electrons. Thus, the lower energy barrier of the (101) facets and a faster rate of formic acid production on this surface can be related to a higher level of CBM (Ma et al. 2016).

PED and the optimized structure of CO2 reduction to formic acid over both facets of TiO2 (101) (right) and (001) (left) (Ma et al. 2016)
4.1.5 Heterocycles
Heterocyclic compounds are an important class of chemicals that possess various biological and pharmacological properties (Joule and Mills 2010). Value-added heterocycles such as carbonates, carbamates, carboxylic acids, and numerous sophisticated heterocyclic rings having “CO2”, “CO”, “CH2” or “CH” ingredients are the products of CO2 reaction with different substrates such as amino groups, hydroxyl groups, and carbon nucleophiles. CO2 incorporation by nucleophiles, as an efficient and green approach in heterocycle synthesis, has been investigated in recent years, frequently (Wang and Xi 2019). Based on the investigations, CO2 incorporation proceeds through three modes including (I) nucleophilic addition to CO2 followed by intramolecular cyclization to carboxylated cycle production; (II) concerted two nucleophilic site attacks on CO2 affording cyclic carbonyl compound; (III) cyclization due to nucleophilic attack on the reduced CO2 (Wang and Xi 2019). Figure 53 represents different approaches of CO2 incorporation cyclization in the formation of the heterocyclic compounds. The carbon atom of CO2 is an electrophile, and therefore, CO2 reaction with strong nucleophiles leads to the development of the C–N, C–O or C–C bond and the negative charge over the O atom of CO2. However, due to CO2 stability, an efficient catalyst is required for CO2 transformation to heterocycles.

Different approaches of CO2 incorporation cyclization in the formation of a heterocyclic compound (Wang and Xi 2019)
4.1.5.1 Cyclic Carbonates
Low vapor pressure, high boiling point, low toxicity, and environment-friendly are appealing characters of cyclic carbonates, which cause extensive applications of this compound. The use of the cyclic carbonates as the high-boiling polar solvents, additives for fuel, and precursor of plastics are the well-known applications of cyclic carbonates. Moreover, these compounds are intermediates for the formation of other valuable compounds like dialkyl carbonates, glycols, carbamates, and pyrimidines (Calabrese et al. 2019). Traditionally, cyclic carbonates are prepared by phosgene as highly toxic, corrosive, and difficult to handle chemicals. Thus, the formation of cyclic carbonates via carbon dioxide and epoxide, as a low-toxicity and sustainable choice, has been attracted very much attention among scientists. Also, this approach is known as a high-yielding catalytic procedure which shows an atom economy of 100%. However, the use of efficient catalysts is an important necessity for this conversion (Calabrese et al. 2019).
Xia and coworkers report an amine-functionalized ionic liquid (AFIL) to produce 3-chloro-1,2-propylene via CO2 cycloaddition with epichlorohydrin (Chen et al. 2019). All the structures of reactants, intermediates, transition states, and products were optimized in the solution phase by employing ωB97X-D/6–31 + G(d) level of calculation. Also, the solvent effects were described by the solvation model based on density (SMD) to improve the accuracy of the calculated results. Based on the obtained results, the Br− of the AFIL acts as a nucleophile. However, hydrogen bond formation between the oxygen atom of the epoxide and a hydrogen atom of the catalyst, as a Lewis acid, is a key interaction to epoxide activation against a nucleophilic addition. As depicted in Fig. 54, the authors described a reaction between AFIL and carbonic acid (H2CO3) to protonation of tertiary amine groups of the ionic liquids (Chen et al. 2019).

PED and optimized structures of protonated AFIL via reaction with H2CO3 (Chen et al. 2019)
Figure 55 shows the PED of the ring-opening step of the reaction of epichlorohydrin facilitated by the hydrogen bond interaction of AFIL. The protonated tertiary amine group of AFIL stabilizes Br− along with the ring-opening reaction of epichlorohydrin through the Br− nucleophilic attack to the carbon atom of epichlorohydrin. 2-buthylimidazole is another moiety of the ionic liquid, which has an important effect in the ring-opening procedure of epoxides via hydrogen bonding interaction. After hydrogen bond formation, due to the nucleophilic attack of Br−, the O–C bond of epichlorohydrin is broken, and yielding Int-b3 consists of the protonated AFIL and ring-opened epichlorohydrin. After coming back the protonated AFIL to the catalytic cycle, Int-b6 is produced due to the nucleophilic reaction between the negatively charged oxygen atom of the Int-b4 and CO2. Int-b6 is an intermediate, which can be considered as the starting material for 3-chloro-1,2-propylene formation in the final step. The obtained results show that the ring-opening process is the RDS of the whole reaction. Figure 56 illustrates the PED of the final step and the optimized structure of the involved species.

PED and the involved compounds in the ring-opening process of epichlorohydrin through the hydrogen bond interaction of AFIL (Chen et al. 2019)

PED and the involved species of 3-chloro-1,2-propylene formation through intramolecular nucleophilic reaction of Int-b6 (Chen et al. 2019)
The MOFs are the next efficient catalysts in CO2 transformation to cyclic carbonate. Park and coworkers reported a joint of experimental and theoretical studies on novel adenine-based Zn-(II)/Cd(II), namely PNU-21 and PNU-22 assisted by tetrabutylammonium bromide (TBAB) as the cocatalyst. Based on structural analysis, both MOFs possess unsaturated Lewis acidic metal centers [Zn(II) and Cd(II)], free basic N atoms of adenine molecules, and auxiliary dicarboxylate ligand (Fig. 57). Experimental results show that both catalysts have similar efficiency in the cycloaddition reaction of CO2 and epichlorohydrin (ECH) as the heterogeneous catalysts. In the theoretical section, a comparative DFT-based investigation was performed on the mechanistic aspects of the cycloaddition reaction in three paths, namely noncatalyzed, TBAB-catalyzed, and PNU-21/TBAB cocatalyzed. For this purpose, meta-hybrid GGA functional M06 was employed, in which 6-31G(d) basis set was applied for H, C, N, O, and Cl atoms, while the heavier atoms (Zn and Br) were studied by LanL2DZ as a “double-ξ” quality basis set. Figure 58 depicts the investigated mechanism for the cycloaddition reaction by PNU-21/TBAB catalytic system (Rachuri et al. 2019).

Bonded ligands to Zn and Cd atoms in PNU-21 (a) and PNU-22 (b) structures (Rachuri et al. 2019)

Proposed mechanism for cycloaddition of CO2 and epichlorohydrin in the presence of PNU-21/TBAB catalytic system (Rachuri et al. 2019)
In the reaction mechanism, firstly, the oxygen atom of the epoxide is coordinated to the Zn site via the oxygen atom of the epoxide. Afterward, due to the nucleophilic attack of the activated Br− anion of TBAB, three-membered epoxide rings are opened. Then, alkyl carbonate anion is formed through another nucleophilic attack of oxide anion of the opened epoxy to the polarized CO2 molecule by one of the −NH2 groups of adenine molecules. Finally, the corresponding cyclic carbonate is produced by an ultimate ring-closure step. The obtained results show that the activation energy of the ring epoxide opening for the noncatalyzed pathway, as the RDS of the cycloaddition reaction, is 61.96 kcal mol−1. This value for TBAB-catalyzed pathways is reduced to 39.60 kcal mol−1. However, as depicted in the PED of PNU-21/TBAB cocatalyzed cycloaddition pathway (Fig. 59), the energy barrier of the ring-opening of epoxide by nucleophilic attack of Br− is 7.58 kcal mol−1.

PED and optimized involved structures of the PNU-21/TBAB-catalyzed cycloaddition (Rachuri et al. 2019)
Organocatalysts are the other kind of the applied catalysts in CO2 transformation to cyclic carbonates by ethylene oxide (Galvan et al. 2014; Aoyagi et al. 2013; Wang et al. 2012; Wong et al. 2008; Girard et al. 2014; Yu et al. 2010; Tsutsumi et al. 2010; Chatelet et al. 2013). We reported a DFT study on the kinetics and mechanism of the cyclic carbonate formation by carbonyl-stabilized phosphonium ylides as a helpful organocatalyst (Fig. 60). M06-2X/6-31G(d,p) level of the theory was employed for geometry optimization of the starting materials, intermediates, products, and transition states. Also, single-point energies were obtained to reinforce the accuracy of the theoretical results, at the MPW1PW91/6–311 + + G(d,p) (Sabet-Sarvestani et al. 2020).

Applied carbonyl-stabilized phosphonium ylides as the efficient catalysts in the cyclic carbonate synthesis (Sabet-Sarvestani et al. 2020)
This transformation can be considered by two mechanisms (Fig. 61). The oxygen atom of the organocatalyst has a nucleophilic character, and the first mechanism is begun via the nucleophilic attack of this atom to the carbon atom of CO2 (mechanism A). This pathway is followed by another nucleophilic attack of the oxygen atom of In1A, as the product of step 1, to the epoxide ring. In the second mechanism (mechanism B), the reaction is initiated by the oxygen attack of organocatalysts to the three-membered ring of ethylene oxide, followed by the next nucleophilic attack to the carbon atom of CO2.

Two investigated mechanisms for cyclic carbonate formation in the presence of phosphonium ylides as the organocatalyst (Sabet-Sarvestani et al. 2020)
The energetic span model (ESM) is a quantum chemistry descriptor to evaluate turnover frequency (TOF) based on the obtained energy profile from the electronic structure calculations, which is used for investigation of the kinetic aspects of the proposed mechanisms (Kozuch and Shaik 2011). In this model, the degree of the TOF control (XTOF) is applied to determine the effects of each involved species in reaction on the TOF values. Then, TOF-determining intermediate (TDI) and the TOF-determining transition state (TDTS) can be identified by the XTOF values (Kheirabadi et al. 2018; Falivene et al. 2018).
Figure 62 illustrates the obtained TDI and TDTS of mechanisms A and B on the PEDs of four typical organocatalysts. The results show that the transition state of In2A formation through In1A in step 2 is TDTS for mechanism A. In the case of mechanism B, the energy barrier of step 1 is the TDTS. Also, In2B is considered as the TDI of the mechanism. Finally, based on the calculations, it can be concluded that linked electron-donating groups to carbonyl-stabilized phosphonium ylides have an efficient effect on the barrier energy of the cyclic carbonate formation.

