
Abstract
This paper deals with the study of the bimetallic Pd–Cu and monometallic Cu, Pd catalyst used in the methanol synthesis reaction. To reveal the influence of the catalyst properties on methanol production, various characterization techniques were used, such as Fourier transform infrared (FTIR) spectroscopy, X-ray diffraction, TPR-H2 and TPD-NH3. The catalytic activity was studied in CO hydrogenation under elevated pressure (4.8 MPa) in a fixed bed high-pressure reactor. The activity results confirmed the promotion effect of palladium and CeO2 on the catalytic activity of copper catalysts. The highest activity and selectivity of Pd–Cu/ZnAl2O4–5 % CeO2 is explained by the Pd–Cu alloy formation during activation process. The presence of adsorption species attributed to b-HCO3–Ce, b-HCOO–Ce and b-HCOO–Ce species on the surface of CeO2 promoted catalysts was confirmed by FTIR method.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Methanol synthesis is one of the basic chemical processes in the world, because it is the substrate for many chemicals e.g., formaldehyde, chloromethane, amines, acetic acid, methyl methacrylate. The increased interest in issues of methanol production methods may be due to the possibility of its use as a fuel (e.g., fuel cells) to produce electricity and its processing into high-octane methyl tert-butyl ether and dimethyl terephthalate [1–4].
In the twenties of the previous century in Germany, there was a growing interest in high-pressure processes (so-called “hydrerungs verfahrung” processes), which contributed to the development of technological processes, including: ammonia synthesis “Haber - Bosch” synthesis, “hydrogen - desulfurization” process, Fischer–Tropsch synthesis and methanol synthesis from synthesis gas.
In 1922, M. Pier produced methanol in BASF’s laboratory. The process required high pressure (250–350 atm) and high temperature (320–450 °C), due to the operating conditions of the process it was named “high-pressure methanol synthesis”. It was a process widely used for over 45 years, in which production of methanol [5, 6] was carried out from synthesis gas obtained by gasification of coal or brown coal, using relatively resistant to poisoning ZnO/Cr2O3 catalyst.
Unfavorable conditions of the “high-pressure methanol synthesis” process led to a search for new, more efficient and selective catalysts for enabling operation of the methanol synthesis in a much milder conditions. The research led to the development of a copper catalyst, which in the 1966 has been improved by the company ICI (Imperial Chemical Industries, nowadays Synetix) by introducing Zn. As it turned out, zinc is an ideal dispersing agent of copper on the catalyst surface [7–9]. Promotion of copper catalyst by zinc caused an increase of the catalytic activity, which allowed to conduct the methanol synthesis under milder conditions, i.e., at a lower pressure 35–55 atm and in the lower temperature range of 200–300 °C. The process was named “low pressure methanol synthesis”. Currently, this is used for the methanol synthesis process on an industrial scale [1, 3, 10].
Typical catalysts used in industrial low-pressure methanol synthesis are systems containing copper, zinc and aluminum oxides. The molar ratio of the catalyst components varies depending on the manufacturer, but the content of CuO is contained in the range 40–80 %, ZnO 10–30 %, Al2O3 from 5 to 10 %. There are also various types of promoters of both structural and textural (e.g., MgO), but a detailed description of promoters and their content is protected by the manufacturers [1].
Many researchers claimed that the oxygen vacancies in the metal oxide support play an important role in the catalysis. Li et al. [11] investigated the adsorption of CO on partially reduced CeO2 by Fourier transform infrared spectroscopy (FTIS). CO adsorbed on the oxygen vacancy of pre-reduced CeO2 is turned into formate species. A possible proposed mechanism is that the activated CO molecules on reduced surface sites interact with the nearby OH groups to form formate species via an unstable intermediate, formyl species [11]. Considering those studies it was expected that the oxygen vacancies of CeO2 could provide effective active sites for CO adsorption during methanol synthesis.
In addition, CeO2 has a high oxygen storage capacity and excellent redox properties [12–14]. However, these properties disappeared at high temperature due to its poor thermal stability [15].
Palladium supported CeO2 catalysts have been reported to be effective for the hydrogenation of CO and CO2 to methanol. Pd/CeO2 also exhibits a more stable reaction activity for methanol synthesis from syngas containing sulfur in comparison to Pd/Al2O3 catalyst [16].
Shen et al. [17] studied the Pd/CeO2 supported catalyst in methanol synthesis reaction from CO and H2 and claimed that this catalytic system exhibited high activity due to the strong interaction between palladium and ceria. The interaction caused a change in the electronic charge density at the Pd surface.
