
Abstract
The present study focuses on the hybrid plasma catalytic process for CO2 methanation. This plasma-catalytic process, based on the combination of a DBD plasma and Ni/CeZrO2 catalyst, has several advantages over conventional catalysis: it operates at ambient conditions and requires no external heating. An optimization of the process considering the effect of the different operational parameters such as voltage, GHSV, catalyst mass, flow rate, discharge length, is herein presented. Moreover, a spectroscopic study, aiming to understand the mechanism of the reaction, is also showed. At temperatures around 270 °C and under adiabatic conditions, CO2 conversion rates of about 80% were measured, with a CH4 selectivity greater than 95%.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
In the last century, the CO2 concentration in the atmosphere has constantly increased from 280 ppm in the beginning of the industrial revolution to ~ 390 ppm in 2010, at a rate of ca. 1% per year [1, 2]. The increasing concern about the consequences of this increasing CO2 atmospheric concentration has led to the implementation of different measures in order to decrease, or at least control, the anthropogenic emissions of this greenhouse gas. Carbon capture and sequestration (CCS) technologies are considered to be an efficient way for CO2 the stabilization of CO2 emissions [3, 4]. The conversion of the captured CO2 into value-added chemicals and liquid fuels is considered as one of the main challenges for the 21st century. The utilization of this waste and its conversion it into a new feedstock not only complies with the framework of sustainable and green chemistry but also fits within the ‘cradle-to-cradle’ concept [5]. By generating useful products out of CO2, we create the possibility to effectively close the carbon loop. However, CO2 is a very stable molecule whose activation requires important amounts of energy.
Among the different routes for the chemical utilization of CO2, its hydrogenation to methane and/or other fuels, offers a good opportunity for sustainable development in the energy and environmental sectors [6, 7]. More concretely, methane has a wide range of applications in the industry and civil use, and is also used to produce some downstream products, such as ethyne, hydrogen, and ammonia [8, 9]. CO2 conversion to methanol and dimethyl ether is still very low (~ 20%) and it is difficult to achieve higher conversion of CO2 [10, 11]. Methane production from CO2 at low temperature represents also an important challenge for this chemical utilization of CO2 [12]. Although CO2 conversion into methane is exothermic and thermodynamically favorable at ambient temperature, a catalyst and high temperatures (> 350 °C) are needed in order to achieve acceptable methane yield [13].
Nickel containing catalysts have been often proposed and considered for this methanation reaction. Different supports have been also used, such as alumina, silica, glasses, clays and zeolites [14, 15]. Among them, the use of ceria–zirconia mixed oxides led to interesting results in terms of activity and selectivity [16]. Indeed, the presence of cerium oxide favors the storage and the mobility of oxygen at the surface [13]. Since the methanation reaction is exothermic, the excessive heat of reaction may induce metal sintering, leading to an important decrease of the catalytic activity. However, even in the presence of a catalysts, CO2 methanation proceeds at very low rates at temperatures lower than 350 °C. Higher temperatures are thus needed in order to obtain acceptable methane yields. Working at temperatures higher than 350 °C is only possible at high pressures, i.e., around 20–30 bar, in order to hinder the side reactions leading to the production of CO.
We have recently proved that the association of a catalyst with a non-thermal plasma, i.e. a dielectric-barrier discharge plasma (DBD), can lead to boosted CO2 methanation [16, 17]. This coupled plasma-catalytic process presents several advantages vis-à-vis conventional catalysis, since it operates at ambient pressure and proceeds in the absence of external heating. The aim of the present work is to study the effect of the modification of different system parameters, such voltage, GHSV, catalyst mass, flow rate, discharge length and discharge configuration (post discharge and in situ discharge), in order to optimize both the methane yield and the energy efficiency of the process.
Experimental setup and diagnosis
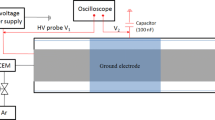
The activity towards CO2 methanation under different reaction conditions, i.e. different applied voltages, different weight of catalyst was tested in an experimental set-up briefly consisting of a quartz tubular cylindrical reactor, a plasma generator and different devices for gas analysis. A schematic of the installation is shown in Fig. 1.