PED of the substituted organocatalysts (G = CF3, CO2Me, Me and NMe2) and corresponding TDI and TDTS of mechanisms A and B (Sabet-Sarvestani et al. 2020)
4.1.5.2 Cyclic Carbamate
This class of heterocyclic rings is found in numerous valuable chemicals. For example, a five-membered carbamate ring (2-oxazolidinone) is a heterocyclic core of antibiotics such as Linezolid, Posizolid, and Tedizolid and also antidepressants like Toloxatone (Brickner 1996; Schaadt et al. 2009; Chen et al. 2017). Traditionally, chemicals such as isocyanates, phosgene, urea, or organic carbonates are used to synthesize cyclic carbamates. However, due to high toxic and expensive starting materials, CO2-based synthetic paths, as sustainable, cost-effective, and highly efficient methods, can be an alternative for the cyclic carbamates synthesis. Thus, the investigation of novel catalysts to overcome the kinetic and thermodynamic stability of the CO2 conversion to cyclic carbamates is of interest to scientists (Niemi 2019).
Lan and coworkers reported a DFT study on the mechanism of CO2 (1 atm) transformation to cyclic carbamates via the copper complex and Togni’s reagent in the presence of DBU (1,8-diazabicyclo[5.4.0]undec-7-ene) (Fig. 63). The calculations are based on B3LYP functional with the standard 6-31G(d) basis set, for normal atoms, and SDD basis set for Cu and I. Also, single-point calculations by the integral equation formalism (IEF) polarizable continuum (PC) model (IEF-PCM) were performed to evaluate the solvent effects (Zhu et al. 2018).

Mechanism of the CO2 conversion to cyclic carbamates via copper complex (Zhu et al. 2018)
Based on obtained results, the reaction is progressed by a mutual conversion of Cu(I)/Cu(II) in the catalytic cycle. As shown in Fig. 63, coordination of the nitrogen atom of 1a to the Cu(I) center of catalyst gives an amine-coordinated intermediate. This step is followed by the deprotonation of amine by the DBU base, as the RDS of the reaction. The next step is an insertion reaction, in which CO2 is inserted between the Cu(I)-N bond of intermediate II, yielding copper(I) carboxylates (III). Then, the Cu (I) atom of the later intermediate is oxidized by Togni reagent (2) to copper (II) dicarboxylate intermediate (IV) and free trifluoromethyl radical. Subsequently, radical intermediate (V) is produced through the trifluoromethyl radical attack to the alkene moiety of IV. Finally, the final oxytrifluoromethylation product and the released active Cu(I) intermediate are produced via a radical–radical cross-coupling reaction. Moreover, regarding the global electrophilicity, ω°, and global nucleophilicity, N°, indices, proposed by Domingo and coworkers, ω° and N° indices of the CF3 radical are 2.36 eV and 0.74 eV, respectively, which suggest that this radical possesses a strong electrophilicity character. Also, based on the local electrophilicity analysis of intermediate IV, the ω° index of the Cu(II) atom is 2.16 eV. Therefore, the combination of the strong electrophile (CF3·) and the electron-deficient Cu center is unfavorable. Thus, electrophilic addition to the C–C double bond of intermediate IV simply occurs to produce the diradical intermediate V.
In another study, the formation of cyclic carbamates from carbon dioxide and amino alcohols, in the presence of p-Toluenesulfonyl Chloride (TsCl) as a hydroxyl protecting agent, was reported (Fig. 64). Some properties, such as mild situations, appropriate yields, high enantiomeric excess, and stereoselectivity, were considered as the advantages of this method. The results show that the tosylation of the alcohol group of the amino alcohols improves the leaving group character, and also, an external base is required for the reaction progress toward the corresponding cyclic carbonates. However, tosylation of the amine group is a side reaction that can reduce the yield of the cyclic carbonate formation (Niemi et al. 2018).

Cyclization of carbon dioxide and amino alcohols in the presence of an external base and N-tosylation reaction, as a disrupting reaction (Niemi et al. 2018)
DFT calculations were carried out based on the dispersion corrected meta-hybrid functional M06-2X by the standard 6–31 + G(d) basis set for all atoms. According to the PED of the reaction (Fig. 65), the energy barrier of the nucleophilic attack of a nitrogen atom to MsCl (30.3 kcal mol−1) is higher than that of nucleophilic attack to CO2 molecule (17.5 kcal mol−1). The linked proton atom to INT1 is abstracted by the carbonate part of Cs2CO3 as an external base. Finally, cyclic carbonate is formed due to the tosylation of the alcohol group and an SN2-type reaction mechanism during the ring-closing stage.

PED of the reaction of amino alcohol 1a and CO2 in the presence of MsCl and other compounds (Niemi et al. 2018)
Kukhtin–Ramirez adducts, as organophosphorus compounds, are other intermediates in carbamate synthesis (Wang and Radosevich 2013). Between the various organophosphorus having distinct valence states, trivalent phosphorus derivatives are known as compounds that undergo an addition reaction with 1,2-dicarbonyl structures, yielding 1:1 adduct, formulated as either dioxaphospholene (A) or oxyphosphonium enolate (B) (Fig. 66) (Zhao et al. 2013). The reaction is named as Kukhtin–Ramirez addition. The reaction of B with the electrophilic agents produces alkoxyphosphonium C, which has an electrophile character for nucleophilic displacement. We reported the thermodynamic and kinetic parameters of the carbamate formation through various phosphorous reagents (PRs), as a Kukhtin–Ramirez addition (Sabet-Sarvestani et al. 2017a). The produced carbamate is a valuable starting material for the formation of oxazolidine-2,4-dione as a bioactive compound. DFT calculations are carried out at the B3LYP/6–31 + G(d,p) level of theory. Also, MPWB95, as a meta-GGA functional, is employed for single-point energy calculations. Figures 67 and 68 show the studied PRs in overall reaction and considered mechanism, respectively (Sabet-Sarvestani et al. 2017a).

Kukhtin–Ramirez addition via trivalent phosphorus derivatives (Sabet-Sarvestani et al. 2017a)

Overall reaction and studied trivalent phosphorus in this reaction (Sabet-Sarvestani et al. 2017a)

Proposed mechanism of Kukhtin–Ramirez addition (Sabet-Sarvestani et al. 2017a)
In 1 is formed through the nucleophilic addition of the phosphorous atom of PR to the carbonyl group of α-keto ester (1), with two isomeric structures of In 1(A) and In 1(B). The corresponding absolute Gibbs energies of these isomeric forms are −1318.207 and −1318.211 (a.u), respectively. Thus, In 1(B) is the most stable isomer of In 1. In step 2, In 2 is produced through the proton abstraction of phenyl carbamic acid (2) by In 1. However, In 1(B) possesses two prochiral centers, re and si faces, which make step 2 able to proceed via re face or si face approaches. Finally, in step 3, the final product 3 is developed via the nucleophilic attack of phenyl carbamate part of In 2 and PRs abstraction as oxide forms (PORs). Kinetic results show that step 1 is RDS of the reaction. Based on quantum chemistry descriptors such as local nucleophilicity indices (Nk), Mulliken atomic spin density (Pk−)(Domingo et al. 2013), and donor–acceptor orbital interactions of the phosphorous atom of the PRs, it has been concluded that the PRs having nitrogen atom such as P(NC4H8)3 and P(NEt)3 are appropriate agents in the reaction, due to larger Pk− values. Finally, it has been concluded that P(NEt)3 is a more efficient phosphorus agent for the reaction, kinetically and thermodynamically.
4.1.5.3 Quiznazoline-2,4(1H,3H)-Dione
Because of various biological activities and being a basic moiety of numerous available drugs, such as Ketanserin, Cloperidone, and Pelanserin this heterocyclic system has attracted the attention of chemists and medicinal chemists (Biswas et al. 2020). The reactions between anthranilamide and phosgene, anthranilic acid and potassium cyanate or chlorosulfonyl isocyanate, aromatic amino nitriles and diethylformamide, methyl anthranilate and various iso(thio)cyanates as well as 2-nitrobenzamide and CO are traditional approaches for the synthesis of quinazoline-2,4(1H,3H)-dione (Nale et al. 2014). Thus, a synthetic methodology based on carbon dioxide as a feedstock is much more admitted than other methods. The majority of the traditional approaches have drawbacks, such as the use of highly toxic and specialized reagents like phosgene. Therefore, researchers focus on designing efficient catalysts for CO2 utilization to various derivatives of this heterocyclic system.
Islam and coworkers reported a heterogeneous catalyst based on incorporated palladium metal to aminically improved graphene oxide (GO). They used Pd(II)EN@GO for the formation of quinazoline-2,4(1H,3H)-dione derivatives through a one-pot reaction of atmospheric carbon dioxide, isocyanides, and 2-iodoaniline (Fig. 69) (Biswas et al. 2020). In the mechanism study, a DFT-based investigation was carried out by the B3LYP function and LanL2DZ basis set. The structural analysis of the Pd(II)EN@GO shows that Pd(II) atoms are stabilized by nitrogen atoms of ethylene diamine moieties on the EN@GO composite. At the first step of reaction, Pd(II)EN@GO is reduced to Pd(0)EN@GO through the elimination of two chlorine atoms as a Cl2 molecule. The next step is an oxidation addition in which intermediate-1 is produced via o-iodoaniline coordinates with Pd(0)EN@GO. Subsequently, due to the tertbutyl isocyanide attack to intermediate-1, the next intermediate is formed. The coordination of the oxygen atom of CO2 with Pd increases the p-accepting capacity of the carbon atom of carbon dioxide. Thus, the carbon atom of CO2 can be readily attacked by the nitrogen atom of the amine group of intermediate-2, yielding intermediate-3, which is converted to intermediate-4 due to the intramolecular rearrangement. Finally, after proton attraction by the DBU as the applied based in the reaction and a ring-closing/ring-opening rearrangement, intermediate-4 is converted to the final product (Fig. 70). Figure 71 depicts the PED of the reaction, in which the attack of tertbutyl isocyanide to intermediate-1 is the RDS of the reaction (Biswas et al. 2020).