A possible way to improve the activity of copper-based catalysts is to create multifunctional catalysts containing intermetallic or alloy compound [18–21] on the catalysts surface. The formation of these alloy or intermetallic compounds had to be carried out directly during activation process of studied systems. These compounds are thought to be a key factor to achieve highly active and selective systems.
Another way to improve the activity of copper catalysts is the introduction of the textural promoter such as CeO2 into the support structure, which may create structural defects to facilitate the mobility of lattice oxygen and change the redox properties.
Based on the above suggestion in this study, we prepared multifunctional systems and investigated the effect of palladium and CeO2 on the catalytic activity and selectivity of Cu/ZnAl2O4 catalyst in the methanol synthesis reaction. Using X-ray diffraction (XRD), FTIR, temperature programmed reduction (TPR)-H2, and NH3-TPD, the chemical and physicochemical properties of the investigated catalysts were characterized and correlated with the activity results in CO hydrogenation.
Experimental section
Catalysts preparation
Copper catalysts were prepared by wet aqueous impregnation. In order to prepare the ZnAl2O4 support, the following molar ratio of Zn:Al = 0.5 zinc and aluminum nitrates were used. Aqueous solutions of 1 M zinc nitrate (Zn(NO3)2·6H2O) and 1 M aluminum nitrate (Al(NO3)3·9H2O) were mixed in appropriate quantity under vigorous stirring at 80 °C. A concentrated ammonia solution was then added by dropwise until the pH reached values of between 10 and 11, and then the mixtures were stirred for another 30 min. The resulting fine precipitates were washed two times in deionized water and then dried at 120 °C for 15 h and calcined for 4 h at 600 °C in air. Calcination at 600 °C was chosen because such high temperature is required for creation of spinel structure and for removal of the nitrate precursor from the catalytic systems.
Supports ZnAl2O4–1 % CeO2 and ZnAl2O4–5 % CeO2 were prepared by the impregnation method. An appropriate quantity of cerium nitrate was taken to obtain relevant content of CeO2 and introduced on the carrier (ZnAl2O4) surface. The obtained material was further dried at 120 °C for 2 h and then calcined in air at 400 °C for 4 h.
Metal phase Cu and Pd was introduced on the previously prepared support surface (ZnAl2O4, ZnAl2O4–1 % CeO2 and ZnAl2O4–5 % CeO2) by wet impregnation using aqueous solutions of copper and palladium nitrates. The supported catalysts were then dried in air at 120 °C for 2 h and finally calcined for 4 h in air at temperatures of 400 °C. Copper and palladium loadings were 20 and 2 wt%, respectively.
Catalysts characterization
Temperature programmed reduction (TPR-H2)
The TPR-H2 measurements were carried out in an automatic TPR system AMI-1 in the temperature range of 25–900 °C with a linear heating rate of 10 °C min−1. Samples (weight about 0.1 g) were reduced in hydrogen stream (5 % H2–95 % Ar) with a volumetric flow rate of 40 cm3 min−1. Hydrogen consumption was monitored by a thermal conductivity detector.
XRD measurements
Room temperature powder XRD patterns were collected using a PANalytical X’Pert Pro MPD diffractometer in Bragg–Brentano reflecting geometry. Copper Cu Kα radiation from a sealed tube was used. Data were collected in the range 5–90° 2θ with a step of 0.0167° and exposure per step of 27 s. Due to the fact that raw diffraction data contain some noise, the background during the analysis was subtracted using Sonneveld, E.J. and Visser algorithm. The data was then smoothed using cubic polynomial. All calculations were done using X’Pert HighScore Plus computer software.
Temperature programmed desorption of ammonia (TPD-NH3)
The procedure of catalyst surface acidity measurement includes the purification of catalyst surface, adsorption of ammonia and TPD. Catalyst sample purification (about 0.15 g), removal of water from the system leading to surface dehydroxylation, was completed in pure argon stream with a volume velocity of 40 cm3 min−1 at 400 °C for 2 h and after cooling sample to 100 °C argon stream was switched into a gaseous ammonia flowing 0.5 h through catalyst sample to entirely saturate its surface. After NH3 adsorption, the excessive amount of weakly and physically adsorbed ammonia on the catalyst surface had been removed by flushing of the catalyst in pure argon stream with volume velocity of 40 cm3 min−1. A TPD-NH3 run was done in a temperature range of 100–500 °C with a linear heating rate of 27 °C min−1 and a thermo-conductivity detector to obtain a TPD-NH3 profile.