Schematic diagram of the experimental setup used in plasma-catalysis methanation of CO2 process
The dielectric barrier discharge (DBD) plasma reactor consists of two coaxial tubes (quartz and alumina tubes). Between the outer tube (10 mm internal diameter, 1 mm wall thickness) and the inner (3 mm diameter) the discharge is sustained in a gap of 2.5 mm with a steel wire wrapped around the outer surface of the quartz tube acting as the ground electrode and grounded via an external capacitor Cext (2 nF). The reactor was loaded with 300 mg of catalyst (grain size < 0.2 mm). The effective length of the plasma-catalytic reactor is approximately 6.5 mm. On both sides of the catalytic bed, glass wool was placed in order to keep the bed fixed within the discharge zone. The catalyst used was a nickel-based catalyst with ceria–zirconia mixed oxide as a support; with a loading of 15 wt% of Ni on a Ce0.58Zr0.42O2 support (15NiCZ5842). This catalyst was chosen since it was already presented as potential catalyst for such kind of process [16, 18]. Its preparation and characterization is presented elsewhere [16, 18]. Conventional thermal reduction of the catalyst was performed in a H2 stream for 2 h with a total gas flow of 40 mL/min (mixture 50% H2/Ar) at temperature 470 °C with a heating rate 5 °C/min [19]. The in situ plasma reduction on the catalyst was also considered and performed in the following conditions: 160 mL/min of pure H2 was passed through the reactor for 20 min while plasma was generated at 15 kV. In all the methanation experiments, a gas mixture of pure hydrogen and carbon dioxide with a H2/CO2 molar ratio of 4:1 was fed to the reactor.
The reaction temperature was measured by a K-type thermocouple placed close to the catalytic bed and with the help of an infrared gun OMEGA directed onto the outer electrode, after regulating the emissivity of this latter using Stefan–Boltzman law of radiation and by comparing the values obtained with the k-thermocouple. The emissivity of the electrode was found equal to 0.38. The reactor was eventually heated (when needed), using a Leister hot air tool directed to the sides of the reactor and over the length of the catalytic bed. Note that all experiments were conducted at atmospheric pressure.
In the hybrid methanation experiments, an alternative current (AC) high voltage (operating frequency: 40–41 kHz) of 14 until 18 kV was applied to the discharging electrode to create non-thermal plasma. The voltage applied to the plasma-catalytic reactor was measured with a digital oscilloscope (LT 342, LeCroy) using a probe (ELDITEST GE 3830). The product stream passed through a short stainless-steel tube into an unheated section of tubing that led to the collector section of the apparatus. Water formed as a product was continuously removed and condensed from the product gas thanks to a condenser. The outlet gas flowrate is different from that of the input, thus it is important to know its value. In our case, it was determined using a bubble flowmeter. A specific sensor was used to analyze CO2, CH4 and H2. A gas chromatograph (Agilent MicroGC 490) equipped with thermal conductivity detector (TCD) has been used to analyze the outlet gas mixture from the reactor. Thanks to this device, we can found the composition of the gas mixture and thus the conversion and yield presented below by knowing the outlet flow rate.
Optical emission spectroscopy (OES) is an in situ method to monitor the excited and ionized molecules and ions in a gas discharge. It allows the partial determination of the plasma composition without exerting any influence over it [20,21,22]. Plasma emission spectra were recorded with a monochromator spectrometer type Czerny Turner with three diffraction gratings (150, 1200 and 3600 grooves/mm) coupled with a CCD camera. The opening of the slit was set at 100 µm and the optical fiber was placed vertically along the plasma with a constant distance of 2 mm from the quartz tube. OES spectra were acquired at wavelengths from 200 to 800 nm and 100 ms as acquisition time. Electric power is an indispensable parameter since it expresses the total amount of charges transferred by the plasma. Due to the plasma is a series of microdischarges of short duration and the current waveform needed to capture this information accurately, thus U–I method is difficult to perform [23]. Lissajou’s method introduced by Manley in 1943 [24] is now recognized to be the most precise method for DBD power determination. This method requires the measurement of the input voltage and the voltage (Uc) across a capacitor displayed in oscilloscope the latter is obtained thanks to a capacity placed in series with the reactor. The capacitor accumulates a charge from the current flowing over the reactor and this can be determined by measuring the voltage on the capacitor (Uc) as shown below:
Thus, by plotting the charge versus high voltage which is known as a Q–U Lissajou’s plot by recording the values for Q(t) and U(t) via a series of regularly sampled points which capture the full cycle of the AC sinusoidal wave (Fig. 2), we can obtain the discharge power dissipated in the reactor which is equal to the area of Q-U Lissajou’s plot [19, 24,25,26].