Formation of quinazoline-2,4(1H,3H)-dione derivatives in the presence of Pd(II)EN@GO (Biswas et al. 2020)

Mechanism of quinazoline-2,4(1H,3H)-dione formation through a cycloaddition reaction using Pd(II)EN@GO and DBU as the catalyst and base, respectively (Biswas et al. 2020)

PED of quinazoline-2,4(1H,3H)-dione formation (Biswas et al. 2020)
In the next report, the water molecule has been considered as a catalyst that activates CO2 as carbonic acid (H2CO3) in the formation of various substituents of quinazoline-2,4(1H,3H)-diones using CO2 and 2-aminobenzonitriles (Ma et al. 2013). Based on the DFT calculations at the M06-2X/D95(d,p) and M06-2X/aug-cc-pVDZ levels of theory, the mechanism of the reaction was investigated. Moreover, the single-point energies were evaluated at the M06-2X/aug-cc-pVTZ level. The CPCM model was applied to investigate the effects of water as the solvent. Figure 72 depicts the considered mechanism for this reaction. H2CO3 is produced via the nucleophilic attack of H2O to CO2. Then, the hydrogen bond formation between the OH moiety of H2CO3 and CN segment of the amino nitrile activates the CN group against the next nucleophilic attack by H2CO3 in step 2. Step 3 consists of continuous rearrangements including oxime group conversion to amide and reorientation of the –COOH group by the C–N bond rotation. Step 4 is an intramolecular nucleophilic addition, which is started by hydrogen bonding formation between the intermediate 6 and new formic acid molecules. Thus, along with the reaction, H2CO3 has two distinct behaviors. First, instead of CO2, H2CO3 interacts with 2-aminobenzonitriles, and second, H2CO3 plays as a proton bridge that improves the nucleophilic addition, efficiently. As depicted in the PED of the reaction (Fig. 73), oxime group rearrangement to amide in step 3 is the RDS of the reaction (Ma et al. 2013).

Studied mechanism for the reaction of activated CO2 molecule as H2CO3 in the synthesis of quinazoline-2,4(1H,3H)-diones (Ma et al. 2013)

PED of the studied mechanism of CO2 and 2-aminobenzonitrile reaction in water (Ma et al. 2013)
Guanidines-based organic superbases (SBs) are the other efficient organocatalysts in the formation of quinazoline-2,4(1H,3H)-diones derivatives (Mizuno et al. 2000a,b; Gao et al. 2010). We investigated the performances of four SBs, including DBU, 1,5- diazabicyclo[4.3.0]non-5-ene (DBN), 1,1,3,3-tetramethylguanidine (TMG), and 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) in the conversion of CO2 and 2-aminobenzonitrile to quinazoline-2,4(1H, 3H)-diones. The DFT study was carried out at the M06-2X/6-31G(d) level, in which some quantum chemistry descriptors were analyzed to justify the obtained kinetic parameters. Mechanisms A and B were investigated for the reaction. In 1A in the mechanism A is produced by the nucleophilic attack of 2-aminobenzonitrile to CO2 and proton adsorption by the SB. Step 2 in the mechanism A is an intramolecular nucleophilic attack to the CN group, yielding In 2A. The formation of isocyanate and the iminol groups in In 3A via intramolecular rearrangement is the output of step 3. Finally, due to the tautomerization of the iminol group to amide and another intramolecular nucleophilic attack, the final product is produced. However, mechanism B is started by the nucleophilic attack of SBs to CO2, and the obtained SB+–COO− intermediates act as the base through the reaction. In step 2 of this mechanism, via another nucleophilic addition reaction, SB molecule is liberated, and In 1B is formed. The other stages of this mechanism are the same as mechanism A; however, the SB+–COO− plays as a base. Figure 74 illustrates the mechanisms A and B in the presence of the SBs as the organocatalysts (Sabet-Sarvestani et al. 2017b).

Studied mechanisms A and B in the presence of SBs as organocatalysts in the formation of quinazoline-2,4(1H,3H)-diones (Sabet-Sarvestani et al. 2017b)
The theoretical results show that mechanism B passes through higher barrier energy than mechanism A. Thus, along with the reaction, the basic character of the SBs is more dominant than the nucleophilic character. The quantum theory of atoms in molecules (QTAIM) (Bader 1990; Henkelman et al. 2006) and NBO analysis (Sabet-Sarvestani et al. 2014) are two powerful quantum chemistry descriptors to describe the behavior of the SBs in mechanism A. In this mechanism, the transition state of step 1 includes the N–H bond diminishing and a new bond developing between the dissociated proton of the N–H bond. Figure 75 depicts the obtained QTAIM graph, the bond critical points (BCPs), and the natural atomic charge of nitrogen atom corresponding to studied SBs at TS1. According to the results, when DBU is the involved base, the H–N developing bond and N–H diminishing bond possess the highest and lowest electron density values, respectively. Also, the natural atomic charge of the nitrogen atom of 2-aminobenzonitrile, when DBU has been used as the base, is the maximum value than the next SBs. Thus, DBU is a more effective base than the other SBs at the TS1 (Sabet-Sarvestani et al. 2017b).

QTAIM graph, the bond critical points (BCPs), and natural atomic charge of nitrogen atom corresponding to studied SBs at TS1 (Sabet-Sarvestani et al. 2017b)
4.1.6 Summary and Outlook
It is undeniable that DFT calculations can be applied in exploring new intermediates, designing the novel catalysts, and determining the reaction pathways. Obviously, identifying the reaction intermediate, reaction steps, RDS of a catalytic cycle and most favorable reaction pathway based on energy barrier for each step of the reaction has remarkable advantages of the DFT methods, which significantly improve our insights about the most favorable conditions for the reaction progress. However, it is noticeable that notwithstanding various theoretical reports, an obvious agreement on a unit reaction mechanism is not achieved by the researchers. Because there are not integrative opinions and principles, which can lead the theoretical and experimental researchers to the same outcomes. In the catalytic CO2 transformation, the high chemical durability of the CO2 molecule is another problem in the mechanism investigation. Indeed CO2 activation is an essential step that shows a high chemical energy barrier in CO2 conversions. Moreover, some issues such as active possible path in a given experimental condition and active site of the catalytic surfaces are the other problems in mechanism investigation of catalytic CO2 transformations. Notably, computational costs are also serious problems in the mechanism studies. For a given accomplished reaction, the mechanism must be determined either by the comparison of the previously reported reactions, which is based on guesses or by systematical investigations based on computational approaches. Due to difficulties and being time-consuming, the computational expense of the calculation of activation energy and the actual pathway of the reaction are limiting issues to employing these approaches. Thus, it can be anticipated that new unexplored mechanism may exist, and more attempts should be carried out along with this aim in the future. However, some critical ignored subjects such as synergetic factors of multiple CO2 adsorptions on a catalytic surface, ligand–ligand interactions on a typical catalyst, and unrealistic assumptions in the reaction conditions are effective factors on the accuracy of computational results.
5 Theoretical Designing of Novel Catalysts Based on DFT Studies
Despite defects such as being expensive and time-consuming, the main outcome of the previous section is that DFT calculations are efficient tools to investigate the mechanism of CO2 transformation to value-added materials. Numerous clear potential advantages of DFT optimization of new catalysts and virtually designing novel procedures in the computer before the experimental evaluations can be considered (Streitwieser 2009). Against the experimental efforts, whenever a mechanism is explored based on DFT calculations, it is readily possible to make changes in the catalytic structures and reactant structures to evaluate the effect of these variations. Virtual exploring attempts with the aim of discovering novel catalysts in CO2 conversion have been rapidly achieved popularity. Because of the importance of CO2 transformation, mechanistic studies often require specific regards (Cheng et al. 2013). Besides the energies and optimized structures of the intermediates and transition states that cannot be achievable experimentally, approaches based on DFT provide a qualitative/quantitative investigation of the new optimized structures that are responsible for the observed kinetic and thermodynamic behaviors (Ahn et al. 2019). Although there are numerous reported catalysts in CO2 transformation, the greater selectivity and reactivity of the catalysts are also considered as the main issues among the researchers (Sakakura et al. 2007; Wang et al. 2011; Centi and Perathoner 2009). Hence, catalyst designing based on the DFT calculations can be regarded as a helpful tool beyond tedious trial-and-error experimental approaches. The opportunities and challenges of this approach have been discussed in the following section.
Nørskov and coworkers are the pioneers in catalyst designing based on the DFT approach (Nørskov et al. 2009, 2011). Considering the explained principles, an intuition into the kinetic aspects of a catalytic reaction, including optimized structures, reaction energies, the energy barrier of each elementary step of the reaction are necessities in designing a catalyst based on the DFT approach. However, to obtain a convenient agreement with the experimental results, the kinetic results must be calculated by an appropriate computational technique (Andersson et al. 2006; Jones et al. 2008). In the kinetic investigation, energies of intermediates, reactants, products, energy barriers of the elementary steps, and the energy of RDS are criteria to evaluate the catalytic activity and selectivity (Nørskov et al. 2009; Sehested et al. 2007). Moreover, quantum chemical descriptors are good criteria in the description of the designed catalysts. In the following, some DFT-based designing of catalysts and prediction of their performances in CO2 transformation are described.
Leitner and coworkers reported a DFT investigation on a catalytic cycle, progressed by ruthenium (II) pincer complexes as catalysts to the direct carboxylation of the C–H bonds of an arene with CO2 (Uhe et al. 2012). They applied the ESM to evaluate the TOF of the designed catalysts and their efficiency in the studied mechanism, in which TOF values in the range of 10–5–10–7 h−1 were predicted for the best systems. Moreover, for modification of the obtained results, the effects of the substrate, the solvent, and a base on the thermodynamic and kinetic aspects of the reaction were investigated, computationally. The DFT calculations carried out based on the Grimme’s B97-D functional that considers the empirical dispersion correction, explicitly. Also, the def2-TZVP basis set (Weigend and Ahlrichs 2005) was employed for normal atoms and the associated ECPs for ruthenium and iodine atoms. Figure 76 depicts the studied mechanism for the direct benzene carboxylation by CO2 (Uhe et al. 2012).