Catalytic activity test
Activity tests in the methanol synthesis reaction were carried out using the high pressure fixed bed reactor using a gas mixture of H2 and CO with molar ratio 2:1. The process was carried out under elevated pressure (4.8 MPa) at 260 °C and products were analyzed by GC (gas chromatograph). Before the activity tests, all catalysts were pre-reduced for 2 h in a flow of 5 % H2–95 % Ar mixture at 300 °C under atmospheric pressure. The steady-state activity measurements were taken after at least 12 h on the stream. The analysis of the reaction products were carried out by an on-line GC equipped with a FID detector and 10 % Carbowax 1500 on Graphpac column. The CO and CO2 concentrations were monitored by a GC chromatograph equipped with TCD detector (120 °C, 130 mA), and Carbosphere 60/80 (65 °C) column. CO conversion was calculated by the following equation:
The selectivities of the products were calculated as follows:
where Pi is the amount of substance of every organic product.
Diffuse reflectance FTIS
IR spectra were recorded with a Thermo Scientific Nicolet 6700 FTIR spectrometer equipped with a liquid nitrogen cooled MCT (mercury cadmium telluride—MCT, HgCdTe) detector. Before analysis copper, palladium and palladium–copper catalysts supported on ZnAl2O4 and ZnAl2O4–5 % CeO2 were reduced at 300 °C in gas reduction mixture 5 % H2–95 % Ar for 1 h. After reduction, the catalysts were cooled down to 120 °C and then the reduction mixture was shifted to a mixture of approximately 1 vol.% CH3OH in argon stream. A resolution of 4.0 cm−1 was used throughout the investigations. 64 scans were taken to achieve a satisfactory signal to noise ratio. The background spectrum was collected at 120 °C after reduction. The adsorption process involved exposure of the reduced catalysts to 1 vol.% CH3OH in argon stream flowing at 40 cm3 min−1 for 30 min under atmospheric pressure. After the adsorption process, the cell was evacuated for 30 min at the same temperature in argon.
Result and discussion
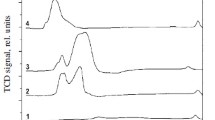
The catalyst reducibility was evaluated by TPR. H2-TPR results for the monometallic Cu and bimetallic Pd–Cu catalysts are shown in Figs. 1 and 2. Monometallic 20 % Cu/ZnAl2O4 supported catalysts exhibited two reduction peaks in the temperature range of 180–380 °C which are attributed to reduction of CuO and Cu2O species [22–27]. TPR curves recorded for copper catalysts promoted by CeO2 also showed two reduction stages, which were slightly shifted towards lower temperature. This indicates that the addition of CeO2 into the copper catalyst improve the copper dispersion and facilitates the reduction of copper species.

TPR profile of Cu/ZnAl2O4 and copper promoted by CeO2 catalysts after calcination in air for 4 h at 400 °C

TPR profile of copper and palladium–copper catalysts after calcination in air for 4 h at 400 °C
The reduction behavior of Pd–Cu supported catalysts was also studied in this work. The comparison of the reduction measurements carried out for unpromoted copper 20 % Cu/ZnAl2O4 supported catalysts and for promoted by Pd and CeO2 is shown in Fig. 2. The TPR-H2 results performed for palladium catalysts showed that all Pd–Cu catalytic systems reduced in three stages. The first effect observed in the temperature range 80–140 °C for all palladium doped catalysts is connected with PdO reduction. The next two hydrogen consumption peaks are assigned to the reduction of CuO and Cu2O copper species reduction according to the following scheme: CuO → Cu2O → Cu [28].
In our previous work, we investigated the reduction behavior of palladium 2 % Pd/ZnAl2O4 catalyst. TPR-H2 of this catalyst showed two hydrogen consumption peaks situated at about 30 and 80 °C. The first hydrogen consumption effect was attributed to the reduction of highly dispersed palladium oxide which is slowly reduced at room temperatures. The second reduction peak indicates the reduction of small palladium crystallites interacting with the surface of ZnAl2O4 [29].
Cubeiro and Fierro [30] also studied the reduction behavior of Pd/ZnO catalysts. They claimed that PdO is an easily reducible oxide, even at room temperature, they observed a low temperature reduction peak situated in the temperature range 30–150 °C, which was assigned to the reduction of PdO to metallic Pd.