Q–U Lissajous plot
Results and discussion
Optimization of experimental parameters
Influence of voltage
Fig. 3 displays the results of the plasma catalytic methanation applying different voltages. In this case methanation was performed using a total flow of 200 mL/min (160 mL/min H2;40 mL/min CO2) and 300 mg of catalysts corresponding to a length of the outer electrode of 6.5 mm. The catalysts was reduced in situ (plasma) during 20 min and under pure H2 (160 mL/min) at a voltage of 15 kV. The results in Fig. 3a show that at 14.5 kV, a conversion of CO2 of approximately 10% was achieved. Conversion sharply increased to 57% at 15 kV, reaching a maximal 71% conversion at 16 kV that remains afterwards approximately constant. We can therefore conclude that, under the reaction conditions considered, the starting point of the methanation reaction is placed between 14.5 and 15 kV.

a Evolution of the conversion of CO2, selectivity of CH4 and the power of the discharge. b The temperature versus applying voltages
Fig. 3b presents the evolution of the temperature as a function of the voltage applied. It can be observed that the temperature increase between 14.5 and 15 kV is more important than for the subsequent 1 kV voltage increases, pointing to the initiation of the exothermic methanation reaction between these two voltages. The increase in temperature improves the plasma energy that can be seen by the increase of the power values that could be directly related with the dissociation of molecules of carbon dioxide [27]. As an electric field is applied on the catalyst, which is related to the complex permittivity value of the sample material [18, 28], the adsorption can be induced in the catalyst by enhancement of this electric field [29]. Besides that, the adsorption–desorption equilibrium of the carbon monoxide and dioxide molecules is strongly affected by the occurrence of micro-discharges in the catalytic bed [30].
The selectivity towards methane formation reaches ca. 100% at 15 and 16 kV. At higher voltage, selectivity decreases, i.e. 82% at 18 kV (337 °C), pointing to CO formation occurring to a certain extent, in agreement with literature [13, 18]. At the lowest reaction temperature (178 °C) selectivity is also low, indicating that the main role of plasma at low temperature may be only related to the direct splitting of CO2 [31]. Although the dissociation of CO has been reported to be the rate-determining step for the CO2 methanation reaction [32, 33], the CO produced may be adsorbed on the surface catalyst and may not be desorbed at very low reaction temperatures, needing more energy to be desorbed from the surface and to be subsequently converted onto CH4.
Influence of the GHSV
The GHSV is the ratio of the volumetric flow rate of the reactants at standard temperature and pressure to the total catalyst volume. Higher GHSV can thus be related to shorter contact times. The results obtained in the plasma-catalytic methanation of CO2 at different GHSV are shown in Fig. 4 and Table 1.

Evolution of the conversion of CO2 and selectivity of CH4 at different GHSVs: a, b the weight of catalyst equal to 200 mg. c, d The weight equal to 100 mg
As expected, lower GHSVs result in a longer contact time and therefore in a higher CO2 conversion. Abate et al. obtained similar results, for the conventional thermo-catalytic methanation of CO2 [34]. Under plasma-catalytic conditions the power communicated to the catalytic reactor depends both on the mass of the catalyst used and of the total gas flow. The power is always lower when higher amounts of catalyst are used and increases with increasing flow rate.
Influence of the specific input energy
The specific input energy (SIE) is defined as the amount of energy spent per liter of reactant or product gas, as shown in Eq. 7.