Studied mechanism for the direct benzene carboxylation with CO2, in which X− corresponds to a monodentate anionic ligand (Uhe et al. 2012)
In the first step of the catalytic cycle, the benzene molecule is replaced by the oxygen atom of a coordinated benzoate ligand, yielding a C–H bond σ-complex (complex 2). Then, through a formed six-membered transition state via σ-bond-metathesis type CH-activation step, Ru–C bond forms (complex 3). Complex 4, as a phenyl hydride complex, is the outcome of leaving the coordinated benzoic acid from complex 3, which is accomplished by deprotonation via a base in the reaction mixture. Subsequently, the CO2 molecule can link in a 2 mode, which results in compound 5. Finally, the C–C bond and complex 1 are formed, due to passing the final step through TS5–1.
Two different complexes A and B having 2,6-bis(di-tert-butylphosphino) methylpyridine ligand, PNP(tBu), and 2,6-bis(diphenylphosphino)methylpyridine, PNP(Ph), respectively, were considered as the catalysts of the reaction (Fig. 77a). Figure 78 depicts the PED of the reaction in the presence of these catalysts. Considering the ESM, the calculated TOF values of PNP(tBu) and PNP(Ph) are 1.6 \(\times\) 10–9 h−1 and 7.9 \(\times\) 10–3 h−1, respectively. This result shows that a small change in catalyst structure alters the TOF by six orders of magnitude. The next studied change is the outcome of the bonded substituents (R) to the phosphorous atom (Fig. 77b), in which the results show that the pyrrolidinyl group has the maximum increase in TOF = 8.6 h−1 relative to the phenyl group. The calculated results of TOF values corresponding to the variation of pincer backbone Y (Fig. 77c) show that the PONOP structure with an electron-releasing residue at the 4-position of the pyridine possesses the maximum TOF values (3.7 × 10–1 h−1). The variation of the anionic ligand X− (Fig. 77d) illustrates that the TOF quantity of the halide systems strongly related to the pKB value of the applied base, and thus, the studies are carried out in trimethylamine and 2,7-substituted 1,8-bis(diethylamino) naphthalenes (BDN). TOF values of Cl− and Br− in the presence of DBN are larger than trimethylamine. Therefore, these anions as the X ligand would be the favorite selection for experimental investigations. Moreover, the calculated TOF values in the gas phase, water, acetonitrile, acetone, and anisole illustrate that acetone strongly affects the TOF (1.6 × 10–2 h−1).

Studied variations in the analysis of the TOF values of the direct carboxylation of the C–H bonds with CO2 (Uhe et al. 2012)

PED of the reaction in the presence of PNP (tBu) and PNP(Ph) ligands (Ge et al. 2016)
As the final result, the most favorable TOF can be provided by the conditions that include substitute R = 1-pyrrolidinyl, backbone Y = PONOP, having an electron-releasing group at the 4-position, X = bromide as an anionic ligand in composition with a powerful base, and acetone as the solvent of reaction. Based on these choices, synthetic conditions could be recognized that leading to efficient catalysts with TOFs in the range of 10–2 to 10–5 h−1.
In another catalyst designing, a class of the PNP cobalt pincer complexes, inspired from the structure of the active site of [Fe]-hydrogenase, was reported for reversible base-free hydrogenation of CO2 to formic acid (Ge et al. 2016). All calculations were carried out by applying the M06-2X/6–31 + + G(d,p) level of theory for all atoms, in which the solvent effects of water on the optimized structures were considered by the integral equation formalism polarizable continuum model (IEFPCM) with SMD atomic radii solvent effect corrections. Figure 79 depicts the optimized structures of 1Co and its isomer 1Co′. Also, Fig. 80 illustrates the studied mechanism of the CO2 hydrogenation (Ge et al. 2016).

Optimized structures of PNP cobalt pincer complexes as the designed catalysts in CO2 conversion to formic acid (Ge et al. 2016)

Studied mechanism of the CO2 hydrogenation by PNP cobalt pincer complex (Ge et al. 2016)
The first step has been considered as a σ-complex formation between hydrogen molecule and cobalt complex, yielding intermediate 2. Then, similar to the FLPs, the ortho oxygen atom of acylmethylpyridinol ligand in intermediate 2 assists the H2 splitting by establishing a Co−Hδ−···Hδ+−O dihydrogen bond. Subsequently, intermediate 4 is produced through the CO2 activation as the formate anion by releasing the hydride from the Co atom. This step is considered as the CO2 insertion process, which is the RDS of the overall reaction. A formic acid molecule in intermediate 5 is produced due to the transformation of hydroxyl proton in acylmethylpyridinol to one of the oxygen atoms. Finally, the catalyst is regenerated by the release of HCOOH from 5. Regarding the principle of microscopic reversibility, the authors reported that the designed catalyst could be applied as an effective catalyst for the hydrogen abstraction of formic acid in organic solvents with a similar energy barrier. Also, this DFT-based design and mechanism investigation produce favorable catalysts for the affordable and high-efficiency reduction of CO2 and hydrogen abstraction of formic acid.
Mizuta and coworkers reported the CO2 conversion to methoxyborane in the presence of the borane molecules and sodium borohydride as a reducing agent, experimentally (Fujiwara et al. 2014). We reported a DFT study, in which the role of other borohydride salts of M[RBH3] (M = Li+, Na+, and R = Ph, CH3, CN, and H), as the reductive reagent, was evaluated (Fig. 81). The optimization of structures was carried out by the B3LYP/6–311 + + G(d,p) level of the theory, and the effect of tetrahydrofuran (THF) as the solvent of the reaction was considered by the CPCM model. Figure 82 depicts the studied mechanism for the CO2 reduction reaction (Sabet-Sarvestani et al. 2016).

Studied borohydride salts in the CO2 reduction reaction (Sabet-Sarvestani et al. 2016)

Proposed mechanism for the CO2 reduction (Sabet-Sarvestani et al. 2016)
In 3 is the product of the sequential CO2 reduction from steps 1 to 3 by hydride ions of the studied salts. In this intermediate, three hydride ions of the salt are replaced by three units of the formate ion. In step 4, In 5 (formate salt) and In 4 are made by the cleavage of the B–O bond of In 3. Then, In 5 is reduced by a borane molecule in step 5. Regarding the role of In 6 as the reducing agent, in step 6, the obtained formaldehyde molecule (In 7) is reduced to In 8, having a linked methoxy group to the boron atom. Steps 7 and 8 proceed through the oxidant role of In 4, in which other methoxy groups are formed. Finally, in step 9, due to hydride ion absorption by In 4 and a ring-closing reaction, (MeOBO)3 is formed (Sabet-Sarvestani et al. 2016).
Based on the roles of the linked substituents to the borohydride salts, the steps of reaction can be classified into three categories. Category 1 consists of steps 1–3, in which M[RB(OCHO)3] is produced due to the replication of hydride ions by formate ion. The results show that steps 5 and 6, classified as category 2, are independent of the substituent groups. These steps are similar for all considered borohydride salts. The steps of category 3 include the sequential oxidation of In 4, which leads to the ultimate product with three methoxy groups, and In 2 category includes steps 7–9. According to the calculated data, the reactions of categories 1 and 3 are influenced by the R substituents, only. Based on the theoretical results, the trend of ∆G≠ values in category 1 for the substituted moieties of the borohydride salt is CN > H > Ph > Me. Cyano and methyl substituents as the electron-withdrawing and electron-donating groups, respectively, have the highest and the lowest value of ∆G≠ in category 1. Also, it can be concluded that in this category, Li[MeBH3] is the best reductive agent for these kinds of reactions, kinetically. However, in category 3, electron-withdrawing substituent increases the tendency of hydride absorption via the boron atom of In 4. When Na[CNBH3] is used, the obtained ∆G≠ values are lower than the other reagents. Finally, from the kinetic and thermodynamic viewpoints of all steps for the general reaction, it is specified that Li[MeBH3] is the best borohydride salts for carbon dioxide transformation to methanol.
The role of the phosphorus ylides (P-ylides) chemistry in the novel synthetic protocols is undeniable. P-ylides are used in numerous reactions, mainly in the formation of natural products, biological and pharmacological compounds (Kolodiazhnyi 2008). In another DFT-based investigation, a new class of the P-ylides was designed for CO2 activation in which B3LYP/6–31 + G(d,p) and B3LYP/6–311 + G(2d,2p) levels were employed for the optimization of the involved compounds. Figure 83 depicts the CO2 activation reaction and the considered P-ylides (Sabet-Sarvestani et al. 2017c).

CO2 activation reaction and the considered P-ylides (Sabet-Sarvestani et al. 2017c)
Based on the obtained results, P-ylides having the aliphatic chains linked to the phosphorous atom have lower ΔG values than the others. The minimum value of the molecular electrostatic potential (MESP) (Ajitha and Suresh 2012) was employed for justification of the calculated ΔG values. MESP, as a quantifiable physical character, can be measured experimentally by the X-ray diffraction tool or theoretically determined by the electron density (Ajitha and Suresh 2012). Figure 84 depicts the obtained MESP isosurfaces and the values of minimum MESP (Vmin) at the bonded carbon atom to the phosphorous atom of P-ylides (C1). The results reveal that P-ylides having aliphatic chains, such as methyl, ethyl, and propyl, show the lower Vmin values. Generally, in the CO2 activation process, P-ylides having a more negative character of Vmin possess the lower ΔG. Thus, P(Me)3CH2, P(Et)3CH2, and P(Propyl)3CH2 with the minimum values of Vmin have the lowest ΔG. As a result, among the studied P-ylides, this class is the most efficient in the CO2 activation.

Calculated MESP isosurfaces and corresponding Vmin values (a.u) at the C1 atom of P-ylides (Sabet-Sarvestani et al. 2017c)
Experimental investigations show that PPh3CH2CO2 adduct, as the product of CO2 activation by P-ylides, can be used as an organocatalyst in CO2 conversion to cyclic α–alkylidene carbonates, oxazolidinone, and N-methylated and N-formylated amines (Zhou et al. 2015). These investigations reveal that in CO2 conversion, PPh3CH2CO2 adduct is a more prosperous catalyst than PPh3CH2 ylides. It is clear that the charge densities of the oxygen atoms of the P-ylide-CO2 adducts are more localized and have a lower interaction with the near orbitals than the carbon atom of P-ylides (C1). Figure 85 depicts the MESP isosurfaces and Vmin values (a.u) at the nucleophilic oxygen atom corresponding to the P(Me)3CH2CO2, P(Et)3CH2CO2, P(Propyl)3CH2CO2, and PPh3CH2CO2. As a result, the oxygen atom of P-ylide-CO2 adducts possesses a greater negative character of the Vmin, which makes them stronger nucleophilic catalysts than the P-ylides. Finally, thermodynamic and kinetic data show that P-ylides having the aliphatic chains, including P(Me)3CH2 and P(Et)3CH2, are the best choices for CO2 transformation.