The reduction behavior of Pd supported on mixtures of zinc oxide, zinc aluminate, and alumina, prepared from γ-alumina and zinc nitrate, were studied by Nilsson et al. [31]. The authors observed H2 consumption peak with its maximum at around 90 °C for all studied palladium catalysts and this hydrogen consumption peak authors ascribed to the formation of metallic Pd and Zn by hydrogen spillover from Pd [31]. Additionally, the Pd catalyst supported on a mixture of ZnO and γ-alumina exhibited a peak at 450 °C. This reduction profile was assigned to the ZnO reduction. The authors have proposed this conclusion based on reduction studies performed for the reducibility of ZnO/γ-Al2O3 bi-oxide. The TPR profile of ZnO/γ-Al2O3, prepared by impregnating alumina with zinc nitrate, exhibits a reduction peak located in this temperature region [31].
The Pd and Pd–Cu catalysts reducibility was evaluated by measuring the TPR by Wang and Lu [32]. The authors claimed that the Pd/Al2O3 catalyst reduced in one reduction stage with the maximum of hydrogen consumption peak situated at a lower temperature of 125 °C. This effect was related to the reduction of PdO, which means that there was a weak interaction between Pd and the support. The TPR profiles recorded for bimetallic Pd–Cu/γ-Al2O3 catalysts showed that the reduction peaks were shifted toward higher temperature, which suggested that the addition of Cu species enhanced the interaction between active Pd species and alumina, meanwhile the presence of Pd facilitated the reduction of copper oxides. This result is believed to arise from an interaction of palladium and copper oxides, which results in the formation of a Pd–Cu alloy.
Canner and Falconer [33] proposed that the enhanced reduction of copper species in the presence of Pd on the catalyst surface can be explained by hydrogen spillover from metal Pd to copper species.
Kugai et al. [34] studied the reduction behavior of monometallic Pd, Cu and bimetallic Pd–Cu catalysts supported on CeO2 using TPR method. TPR measurements of those systems indicate that Pd catalysts reduced at 130 °C. In contrast, the monometallic Cu catalyst exhibited two reduction effects with the maximum of hydrogen consumption peaks positioned at around 135 and 160 °C. These peaks were attributed to the reduction in highly dispersed CuO cluster interacting with ceria and/or bulk copper species. In the case of copper catalysts containing 25 wt% of Cu, a new third effect situated at 180 °C appeared and this effect has been assigned to bulk CuO species reduction. The authors noticed that the third peak occurs at lower temperature and compared it to non-supported bulk CuO as suggested by Fierro et al. [35], which means that even bulk CuO species is under strong influence of ceria support. The reduction effects positioned at 135 and 160 °C were assigned to two types of dispersed CuO species closely interacting with ceria to different extents or dispersed CuO species and isolated Cu2+ ion directly interacting with ceria. In the case of Pd–Cu catalysts on the TPR curve, only a single peak was observed at around 135 °C. The interaction between Pd and Cu led to the metal species more reducible than Cu and less reducible than Pd. The broadness of the peak indicates a certain variation in Pd–Cu species, such as variation in the Cu/Pd ratio on the microscopic level or in alloy crystallite size, during reduction [34].
The phase composition studies of unpromoted and promoted by Pd and CeO2 Cu/ZnAl2O4 supported catalysts after calcination and reduction were carried out to elucidate the interaction between the active component and support. Fig. 3 displays XRD patterns of copper catalysts supported on ZnAl2O4–1 % CeO2 after calcination in air atmosphere at 400, 700 and 900 °C for 4 h. The diffraction peaks of the oxide phase in the case of copper catalyst calcined at 400 °C are attributed to ZnAl2O4, ZnO, γ-Al2O3 and CuO phases [22, 23].

XRD patterns for 20 % Cu/ZnAl2O4–1 % CeO2 after calcination in air atmosphere at 400, 700 and 900 °C for 4 h
The increase of the calcination temperature caused the increase of the crystallinity degree for studied system. In addition, the appearance of CeO2 phase in the case of copper catalysts being after calcination at 900 °C was confirmed.
The phase composition studies of CuO·ZnO·ZnAl2O4 multi metal oxide was investigated using XRD patterns by Abdus Subhana et al. [36]. The authors observed sharp diffraction peaks attributed to crystalline copper oxide (CuO), zinc oxide (ZnO) and ZnAl2O4 [37].
In Fig. 4, the phase composition studies of copper catalysts calcined at 400, 700 and 900 °C promoted by 5 % CeO2 are given. On the XRD curves recorded for this system, we can easily observe the same oxidic phases as in the case of copper catalysts promoted by 1 % CeO2.