This specific input energy (SIE) is one of the leading parameters that determines the reactor performance in methanation using DBD plasma [35]. In this case, the total flow rate is kept constant while the input power was varied from 14.5 to 18 kV. Indeed, the input voltage is one of the significant factor for generating reactive species such as radicals, ions and excited species that should be under consideration. Higher SIEs at constant flow rate will then increase the strength of the discharge (and the energy level of the provided electrons), thus leading to a greater extent of molecular fragmentation [36]. Moreover, increasing the SIE can directly increase the reactor temperature (see Fig. 5). This is due to more intense collisions between the gas particles taking place and to the presence of more energetic electrons, resulting therefore in higher conversion of CO2 with increasing SIE. However, a decrease of CH4 selectivity is observed at high temperatures due to the formation of CO (from RWGS reaction or even direct CO2 splitting) [13, 18].

Evolution of conversion of CO2, selectivity of CH4 (a) and temperature (b) with the specific input energy
Effect of reactor configuration
In order to optimize our plasma/catalytic system, we decided to study the influence of the contact of plasma with catalyst bed. Fig. 6 presents the two main configurations considered. In the so-called two-stage configuration, Fig. 6a, the catalyst is spatially separated from the plasma region, and generally placed downstream. In the second, the so-called one-stage configuration Fig. 6b, the catalyst is placed within the discharge region. In a traditional thermal catalysis experiment, molecules are dissociatively adsorbed onto the catalyst with the energy being supplied in the form of heat.

Plasma-catalytic configurations
As commented before, in plasma-assisted catalysis, species are activated by the plasma due to excitation, ionization or dissociation by electrons in the gas phase or on the catalyst surface [23, 31]. The major difference between the one-stage and two-stage configuration is the kind of species the catalyst can be exposed to. In the two-stage configuration, the end-products and the long-life intermediates will interact with the catalyst, while in the one-stage configuration, the catalyst can also interact with all the short-life species, including excited species, radicals, photons and electrons. In the two-stage, off-plasma configuration, the contact between the catalyst and short-living active species is avoided as well as the contact with plasma itself, which can affect the surface of the catalyst [37] and modify important properties such as its adsorption capacity [38], active sites, their stability or activity.
The influence of the type of configuration was studied under the already mentioned conditions, i.e. total flow of 200 mL/min (160 mL/min H2;40 mL/min CO2) and 300 mg of catalysts corresponding to a length of the outer electrode of 6.5 mm. The catalyst was again reduced in situ (plasma) during 20 min and under pure H2 (160 mL/min) at a voltage of 15 kV. Fig. 7 depicts the conversion of CO2, selectivity of CH4 and the power of the discharge as a function of the voltage applied, for this in-plasma configuration. The results obtained have been already discussed within this text. Briefly, conversion increases with the applied voltage, remaining then more or less stable, while power increases constantly with increasing voltage.

Evolution of the conversion of CO2, selectivity of CH4 and the power of the discharge versus different voltages for the in-plasma configuration
Fig. 8 presents the optical emission spectra of the CO2/H2 plasmas registered during one of this in-plasma configuration experiments. Excited CO and Hα species can be detected at 297.7 and 656.3 nm, respectively [39].

Emission spectra of the CO2/H2 plasmas
Fig. 9 presents the results obtained when using the two-stage, off-plasma configuration. The resulting power communicated to the system notably differs from that measured for the in-plasma configuration. The discharge can more easily take place in an empty reactor instead of in the bulk of a solid catalytic bed, and this will moreover depend on the porosity and particle size of the catalyst [27]. In the in-plasma system, the presence of catalyst seems to improve significantly the conversion and the selectivity towards methane. Inside the catalytic bed, the formation of hot spots inside the catalytic bed which can contribute to the improvement of CO2 ionization. Using the off-plasma configuration at 15 kV lower CO2 conversion and CH4 selectivity are obtained, in comparison to those obtained in the in-plasma system. However, at higher voltages, i.e. 16 kV, the results are approximately the same except for the power, which is higher for the off-plasma configuration.