MESP isosurfaces and the corresponding Vmin values (a.u) at the nucleophilic oxygen atom of P-ylide-CO2 adducts (Sabet-Sarvestani et al. 2017c)
5.1 Theoretical Designing: Problems and Opportunities
Several studies involving mechanism investigations and DFT-based designing have been presented in this chapter. Nowadays, computational molecular modeling and catalyst designing have matured and become an inseparable part of investigations. The developments in creating a realistic model for exploring the complex mechanisms and catalytic cycles have been significantly increased, and the field has been grown considerably. Regarding many reported and successful instances, it can be concluded that computational techniques can be applied in an anticipating sense to the logical design and optimization of novel categories of efficient catalysts (Ahn et al. 2019). However, the investigation of numerous possibilities in a DFT-based design is a challenge ahead.
In a catalytic CO2 transformation, depending on the given experimental conditions, various reaction paths might be involved and compete with each other. Thus, kinetic investigation for all possible paths, linking between the composition factors of the reactant, size, and the shape of the designed catalyst to predict the optimal catalytic conditions, needs a perfect evaluation. Hence, the obtained DFT predictions need to be validated by a complete experimental procedure (Bell et al. 2008). Unfortunately, the validation of the DFT-based design has been investigated very partially in the experimental reports, and more comprehensive investigations are remarkably required.
In an experimental study, the catalyst structures are critically significant for the design of a new catalytic cycle. Also, in a typical theoretical design, the prediction of the properties of the structure, size, composition, and shape of the catalysts has important in successful designing (Bell et al. 2008; Guo and Wang 2011; Cuenya 2010). Thus, the reported experimental catalytic structures based on characterization techniques, such as vibrational spectroscopy, X-ray crystallography, nuclear magnetic resonance (NMR), X-ray photoelectron spectroscopy (XPS), scanning electron microscopy (SEM) and transmission electron microscopy (TEM) are good inspirational criteria for DFT-based catalytic design. The combination of the developed theoretical and experimental characterization techniques will finally produce new visions into the design of more efficient catalysts for CO2 transformation (Somorjai and Li 2010).
The efficiency and the cost of the catalyst are also important issues in catalyst efficiency. In a DFT design, higher activity and lower cost must be considered in exploring more economical and efficient pathways. Especially, regarding industrial applications, it would be crucial to design non-expensive-metal catalysts instead of expensive-metal ones. Considering the global issue that the catalysts are used to solve, cost-effectiveness and higher activity are critical purposes in exploring and designing more economical and efficient paths. Moreover, the selectivity of the catalyst is the next important issue. This means that an increase in the yield of the reaction, energy efficiency, and higher selectivity decreases the operating costs of the catalytic cycle and produced by-products. Thus, the designing of the catalysts having efficient active sites is a method to increase the selectivity.
6 Conclusion
Global warming is a serious problem for humanity, and the CO2 transformation into fuels and value-added chemicals is a valuable approach for the limitation of atmospheric CO2 as a greenhouse gas. Furthermore, CO2 transformation can be considered as a cost-effective method for the manufacture of hydrocarbons and thus to attain sustainable CO2 recycling. Unfortunately, experimental catalytic cycles have drawbacks, such as low activity and selectivity of the applied catalysts. These issues are the main challenges in the CO2 utilization process. Accordingly, deep insights into conversion processes and novel catalyst evaluations are an undeniable requirement in this field. It can be concluded that understanding the reaction mechanisms of the catalytic conversion and the logical design of efficient catalysts are two essential concepts in CO2 conversions. Experimental approaches, based on trial-and-error methodologies, are very tedious, time-consuming, and frustrating. DFT-based techniques are promising strategies to overcome the disadvantages of the experimental approaches which could significantly improve the experimental sampling.
In this chapter, recent developed computational studies on the catalytic CO2 transformation to value-added materials, such as CO, CH3OH, CH4, HCOOH, and heterocyclic compounds, have been considered. The investigated catalysts include heterogeneous and homogeneous forms, organocatalysts, and also, the photo- and electro- catalysts. DFT calculations are a successful tool in specifying intermediates, exploring the efficient catalysts, and finding the probable reaction paths. Generally, the goals of a DFT study can be considered as an investigation of the kinetic aspects of a catalytic cycle, providing the optimized structures of the catalyst and helpful descriptors to explore the activity and selectivity of a catalyst. The validity of DFT results can be evaluated through the characterization of the catalytic structures and performing a controlled synthesis.
It can be anticipated that the future of using computational techniques in CO2 conversion reactions is brighter than ever. Improved software, availability of the required hardware, and development in the efficient electronic structure programs based on modern quantum chemistry are the effective factors in this field.
Abbreviations
- AFIL:
-
Amine-functionalized ionic liquid
- BDN:
-
1,8-Bis(diethylamino) naphthalenes
- PNP(Ph):
-
2,6-Bis(diphenylphosphino) methylpyridine
- PNP(tBu):
-
2,6-Bis(di-tert-butylphosphino) methylpyridine
- BCP:
-
Bond critical point
- CBM:
-
Conduction band minimum
- DFT:
-
Density functional theory
- DBN:
-
Diazabicyclo[4.3.0]non-5-ene
- DBU:
-
1,8-Diazabicyclo[5.4.0]undec-7-ene
- DNP:
-
Double numeric plus polarization
- ECP:
-
Effective core potentials
- ESM:
-
Energetic span model
- EGR:
-
Enhanced gas recovery
- EGS:
-
Enhanced geothermal systems
- EOR:
-
Enhanced oil recovery
- FLP:
-
Frustrated Lewis pair
- GGA:
-
Generalized gradient approximation
- GO:
-
Graphene oxide
- HOMO:
-
High occupied molecular orbital
- HER:
-
Hydrogen evolution reaction
- HFC:
-
Hydrofluorocarbon
- IEF:
-
Integral equation formalism
- IEFPCM:
-
Integral equation formalism polarizable continuum model
- IPCC:
-
International Panel on Climate Change
- N k :
-
Local nucleophilicity indices
- LUMO:
-
Low unoccupied molecular orbital
- MOF:
-
Metal−organic framework
- MESP:
-
Molecular electrostatic potential
- MO:
-
Molecular orbitals
- \(P_{k}^{ - }\) :
-
Mulliken atomic spin density
- NBO:
-
Natural bond orbital
- NHC:
-
N-heterocyclic carbine
- NHO:
-
N-heterocyclic olefin
- NHE:
-
Normal hydrogen electrode
- NMR:
-
Nuclear magnetic resonance
- PAW:
-
Projector-augmented wave
- PBE:
-
Perdew−Burke−Ernzerhof
- PW:
-
Perdew–Wang
- PFC:
-
Perfluorocarbon
- PR:
-
Phosphorous reagent
- P-ylide:
-
Phosphorus ylide
- PCM:
-
Polarizable continuum model
- PED:
-
Potential energy diagram
- TsCl:
-
P-Toluenesulfonyl chloride
- QTAIM:
-
Quantum theory of atoms in molecules
- RRKJ:
-
Rappe Rabe Kaxiras Joannopoulos
- RDS:
-
Rate-determining step
- RWGS:
-
Reverse water gas shift
- SEM:
-
Scanning electron microscopy
- SMD:
-
Solvation model based on density
- SFLP:
-
Surface frustrated Lewis pairs
- SB:
-
Superbase
- TBAB:
-
Tetrabutylammonium bromide
- THF:
-
Tetrahydrofuran
- TMG:
-
1,1,3,3-Tetramethylguanidine
- TDI:
-
TOF-determining intermediate
- TDTS:
-
TOF-determining transition state
- TEM:
-
Transmission electron microscopy
- TBD:
-
1,5,7-Triazabicyclo[4.4.0]dec-5-ene
- TOF:
-
Turnover frequency
- VSEPR:
-
Valence shell electron pair repulsion theory
- vdW:
-
Van der Waals
- WGSR:
-
Water gas shift reaction
- XPS:
-
X-ray photoelectron spectroscopy
References
Abeydeera UW, Hewage L, Wadu Mesthrige J, Samarasinghalage TI (2019) Global Research on Carbon Emissions: A Scientometric Review. Sustainability 11(14):3972
Ahn S, Hong M, Sundararajan M, Ess DH, Baik M-H (2019) Design and optimization of catalysts based on mechanistic insights derived from quantum chemical reaction modeling. Chem Rev 119(11):6509–6560
Ajitha MJ, Suresh CH (2012) Assessment of stereoelectronic factors that influence the CO2 fixation ability of N-heterocyclic carbenes: A DFT study. J Org Chem 77(2):1087–1094
Álvarez A, Borges M, Corral-Pérez JJ, Olcina JG, Hu L, Cornu D, Huang R, Stoian D, Urakawa A (2017) CO2 activation over catalytic surfaces. ChemPhysChem 18(22):3135–3141
Andersson MP, Bligaard T, Kustov A, Larsen KE, Greeley J, Johannessen T, Christensen CH, Nørskov JK (2006) Toward computational screening in heterogeneous catalysis: pareto-optimal methanation catalysts. J Catal 239(2):501–506