XRD patterns for 20 % Cu/ZnAl2O4–5 % CeO2 after calcination in air atmosphere mixture at 400, 700 and 900 °C for 4 h

XRD patterns for 2 % Pd–20 % Cu/ZnAl2O4 after calcination in air atmosphere at 400, 700 and 900 °C for 4 h
The phase composition studies of 2 % Pd–20 % Cu/ZnAl2O4 catalyst calcined at various temperatures (400, 700 and 900 °C) confirmed the presence of the following oxidic phases: ZnAl2O4, ZnO, PdO, γ-Al2O3 and CuO (see Fig. 5). The appearance PdO and CuO phases in the investigated catalytic systems confirmed the reduction behavior of copper palladium doped catalyst. In the next step of our XRD investigation, we carried out the diffraction measurements for copper palladium catalysts promoted by 5 % CeO2, the results of these studies are shown on Fig. 6.

XRD patterns for 2 % Pd–20 % Cu/ZnAl2O4–5 % CeO2 after calcination in air atmosphere at 400, 700 and 900 °C for 4 h
On the diffractograms recorded for this system, the same oxidic phases were observed as in the case of unpromoted palladium–copper catalysts. The only difference observed in this case was the appearance of additional CeO2 phase, which was visible on the XRD curves starts from 400 °C. To understand this better and to elucidate the reduction results obtained for palladium–copper promoted by cerium oxide, we performed the analogical diffraction measurements for copper and bimetallic Pd–Cu catalysts being after reduction at 300 and 900 °C. The phase composition studies performed for those systems are presented on Figs. 7 and 8. The XRD analysis of 20 % Cu/ZnAl2O4–5 % CeO2 copper catalyst reduced in H2 atmosphere are given in Fig. 7. Only ZnAl2O4 and metallic copper phases were visible on the diffraction patterns. The increase of the reduction temperature caused only the growth of the degree of crystallinity of investigated system. We did not observe any alloying process between the individual components of this system. In contrast, the analogous measurements carried out for palladium–copper catalysts 2 % Pd–20 % Cu/ZnAl2O4–5 % CeO2 (see Fig. 8) confirmed the alloy formation between Pd and Cu and the presence of spinel structure ZnAl2O4 phases. The confirmation of the alloy formation between those two components was the shift of the reflections coming from metallic copper and palladium in comparison to the same reflections observed in the case of monometallic copper supported catalysts. The shift and broadening of the observed reflections attributed to the metallic copper confirmed the Pd–Cu alloy formation for the Pd–Cu catalyst after reduction at 300 and 900 °C for 2 h in 5 % H2–95 % Ar atmosphere.

XRD patterns for 20 % Cu/ZnAl2O4–5 % CeO2 after reduction in 5 % H2–95 % Ar atmosphere at 300 and 900 °C for 2 h

XRD patterns for 2 % Pd–20 % Cu/ZnAl2O4–5 % CeO2 after reduction in 5 % H2–95 % Ar atmosphere at 300 and 900 °C for 2 h
The morphology of the Pd–Cu/CeO2 catalysts using SEM–EDS techniques was investigated by Kugai et al. [38]. All studied bimetallic supported catalysts were reduced in H2 flow at 260 °C and exposed to oxygen-enhanced water–gas shift condition “ex situ” for about 2 h. The SEM–EDS analysis showed that Pd–Cu alloy was dispersed on CeO2, whose composition was not elucidated. Additionally, the same authors [34] performed EXAFS studies of ceria-supported bimetallic Pd–Cu catalyst, and they confirmed that Cu forms alloy with Pd upon reduction and claimed that Pd helps to keep Cu in reduced state during the reaction.
It is worth noticing that the alloying process that occurs between Pd and Cu was observed at 300 °C, which means that the high activity and selectivity to methanol production of palladium promoted catalysts in the methanol synthesis reaction can be explained by the alloy formation during activation process (reduction at 300 °C) of this system before the activity tests.
PdCu bimetallic catalysts supported on alumina prepared by different synthesis routes were investigated in trichloroethylene (TCE) hydrodechlorination to ethylene by Meshesha et al. [39]. The bimetallic supported catalysts were studied to elucidate the relationship between structure, interaction between active phase component and catalytic properties in the hydrodechlorination of TCE in gas phase. The authors confirmed that the alloy phase formation between Pd and Cu leads to high activity and selectivity of palladium–copper supported catalysts in studied reaction.