Evolution of the conversion of CO2, selectivity of CH4 and the power of the discharge versus different applying voltages
Additional experiments were performed covering only half of the length of the catalytic bed with the outer electrode. The results obtained were intermediate of the in- and off-plasma configurations. Moreover, optical emission spectroscopy (OES) showed very similar results in terms of the excited species produced. Both CO and Hα species were detected at 297.7 and 656.3 nm respectively [39], but no remarkable difference was observed. Since the plasma discharge may be only promoting CO formation through CO2 splitting, the results obtained indicate that the main role of the catalyst is to direct the selectivity of this process, i.e. from non-selective one (Non thermal plasma) to a selective one (transform CO2 dissociated species to CH4). The lowest values of conversion and selectivity in the post-plasma configuration at 15 kV point to the discharge not being intense enough in order to create the different excited species (radicals, ions…), thus the CO2 is not highly splitted and interacts with H2 on the surface of the catalyst to a lower extent.
Proposed mechanism of the plasma-catalytic methanation reaction
Fig. 10 shows the CO and Hα regions of the OES spectra acquired in the presence of the DBD plasma in an empty reactor (A), in the presence of the CeO2–ZrO2 support (B) and of the 15NiCZ5842 catalysts (C).

CO and Hα regions in the optical emission spectra of the CO2/H2 plasmas, in a empty reactor, b CeO2–ZrO2 support, and c Ni-containing catalysts
The generation of a CO2/H2 plasma into the empty reactor results itself in the formation of several excited species in the gaseous phase which increase the reaction rate at low temperature, i.e. CO (297.7 nm, Fig. 10a), Hα (656.3 nm, Fig. 10a) and O (777.5 nm, not shown). In the presence of the Ni-containing catalyst 15NiCZ5842, the OES peaks for excited CO and Hα almost disappear (Fig. 10c) pointing to its adsorption or reaction on the surface of the solid material. The ceria–zirconia support is also able to interact to a certain extent with some of the excited species created by the plasma. However, only CO adsorption is observed, whereas the Hα peak remains visible. Indeed, the adsorption and further conversion of Hα excited species is only possible in the presence of the Ni-active sites. Although the ceria–zirconia support contributes to the adsorption of CO and/or CO2 related species and may contribute as well to oxygen transfer, the presence of Ni is crucial for the methanation reaction. Moreover, under conventional thermo-catalytic methanation, CO2 is adsorbed and dissociated on the catalyst surface, forming adsorbed CO species (or mono-carbonyl species). This step has been demonstrated several times using infrared spectroscopy and X-ray photoelectron spectroscopy [18, 37, 40]. This adsorption–dissociation step is thermodynamically favored even at ambient temperature (ΔG0 = − 130.8 kJ/mol) [15]. However, the reduction of the fully oxidized carbon to methane is an eight-electron process with significant kinetic limitations that requires a good catalyst that can achieve acceptable rates and selectivity [17].
According to these observations, the following mechanism can be proposed: (1) Since plasma can dissociate CO2, already at low temperatures adsorbed CO-active species will be formed on the catalyst surface, (2) the DBD plasma can also produce excited H species that will adsorb on the Ni0 sites, finally (3) the adsorbed CO and H species will react producing CH adsorbed species that will subsequently incorporate (4) one more H atom, and (5) a third one, leading to the formation of methane that will finally leave the catalyst surface.
The dissociation of CO2 is irreversible owing to rapid removal of surface O by hydrogenation. Step 5 of methane desorption is also irreversible. Both Step 3 and step 4 occurring after the rate-determining steps, as pointed out by Choe et al. [40]. Though the present mechanism explains the positive effect of the plasma-catalytic coupling, as stated before, the DBD plasma may also modify the properties of the catalytic surface, and more detailed studies are needed considering this fact.
Conclusions
In the present work, a hybrid plasma catalytic system for CO2 methanation based on the combination of a DBD plasma and Ni/CeZrOx (15NiCZ5842) catalyst was presented. The hybrid plasma catalytic process was active at low temperature (< 270 °C) on the selective conversion of CO2 into methane. At low temperature and in the absence of plasma, the conversion of CO2 and selectivity to CH4 were about 30%, but they were drastically enhanced and in the presence of plasma reaching 73–75% of conversion, and 100% of selectivity at temperatures between 200 and 300 °C. In the presence of a plasma discharge, the CO2 is activated yielding both CO* and O* species, even at low temperatures, which are able to interact with the catalyst surface as well as with excited H* species, also adsorbed on Ni0 sites. The most effective results were obtained at voltages between 15 and 16 kV and under adiabatic conditions, i.e. without any external heating. Avoiding external heating and reducing the used voltage can have a huge positive economic impact in the reaction, especially at a large scale.