Aoyagi N, Furusho Y, Endo T (2013) Effective synthesis of cyclic carbonates from carbon dioxide and epoxides by phosphonium iodides as catalysts in alcoholic solvents. Tetrahedron Lett 54(51):7031–7034
Aresta M (2010) Carbon dioxide as chemical feedstock, Wiley Online Library
Aresta M, Dibenedetto A (2007) Utilisation of CO2 as a chemical feedstock: opportunities and challenges. Dalton Trans 28:2975–2992
Aresta M, Nobile CF, Albano VG, Forni E, Manassero M (1975) New nickel–carbon dioxide complex: synthesis, properties, and crystallographic characterization of (carbon dioxide)-bis (tricyclohexylphosphine) nickel. J Chem Soc. Chem Commun 15:636–637
Aresta M, Dibenedetto A, Quaranta E (2016) Reaction mechanisms in carbon dioxide conversion. Springer, Heidelberg
Artz J, Müller TE, Thenert K, Kleinekorte J, Meys R, Sternberg A, Bardow A, Leitner W (2018) Sustainable conversion of carbon dioxide: an integrated review of catalysis and life cycle assessment. Chem Rev 118(2):434–504
Aziz M, Jalil A, Triwahyono S, Ahmad A (2015) CO2 methanation over heterogeneous catalysts: recent progress and future prospects. Green Chem 17(5):2647–2663
Richard F, Bader R (1990) Atoms in molecules: a quantum theory, Oxford University Press
Bartlett RJ, Musiał M (2007) Coupled-cluster theory in quantum chemistry. Rev Mod Phys 79(1):291
Bell AT, Gates BC, Ray D, Thompson MR (2008) Basic research needs: catalysis for energy. The U.S. Department of Energy's Office of Scientific and Technical Information
Ben-Nun M, Martínez TJ (2002) Ab initio quantum molecular dynamics. Adv Chem Phys 121:439–512
Bensaid S, Centi G, Garrone E, Perathoner S, Saracco G (2012) Towards artificial leaves for solar hydrogen and fuels from carbon dioxide. Chemsuschem 5(3):500–521
Bertau M, Offermanns H, Menges G, Keim W, Effenberger F (2010) Methanol needs more attention as a fuel and raw material for the future. Chem Ing Tech (weinh) 82(12):2055–2058
Biswas S, Khatun R, Dolai M, Biswas IH, Haque N, Sengupta M, Islam MS, Islam SM (2020) Catalytic formation of N3-substituted quinazoline-2, 4 (1H, 3H)-diones by Pd (ii) EN@ GO composite and its mechanistic investigations through DFT calculations. New J Chem 44(1):141–151
Bontemps S (2016) Boron-mediated activation of carbon dioxide. Coord Chem Rev 308:117–130
Brickner SJ (1996) Oxazolidinone antibacterial agents. Curr Pharm Des 2(2):175–194
RJ BS, Loganathan M, Shantha MS (2010) A review of the water gas shift reaction kinetics. Int J Chem React Eng 8(1):1–32
Calabrese C, Giacalone F, Aprile C (2019) Hybrid catalysts for CO2 conversion into cyclic carbonates. Catalysts 9(4):325
Cao Z, Guo L, Liu N, Zheng X, Li W, Shi Y, Guo J, Xi Y (2016) Theoretical study on the reaction mechanism of reverse water–gas shift reaction using a Rh–Mo 6 S 8 cluster. RSC Adv 6(110):108270–108279
Centi G, Perathoner S (2009) Opportunities and prospects in the chemical recycling of carbon dioxide to fuels. Catal Today 148(3–4):191–205
Centi G, Quadrelli EA, Perathoner S (2013) Catalysis for CO2 conversion: a key technology for rapid introduction of renewable energy in the value chain of chemical industries. Energy Environ Sci 6(6):1711–1731
Chatelet B, Joucla L, Dutasta J-P, Martinez A, Szeto KC, Dufaud V (2013) Azaphosphatranes as structurally tunable organocatalysts for carbonate synthesis from CO2 and epoxides. J Am Chem Soc 135(14):5348–5351
Chen C-S, Cheng W-H, Lin S-S (2000) Mechanism of CO formation in reverse water–gas shift reaction over Cu/Al2O3 catalyst. Catal Lett 68(1–2):45–48
Chen Y, Hone CA, Gutmann B, Kappe CO (2017) Continuous flow synthesis of carbonylated heterocycles via Pd-catalyzed oxidative carbonylation using CO and O2 at elevated temperatures and pressures. Org Process Res Dev 21(7):1080–1087
Chen J, Gao H, Ding T, Ji L, Zhang JZ, Gao G, Xia F (2019) Mechanistic studies of CO2 cycloaddition reaction catalyzed by amine-functionalized ionic liquids. Front Chem 7:615
Cheng D, Negreiros FR, Apra E, Fortunelli A (2013) Computational approaches to the chemical conversion of carbon dioxide. Chemsuschem 6(6):944–965
Chiang C-L, Lin K-S, Chuang H-W (2018) Direct synthesis of formic acid via CO2 hydrogenation over Cu/ZnO/Al2O3 catalyst. J Clean Prod 172:1957–1977
Choi S, Sang B-I, Hong J, Yoon KJ, Son J-W, Lee J-H, Kim B-K, Kim H (2017) Catalytic behavior of metal catalysts in high-temperature RWGS reaction: In-situ FT-IR experiments and first-principles calculations. Sci Rep 7:41207
Chorkendorff I, Niemantsverdriet JW (2017) Concepts of modern catalysis and kinetics. John Wiley & Sons
Cokoja M, Bruckmeier C, Rieger B, Herrmann WA, Kühn FE (2011) Transformation of carbon dioxide with homogeneous transition-metal catalysts: a molecular solution to a global challenge? Angew Chem Int Ed 50(37):8510–8537
Costentin C, Robert M, Savéant J-M (2013) Catalysis of the electrochemical reduction of carbon dioxide. Chem Soc Rev 42(6):2423–2436
Cuenya BR (2010) Synthesis and catalytic properties of metal nanoparticles: Size, shape, support, composition, and oxidation state effects. Thin Solid Films 518(12):3127–3150
Dai Z, Stauffer PH, Carey JW, Middleton RS, Lu Z, Jacobs JF, Hnottavange-Telleen K, Spangler LH (2014) Pre-site characterization risk analysis for commercial-scale carbon sequestration. Environ Sci Technol 48(7):3908–3915
Dang S, Yang H, Gao P, Wang H, Li X, Wei W, Sun Y (2019) A review of research progress on heterogeneous catalysts for methanol synthesis from carbon dioxide hydrogenation. Catal Today 330:61–75
Darensbourg DJ (2010) Chemistry of carbon dioxide relevant to its utilization: a personal perspective. Inorg Chem 49(23):10765–10780
Domingo LR, Pérez P, Sáez JA (2013) Understanding the local reactivity in polar organic reactions through electrophilic and nucleophilic Parr functions. RSC Adv 3(5):1486–1494
Dong X, Liu X, Chen Y, Zhang M (2018) Screening of bimetallic M-Cu-BTC MOFs for CO2 activation and mechanistic study of CO2 hydrogenation to formic acid: A DFT study. J CO2 Util 24:64–72
Falivene L, Kozlov SM, Cavallo L (2018) Constructing bridges between computational tools in heterogeneous and homogeneous catalysis. ACS Catal 8(6):5637–5656
Fang S, Chen H, Wei H (2018) Insight into catalytic reduction of CO2 to methane with silanes using Brookhart’s cationic Ir (iii) pincer complex. RSC Adv 8(17):9232–9242
Filonenko GA, Vrijburg WL, Hensen EJ, Pidko EA (2016) On the activity of supported Au catalysts in the liquid phase hydrogenation of CO2 to formates. J Catal 343:97–105
Fiorani G, Guo W, Kleij AW (2015) Sustainable conversion of carbon dioxide: the advent of organocatalysis. Green Chem 17(3):1375–1389
Frontera P, Macario A, Ferraro M, Antonucci P (2017) Supported catalysts for CO2 methanation: a review. Catalysts 7(2):59
Fujiwara K, Yasuda S, Mizuta T (2014) Reduction of CO2 to Trimethoxyboroxine with BH3 in THF. Organometallics 33(22):6692–6695
Galvan M, Selva M, Perosa A, Noè M (2014) Toward the design of halide-and metal-free ionic-liquid catalysts for the cycloaddition of CO2 to epoxides. Asian J Org Chem 3(4):504–513
Gao J, He L-N, Miao C-X, Chanfreau S (2010) Chemical fixation of CO2: efficient synthesis of quinazoline-2, 4 (1H, 3H)-diones catalyzed by guanidines under solvent-free conditions. Tetrahedron 66(23):4063–4067
Gattrell M, Gupta N, Co A (2007) Electrochemical reduction of CO2 to hydrocarbons to store renewable electrical energy and upgrade biogas. Energy Convers Manag 48(4):1255–1265
Ge H, Jing Y, Yang X (2016) Computational design of cobalt catalysts for hydrogenation of carbon dioxide and dehydrogenation of formic acid. Inorg Chem 55(23):12179–12184
Gershikov A, Spiridonov V (1983) Anharmonic force field of CO2 as determined by a gas-phase electron diffraction study. J Mol Struct 96(1–2):141–149
Girard A-L, Simon N, Zanatta M, Marmitt S, Gonçalves P, Dupont J (2014) Insights on recyclable catalytic system composed of task-specific ionic liquids for the chemical fixation of carbon dioxide. Green Chem 16(5):2815–2825
Guharoy U, Ramirez Reina T, Gu S, Cai Q (2019) Mechanistic Insights into Selective CO2 Conversion via RWGS on Transition Metal Phosphides: A DFT Study. J Phys Chem C 123(37):22918–22931
Guo S, Wang E (2011) Noble metal nanomaterials: controllable synthesis and application in fuel cells and analytical sensors. Nano Today 6(3):240–264
Han X, Yang J, Han B, Sun W, Zhao C, Lu Y, Li Z, Ren J (2017) Density functional theory study of the mechanism of CO methanation on Ni4/t-ZrO2 catalysts: roles of surface oxygen vacancies and hydroxyl groups. Int J Hydrog Energy 42(1):177–192
Henkelman G, Arnaldsson A, Jónsson H (2006) A fast and robust algorithm for Bader decomposition of charge density. Comput Mater Sci 36(3):354–360
Hofmann DJ, Butler JH, Tans PP (2009) A new look at atmospheric carbon dioxide. Atmospheric Environ 43(12):2084–2086
Hu B, Guild C, Suib SL (2013) Thermal, electrochemical, and photochemical conversion of CO2 to fuels and value-added products. J CO2 Util 1:18–27
Huang C-H, Tan C-S (2014) A review: CO2 utilization. Aerosol Air Qual Res 14(2):480–499
Hunt AJ, Sin EH, Marriott R, Clark JH (2010) Generation, capture, and utilization of industrial carbon dioxide. Chemsuschem 3(3):306–322
Inoue Y, Izumida H, Sasaki Y, Hashimoto H (1976) Catalytic fixation of carbon dioxide to formic acid by transition-metal complexes under mild conditions. Chem Lett 5(8):863–864
Jing H, Li Q, Wang J, Liu D, Wu K (2018) Theoretical study of the reverse water gas shift reaction on copper modified β-Mo2C (001) surfaces. J Phys Chem C 123(2):1235–1251
Jones G, Jakobsen JG, Shim SS, Kleis J, Andersson MP, Rossmeisl J, Abild-Pedersen F, Bligaard T, Helveg S, Hinnemann B (2008) First principles calculations and experimental insight into methane steam reforming over transition metal catalysts. J Catal 259(1):147–160
Joule JA, Mills K (2010) Heterocyclic chemistry, 5th (Ed.), John Wiley & Sons, Publication Ltd., Blackwell Publishing Ltd, West Sussex, UK
Kayaki Y, Yamamoto M, Ikariya T (2009) N-heterocyclic carbenes as efficient organocatalysts for CO2 fixation reactions. Angew Chem Int Ed 48(23):4194–4197
Keim W, Offermanns H (2010) Beyong oil and gas: early visions of the natives of Aachen. Nachr Chem 58(4):434–435
Kheirabadi R, Izadyar M, Housaindokht MR (2018) Computational kinetic modeling of the catalytic cycle of glutathione peroxidase nanomimic: effect of nucleophilicity of thiols on the catalytic activity. J Phys Chem A 122(1):364–374
Kohn W (1999) Nobel Lecture: Electronic structure of matter—wave functions and density functionals. Rev Mod Phys 71(5):1253
Kohn W, Sham LJ (1965) Self-consistent equations including exchange and correlation effects. Phys Rev 140(4A):A1133
Kolodiazhnyi OI (2008) Phosphorus ylides: chemistry and applications in organic synthesis. John Wiley & Sons
Kozuch S, Shaik S (2011) How to conceptualize catalytic cycles? The energetic span model. Acc Chem Res 44(2):101–110
Kwon IS, Debela TT, Kwak IH, Seo HW, Park K, Kim D, Yoo SJ, Kim J-G, Park J, Kang HS (2019) Selective electrochemical reduction of carbon dioxide to formic acid using indium–zinc bimetallic nanocrystals. J Mater Chem A 7(40):22879–22883
Lapidus A, Gaidai N, Nekrasov N, Tishkova L, Agafonov YA, Myshenkova T (2007) The mechanism of carbon dioxide hydrogenation on copper and nickel catalysts. Pet Chem 47(2):75–82
Lee S-Y, Park S-J (2015) A review on solid adsorbents for carbon dioxide capture. J Ind Eng Chem 23:1–11
Liu Q, Wu L, Jackstell R, Beller M (2015) Using carbon dioxide as a building block in organic synthesis. Nat Commun 6:5933
Liu M, Yi Y, Wang L, Guo H, Bogaerts A (2019) Hydrogenation of carbon dioxide to value-added chemicals by heterogeneous catalysis and plasma catalysis. Catalysts 9(3):275
Luther Iii GW (2004) Kinetics of the reactions of water, hydroxide ion and sulfide species with CO2, OCS and CS2: frontier molecular orbital considerations. Aquat Geochem 10(1–2):81–97
Ma J, Sun N, Zhang X, Zhao N, Xiao F, Wei W, Sun Y (2009) A short review of catalysis for CO2 conversion. Catal Today 148(3–4):221–231
Ma J, Hu J, Lu W, Zhang Z, Han B (2013) Theoretical study on the reaction of CO2 and 2-aminobenzonitrile to form quinazoline-2, 4 (1 H, 3 H)-dione in water without any catalyst. Phys Chem Chem Phys 15(40):17333–17341
Ma S, Song W, Liu B, Zhong W, Deng J, Zheng H, Liu J, Gong X-Q, Zhao Z (2016) Facet-dependent photocatalytic performance of TiO2: A DFT study. Appl Catal B 198:1–8
Maslin M (2008) Global warming: a very short introduction. Oxford University Press
Mikkelsen M, Jørgensen M, Krebs FC (2010) The teraton challenge. A review of fixation and transformation of carbon dioxide. Energy Environ Sci 3(1):43–81
Mizuno T, Okamoto N, Ito T, Miyata T (2000a) Synthesis of 2, 4-dihydroxyquinazolines using carbon dioxide in the presence of DBU under mild conditions. Tetrahedron Lett 41(7):1051–1053
Mizuno T, Okamoto N, Ito T, Miyata T (2000b) Synthesis of quinazolines using carbon dioxide (or carbon monoxide with sulfur) under mild conditions. Heteroat Chem 11(6):428–433
Mukherjee A, Okolie JA, Abdelrasoul A, Niu C, Dalai AK (2019) Review of post-combustion carbon dioxide capture technologies using activated carbon. J Environ Sci 83:46–63
Muradov N (2014) Industrial utilization of CO2: a win–win solution. In: liberating energy from carbon: introduction to decarbonization. Springer, pp 325–383
Murphy LJ, Robertson KN, Kemp RA, Tuononen HM, Clyburne JA (2015) Structurally simple complexes of CO2. Chem Commun 51(19):3942–3956
Nakamura S, Hatakeyama M, Wang Y, Ogata K, Fujii K (2015) A basic quantum chemical review on the activation of CO2. In: advances in CO2 capture, sequestration, and conversion. ACS Publications, Chapter 5, pp 123–134
Nale DB, Saigaonkar SD, Bhanage BM (2014) An efficient synthesis of quinazoline-2, 4 (1H, 3H)-dione from CO2 and 2-aminobenzonitrile using [Hmim] OH/SiO2 as a base functionalized supported ionic liquid phase catalyst. J CO2 Util 8:67–73
Niemi T, Repo T (2019) Antibiotics from carbon dioxide: sustainable pathways to pharmaceutically relevant cyclic carbamates. Eur J Org Chem 2019(6):1180–1188
Niemi T, Fernández I, Steadman B, Mannisto JK, Repo T (2018) Carbon dioxide-based facile synthesis of cyclic carbamates from amino alcohols. Chem Commun 54(25):3166–3169
Nørskov JK, Bligaard T, Rossmeisl J, Christensen CH (2009) Towards the computational design of solid catalysts. Nat Chem 1(1):37
Nørskov JK, Abild-Pedersen F, Studt F, Bligaard T (2011) Density functional theory in surface chemistry and catalysis. Proc Natl Acad Sci USA (PNAS) 108(3):937–943
IEA OECD (2016) Energy and air pollution: world energy outlook (Special Report)
Ola O, Maroto-Valer MM, Mackintosh S (2013) Turning CO2 into valuable chemicals. Energy Procedia 37:6704–6709
Olah GA, Goeppert A, Prakash GS (2009) Chemical recycling of carbon dioxide to methanol and dimethyl ether: from greenhouse gas to renewable, environmentally carbon neutral fuels and synthetic hydrocarbons. J Org Chem 74(2):487–498
Olah GA, Goeppert A, Prakash GS (2018) Beyond oil and gas: the methanol economy. Third updated and enlarged edition, John Wiley & Sons (US)
Ou L, Chen J, Chen Y, Jin J (2019) Mechanistic study on Cu-catalyzed CO2 electroreduction into CH4 at simulated low overpotentials based on an improved electrochemical model. Phys Chem Chem Phys 21(28):15531–15540
Paparo A, Okuda J (2017) Carbon dioxide complexes: bonding modes and synthetic methods. Coord Chem Rev 334:136–149
Peng G, Sibener S, Schatz GC, Mavrikakis M (2012) CO2 hydrogenation to formic acid on Ni (110). Surf Sci 606(13–14):1050–1055
Podrojkova N, Sans V, Orinak A, Orinakova R (2020) Recent developments in heterogeneous catalysts modelling for CO2 conversion to chemicals. ChemCatChem 12(7):1802–1825
Pople JA (1999) Nobel lecture: Quantum chemical models. Rev Mod Phys 71(5):1267
Qiu M, Tao H, Li R, Li Y, Huang X, Chen W, Su W, Zhang Y (2016) Insight into the mechanism for the methanol synthesis via the hydrogenation of CO2 over a Co-modified Cu (100) surface: A DFT study. J Chem Phys 145(13):134701
Rachuri Y, Kurisingal JF, Chitumalla RK, Vuppala S, Gu Y, Jang J, Choe Y, Suresh E, Park D-W (2019) Adenine-based Zn (II)/Cd (II) metal-organic frameworks as efficient heterogeneous catalysts for facile CO2 fixation into cyclic carbonates: a DFT-supported study of the reaction mechanism. Inorg Chem 58(17):11389–11403
Rafiee A, Khalilpour KR, Milani D, Panahi M (2018) Trends in CO2 conversion and utilization: a review from process systems perspective. J Environ Chem Eng 6(5):5771–5794
Rashidi NA, Yusup S (2016) An overview of activated carbons utilization for the post-combustion carbon dioxide capture. J CO2 Util 13:1–16
Rawat KS, Mahata A, Pathak B (2017) Thermochemical and electrochemical CO2 reduction on octahedral Cu nanocluster: role of solvent towards product selectivity. J Catal 349:118–127
Ren J, Guo H, Yang J, Qin Z, Lin J, Li Z (2015) Insights into the mechanisms of CO2 methanation on Ni (111) surfaces by density functional theory. Appl Surf Sci 351:504–516
Riduan SN, Zhang Y (2010) Recent developments in carbon dioxide utilization under mild conditions. Dalton Trans 39(14):3347–3357
Sabet-Sarvestani H, Eshghi H, Bakavoli M, Izadyar M, Rahimizadeh M (2014) Theoretical investigation of the chemoselectivity and synchronously pyrazole ring formation mechanism from ethoxymethylenemalononitrile and hydrazine hydrate in the gas and solvent phases: DFT, meta-GGA studies and NBO analysis. RSC Adv 4(82):43485–43495
Sabet-Sarvestani H, Eshghi H, Izadyar M, Noroozi-Shad N, Bakavoli M, Ziaee F (2016) Borohydride salts as high efficiency reducing reagents for carbon dioxide transformation to methanol: Theoretical approach. Int J Hydrog Energy 41(26):11131–11140
Sabet-Sarvestani H, Eshghi H, Izadyar M (2017a) Understanding the mechanism, thermodynamic and kinetic features of the Kukhtin-Ramirez reaction in carbamate synthesis from carbon dioxide. RSC Adv 7(3):1701–1710