The optimum reaction temperature, at which the highest conversion and selectivity was reached, was found to be between 230 and 270 °C. Below 230 °C, the conversion and the selectivity of CH4 are rather low, due the strong adsorption of the CO produced in the presence of a plasma, needing higher temperatures for desorption and conversion onto CH4. At temperatures higher than 300 °C, side reactions such as the reverse water gas shift reaction (RWGS), start contributing to CO formation and result in decreased selectivity. This study also showed that at high voltages the reaction temperature does not have a huge impact on the methane yield. At low voltages, increasing the reactor temperature by 30–60 °C can make a significant difference. Moreover, the GHSV impact was also evidenced. At 16 kV conversion and selectivity become maximal at the lowest GHSV. Post-plasma configuration resulted in lower methane yield, though the power measured was several times lower than for the in-plasma configuration. Optical emission spectroscopy evidenced the formation of CO, H and O excited species. CO excited species were able to interact both with the ceria–zirconia support and with the Ni-containing catalysts. H excited species were only adsorbed on the surface of the Ni-catalyst. At the sight of these results we proposed a reaction mechanism involving the reaction of both adsorbed CO and H species.
References
Wang W, Wang S, Ma X, Gong J (2011) Chem Soc Rev 40:3703–3727. https://doi.org/10.1039/C1CS15008A
Xiaoding X, Moulijn JA (1996) Energy Fuels 10:305–325. https://doi.org/10.1021/ef9501511
Perry RJ, O’Brien MJ (2011) Energy Fuels 25:1906–1918. https://doi.org/10.1021/ef101564h
Wang J, Huang L, Yang R, Zhang Z, Wu J, Gao Y, Wang Q, O’Hare D, Zhong Z (2014) Energy Environ Sci 7:3478–3518. https://doi.org/10.1039/c4ee01647e
McDonough W, Braungart M, Anastas P, Zimmerman J (2003) Environ Sci Technol 37:434A–441A. https://doi.org/10.1021/es0326322
Jessop PG, Joo F, Tai CC (2004) Coord Chem Rev 2004248:2425–2442. https://doi.org/10.1016/j.ccr.2004.05.019
Qin Z, Zhou Y, Jiang Y, Liu Z, Ji H (2017) Recent advances in heterogeneous catalytic hydrogenation of CO2 to methane. N Adv Hydrog Process Fundam Appl 2:5. https://doi.org/10.5772/65407
Bai M, Zhang Z, Bai M, Bai X, Gao H (2008) Plasma Chem Plasma Process 28:405–414. https://doi.org/10.1007/s11090-008-9132-4
Zeng Y, Wang G (2005) Chem Eng Oil Gas 34:89–93. https://doi.org/10.1039/b614276a
Bonura G, Cordaro M, Cannilla C, Arena F, Frusteri F (2014) Appl Catal B 152–153:152–161. https://doi.org/10.1016/j.apcatb.2014.01.035
R-w Liu, Z-z Qin, H-b Ji, Su T-m (2013) Ind Eng Chem Res 52:16648–16655. https://doi.org/10.1021/ie401763g
Beuls A, Swalus C, Jacquemin M, Heyen G, Karelovic A, Ruiz P (2012) Appl Catal B 113–114:2–10. https://doi.org/10.1016/j.apcatb.2011.02.033
Ocampo F, Louis B, Roger A-C (2009) Appl Catal Gen 369:90–96. https://doi.org/10.1016/j.apcata.2009.09.005
Cerveny L (1986) Catalytic hydrogenation. In: Delmon B, Yates JT (eds) Studies in surface science and catalysis. Elsevier, Amsterdam. ISBN 0-444-42682-5