Sabet-Sarvestani H, Eshghi H, Izadyar M (2017b) A theoretical study on the efficiency and role of guanidines-based organic superbases on carbon dioxide utilization in quinazoline-2, 4 (1H, 3H)-diones synthesis. Struct Chem 28(3):675–686
Sabet-Sarvestani H, Izadyar M, Eshghi H (2017) Phosphorus ylides as a new class of compounds in CO2 activation: Thermodynamic and kinetic studies. J CO2 Util 21:459–466
Sabet-Sarvestani H, Izadyar M, Eshghi H, Noroozi-Shad N, Bakavoli M (2018) Proton sponge as a new efficient catalyst for carbon dioxide transformation to methanol: theoretical approach. Fuel 221:491–500
Sabet-Sarvestani H, Izadyar M, Eshghi H, Norozi-Shad N (2020) Evaluation and understanding the performances of various derivatives of carbonyl-stabilized phosphonium ylides in CO2 transformation to cyclic carbonates. Phys Chem Chem Phys 22(1):223–237
Saeidi S, Amin NAS, Rahimpour MR (2014) Hydrogenation of CO2 to value-added products—a review and potential future developments. J CO2 Util 5:66–81
Saeidi S, Najari S, Fazlollahi F, Nikoo MK, Sefidkon F, Klemeš JJ, Baxter LL (2017) Mechanisms and kinetics of CO2 hydrogenation to value-added products: a detailed review on current status and future trends. Renew Sust Energ Rev 80:1292–1311
Sakakura T, Choi J-C, Yasuda H (2007) Transformation of carbon dioxide. Chem Rev 107(6):2365–2387
Saxena R, Singh VK, Kumar EA (2014) Carbon dioxide capture and sequestration by adsorption on activated carbon. Energy Procedia 54:320–329
Schaadt R, Sweeney D, Shinabarger D, Zurenko G (2009) In vitro activity of TR-700, the active ingredient of the antibacterial prodrug TR-701, a novel oxazolidinone antibacterial agent. Antimicrob Agents Chemother 53(8):3236–3239
Schilling W, Das S (2018) CO2-catalyzed/promoted transformation of organic functional groups. Tetrahedron Lett 59(43):3821–3828
Scibioh M, Viswanathan B (2018) Chapter 5—heterogeneous hydrogenation of CO2. Carbon Dioxide Chem Fuels 191–253
Sehested J, Larsen KE, Kustov AL, Frey AM, Johannessen T, Bligaard T, Andersson MP, Nørskov JK, Christensen CH (2007) Discovery of technical methanation catalysts based on computational screening. Top Catal 45(1–4):9–13
Shih CF, Zhang T, Li J, Bai C (2018) Powering the future with liquid sunshine. Joule 2(10):1925–1949
Siqueira RM, Freitas GR, Peixoto HR, do Nascimento JF, Musse APS, Torres AE, Azevedo DC, Bastos-Neto M (2017) Carbon dioxide capture by pressure swing adsorption. Energy Procedia 114:2182–2192
Sirijaraensre J, Limtrakul J (2016) Hydrogenation of CO2 to formic acid over a Cu-embedded graphene: A DFT study. Appl Surf Sci 364:241–248
Somorjai GA, Li Y (2010) Major successes of theory-and-experiment-combined studies in surface chemistry and heterogeneous catalysis. Top Catal 53(5–6):311–325
Song Q-W, Zhou Z-H, He L-N (2017) Efficient, selective and sustainable catalysis of carbon dioxide. Green Chem 19(16):3707–3728
Spigarelli BP, Kawatra SK (2013) Opportunities and challenges in carbon dioxide capture. J CO2 Util 1:69–87
Spinner NS, Vega JA, Mustain WE (2012) Recent progress in the electrochemical conversion and utilization of CO2. Catal Sci Technol 2(1):19–28
Streitwieser A (2009) Perspectives on computational organic chemistry. J Org Chem 74(12):4433–4446
Su X, Yang X, Zhao B, Huang Y (2017) Designing of highly selective and high-temperature endurable RWGS heterogeneous catalysts: recent advances and the future directions. J Energy Chem 26(5):854–867
Topham S, Bazzanella A, Schiebahn S, Luhr S, Zhao L, Otto A, Stolten D (2000) Carbon dioxide. Ullmann’s encyclopedia of industrial chemistry, wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany
Tsutsumi Y, Yamakawa K, Yoshida M, Ema T, Sakai T (2010) Bifunctional organocatalyst for activation of carbon dioxide and epoxide to produce cyclic carbonate: betaine as a new catalytic motif. Org Lett 12(24):5728–5731
Uhe A, Hoelscher M, Leitner W (2012) Carboxylation of arene C. H Bonds with CO2: aDFT‐based approach to catalyst design. Chem–A Eur J 18(1):170–177
Wang SR, Radosevich AT (2013) Reductive homocondensation of benzylidene-and alkylidenepyruvate esters by a P (NMe2) 3-mediated tandem reaction. Org Lett 15(8):1926–1929
Wang S, Xi C (2019) Recent advances in nucleophile-triggered CO2-incorporated cyclization leading to heterocycles. Chem Soc Rev 48(1):382–404
Wang W, Wang S, Ma X, Gong J (2011) Recent advances in catalytic hydrogenation of carbon dioxide. Chem Soc Rev 40(7):3703–3727
Wang J-Q, Dong K, Cheng W-G, Sun J, Zhang S-J (2012) Insights into quaternary ammonium salts-catalyzed fixation carbon dioxide with epoxides. Catal Sci Technol 2(7):1480–1484
Wang Y-B, Wang Y-M, Zhang W-Z, Lu X-B (2013) Fast CO2 sequestration, activation, and catalytic transformation using N-heterocyclic olefins. J Am Chem Soc 135(32):11996–12003
Wang L, Sun W, Liu C (2018a) Recent advances in homogeneous carbonylation using CO2 as CO surrogate. Chinese J Chem 36(4):353–362
Wang L, Ghoussoub M, Wang H, Shao Y, Sun W, Tountas AA, Wood TE, Li H, Loh JYY, Dong Y (2018b) Photocatalytic hydrogenation of carbon dioxide with high selectivity to methanol at atmospheric pressure. Joule 2(7):1369–1381
Wang W-H, Feng X, Bao M (2018) Transformation of CO2 to formic acid or formate with homogeneous catalysts. In: transformation of carbon dioxide to formic acid and methanol. Springer, pp 7–42
Wei W, Jinlong G (2011) Methanation of carbon dioxide: an overview. Front Chem Sci Eng 5(1):2–10
Weigend F, Ahlrichs R (2005) Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys Chem Chem Phys 7(18):3297–3305
Wong WL, Chan PH, Zhou ZY, Lee KH, Cheung KC, Wong KY (2008) A robust ionic liquid as reaction medium and efficient organocatalyst for carbon dioxide fixation. Chemsuschem 1(1–2):67–70
Wu Z, Cole J, Fang HL, Qin M, He Z (2017) Revisiting catalyst structure and mechanism in methanol synthesis. J Adv Nanomater (JAN) 2(1):1–10
Wu P, Zaffran J, Yang B (2019) Role of surface species interactions in identifying the reaction mechanism of methanol synthesis from CO2 hydrogenation over intermetallic PdIn (310) steps. J Phys Chem C 123(22):13615–13623
Yaashikaa P, Kumar PS, Varjani SJ, Saravanan A (2019) A review on photochemical, biochemical and electrochemical transformation of CO2 into value-added products. J CO2 Util 33:131–147
Yan X, Guo H, Yang D, Qiu S, Yao X (2014) Catalytic hydrogenation of carbon dioxide to fuels. Curr Org Chem 18(10):1335–1345
Yang Z-Z, He L-N, Zhao Y-N, Li B, Yu B (2011) CO2 capture and activation by superbase/polyethylene glycol and its subsequent conversion. Energy Environ Sci 4(10):3971–3975
Yang L, Pastor-Pérez L, Gu S, Sepúlveda-Escribano A, Reina T (2018) Highly efficient Ni/CeO2-Al2O3 catalysts for CO2 upgrading via reverse water-gas shift: effect of selected transition metal promoters. Appl Catal B 232:464–471
Yaumi A, Bakar MA, Hameed B (2017) Recent advances in functionalized composite solid materials for carbon dioxide capture. Energy 124:461–480
Yu KK, Curcic I, Gabriel J, Morganstewart H, Tsang SC (2010) Catalytic coupling of CO2 with epoxide over supported and unsupported amines. J Phys Chem A 114(11):3863–3872
Zhang Y-J, Da Y-B (2015) The decomposition of energy-related carbon emission and its decoupling with economic growth in China. Renew. Sust. Energ. Rev. 41:1255–1266
Zhang Q, Guo L (2018) Mechanism of the reverse water-gas shift reaction catalyzed by Cu12TM bimetallic nanocluster: a density functional theory study. J Clust Sci 29(5):867–877
Zhang X, Liu J-X, Zijlstra B, Filot IA, Zhou Z, Sun S, Hensen EJ (2018a) Optimum Cu nanoparticle catalysts for CO2 hydrogenation towards methanol. Nano Energy 43:200–209
Zhang W, Wang S, Zhao Y, Ma X (2018b) Hydrogenation of CO2 to formic acid catalyzed by heterogeneous Ru-PPh3/Al2O3 catalysts. Fuel Process Technol 178:98–103
Zhao W, Fink DM, Labutta CA, Radosevich AT (2013) A Csp3–Csp3 bond forming reductive condensation of α-Keto esters and enolizable carbon pronucleophiles. Org Lett 15(12):3090–3093
Zhong J, Yang X, Wu Z, Liang B, Huang Y, Zhang T (2020) State of the art and perspectives in heterogeneous catalysis of CO2 hydrogenation to methanol. Chem Soc Rev 49(5):1385–1413
Zhou H, Wang G-X, Zhang W-Z, Lu X-B (2015) CO2 adducts of phosphorus ylides: highly active organocatalysts for carbon dioxide transformation. ACS Catal 5(11):6773–6779
Zhu L, Ye J-H, Duan M, Qi X, Yu D-G, Bai R, Lan Y (2018) The mechanism of copper-catalyzed oxytrifluoromethylation of allylamines with CO2: a computational study. Inorg Chem Front 5(4):633–639
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Sabet-Sarvestani, H., Izadyar, M., Eshghi, H., Noroozi-Shad, N. (2022). Theoretical Approaches to CO2 Transformations. In: Inamuddin, Boddula, R., Ahamed, M.I., Khan, A. (eds) Carbon Dioxide Utilization to Sustainable Energy and Fuels. Advances in Science, Technology & Innovation. Springer, Cham. https://doi.org/10.1007/978-3-030-72877-9_8
Download citation
DOI: https://doi.org/10.1007/978-3-030-72877-9_8
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-72876-2
Online ISBN: 978-3-030-72877-9
eBook Packages: Earth and Environmental ScienceEarth and Environmental Science (R0)