Krämer M et al (2009) The impact of dopants on the activity and selectivity of a Ni-based methanation catalyst. Appl Catal A 369:42–52. https://doi.org/10.1016/j.apcata.2009.08.027
Nizio M, Albarazi A, Cavadias S, Amouroux J, Galvez ME, Da Costa P (2016) Int J Hydrog Energy 41:11584–11592. https://doi.org/10.1016/j.ijhydene.2016.02.020
Jwa E, Lee SB, Lee HW, Mok YS (2013) Fuel Process Technol 108:89–93. https://doi.org/10.1016/j.fuproc.2012.03.008
Aldana PAU et al (2013) Catal Today 215:201–207. https://doi.org/10.1016/j.cattod.2013.02.019
Peeters FJJ, Sanden MC (2015) Plasma Sources Sci Technol 24(1):015016. https://doi.org/10.1016/j.cej.2016.02.047
Laux CO et al (2003) Plasma Sources Sci Technol 12:125. https://doi.org/10.1088/0963-0252/12/2/301
Gaydon AG, Pearse RWB (1976) The identification of molecular spectra. Springer, Dordrecht. ISBN 978-94-009-5760-2
Zhang J-F et al (2009) Chin Phys Lett 26:035203. https://doi.org/10.1088/0256-307X/26/3/035203
Fridman A (2008) Plasma chemistry. Cambridge University Press, Cambridge. https://doi.org/10.1017/CBO9780511546075
Manley TC (1943) The electric characteristics of the ozonator discharge. Trans Electrochem Soc 84:83–96. https://doi.org/10.1149/1.3071556
Xin Tu et al (2011) J Phys D 44:274007. https://doi.org/10.1088/0022-3727/44/27/274007
Valdivia-Barrientos R et al (2006) Plasma Sources Sci Technol 15:237. https://doi.org/10.1088/0963-0252/15/2/008
Zhang Y-P, Ma P-S, Zhu X, Liu C-J, Shen Y (2004) Catal Commun 5:35–39. https://doi.org/10.1016/j.catcom.2003.11.006
Chen HL, Lee HM, Chen SH, Chao Y, Chang MB (2008) Catal B 85:1–9. https://doi.org/10.1016/j.apcatb.2008.06.021
Amouroux J, Cavadias S (2017) J Phys D 50:46550. https://doi.org/10.1088/1361-6463/aa8b56
Brooks KP et al (2007) Chem Eng Sci 62:1161–1170. https://doi.org/10.1016/j.ces.2006.11.020
Snoeckx R, Bogaerts A (2016) Chem Soc Rev 46:5805–5863. https://doi.org/10.1039/c6cs00066e
Schaaf T, Grünig J, Schuster MR, Rothenfluh T, Orth A (2014) Energy Sustain Soc 4:1–14. https://doi.org/10.1186/s13705-014-0029-1
Kogelschatz U (2003) Plasma Chem Plasma Process 23:1. https://doi.org/10.1023/A:1022470901385
Abate S, Mebrahtu C, Giglio E, Deorsola F, Bensaid S, Perathoner S, Pirone RN, Centi G (2016) Ind Eng Chem Res 55:4451–4460. https://doi.org/10.1021/acs.iecr.6b00134
Jiang T, Li Y, Liu C-J, Xu G-H, Eliasson B, Xue B (2002) Catal Today 72:229–235. https://doi.org/10.1016/S0920-5861(01)00497-7
Chung W-C, Chang M-B (2016) Energy Convers Manag 124:305–314. https://doi.org/10.1016/j.enconman.2016.07.023
Beuls A et al (2012) Appl Catal B 113–114:2–10. https://doi.org/10.1016/j.apcatb.2011.02.033
Neyts EC, Bogaerts A (2014) J Phys D 47:224010. https://doi.org/10.1088/0022-3727/47/22/224010
Herzberg G (1944) Atomic spectra and atomic structure. Dover Publications, New York. ISBN 0-486-60115-3
Choe SJ et al (2005) Bull Korean Chem Soc 26:1682–1688. https://doi.org/10.5012/bkcs.2005.26.11.168
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Mikhail, M., Wang, B., Jalain, R. et al. Plasma-catalytic hybrid process for CO2 methanation: optimization of operation parameters. Reac Kinet Mech Cat 126, 629–643 (2019). https://doi.org/10.1007/s11144-018-1508-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11144-018-1508-8