The CO hydrogenation to methanol was also studied in this work. The results of activity tests carried out for mono- and bimetallic Pd–Cu supported on spinel structure catalysts promoted by CeO2 in methanol synthesis expressed as CO conversion and in \({\text{g}}\;{\text{CH}}_{3} {\text{OH}}\;{\text{kg}}_{{{\text{cat}}.}}^{ - 1} \;{\text{h}}^{ - 1}\) are shown in Table 1. Additionally, the selectivities of studied catalysts to methanol production and other organic products are also given in the same table. The activity results clearly showed that methanol and methane were the only organic products formed during the reaction. The highest methanol formation (238 \({\text{g}}\;{\text{CH}}_{3} {\text{OH}}\;{\text{kg}}_{{{\text{cat}}.}}^{ - 1} \;{\text{h}}^{ - 1}\)) and carbon monoxide conversion (24.5 %) was observed in the 2 % Pd–20 % Cu/ZnAl2O4–5 % CeO2 system. Lower values of CO conversion and yield of methanol production were exhibited by copper catalysts promoted by CeO2. The lowest activity was shown for the monometallic Cu/ZnAl2O4 catalyst. The improvement of the catalytic activity observed for CeO2 and/or Pd modified systems can be explained by the presence of alloy compound PdCu formed during the activation process and by increasing the mobility of lattice oxygen and by the redox properties after CeO2 addition. The mobile oxygen coming from CeO2 oxide can oxidize the metallic copper phase and contribute to the formation of Cu+ ions on the catalyst surface increasing thereby the number of active centers in the studied reaction. The occurrence of PdCu alloy on the catalyst surface increases the CO adsorption, which leads directly to an increase in carbon monoxide conversion. Based on the results of this work, we could assume that the role of Pd in the promotion effect is the promotion of hydrogen activation due to the hydrogen spillover effect and increase the adsorption of CO during the reaction.
In order to explain the differences between the activity results observed for 20 % Cu/ZnAl2O4, 20 % Cu/ZnAl2O4–5 % CeO2 and 2 % Pd–20 % Cu/ZnAl2O4–5 % CeO2 catalysts, the surface adsorbed species formed during methanol adsorption were investigated by FTIR technique. Fig. 9 displays the IR spectra taken after the exposure of investigated catalysts to a 1 vol.% CH3OH in argon stream at 120 °C under atmospheric pressure. The IR spectra collected for 20 % Cu/ZnAl2O4 catalyst (Fig. 9a) showed that copper formate (HCOO–Cu: 2925, 2850, 1620, 1364, and 1350 cm−1), zinc formate (HCOO–Zn: 2970, 2880, 2700, 1591, 1372, and 1365 cm−1), carbonate (\({\text{CO}}_{3}^{2 - } :\) 1680, 1620, 1570–1440, and 1220 cm−1), zinc methoxide (CH3O–Zn: 2936, 2919, 2825, 1470, 1230–1200 and 1060 cm−1), copper methoxide (CH3O–Cu: 1,443 and 1,350 cm−1), and aluminum methoxide (CH3O–Al: 2995, 1020–1100 cm−1) species were adsorbed on its surface [22, 23].

Infrared spectra of adsorbed species taken after reduction at 300 °C in the 5 % H2–95 % Ar mixture and exposure of the following catalytic systems: (a) 20 % Cu/ZnAl2O4, (b) 20 % Cu/ZnAl2O4–5 % CeO2, and (c) 2 % Pd–20 % Cu/ZnAl2O4–5 % CeO2 to a 1 vol.% methanol–argon mixture at 120 °C
Analogous measurements were performed for promoted systems. The FTIR results confirmed the presence of the same adsorbed surface species after exposure of 1 vol.% CH3OH in argon mixture (see Fig. 9b and c). The observed difference in the FTIR spectrum in the case of promoted systems was the appearance of additional bands attributed to following species: 1,380 cm−1: b-HCO3–Ce, 1,460 cm−1 and 1,465 cm−1: b-HCOO–Ce and 1,550, 1,575 cm−1: b-HCOO–Ce. The occurrence of these surface species on the catalyst surface after methanol exposure confirms the promotion effect of CeO2 on CO hydrogenation towards methanol formation. These adsorption centers are involved in methanol synthesis reaction and contribute to increase the catalytic activity of copper promoted catalysts. It is worth to notice, that palladium promoted systems exhibited also the more intensive bands recorded for adsorbed species over copper species after methanol exposure in comparison to 20 % Cu/ZnAl2O4 catalyst. The higher intensity of the adsorption bands can be explained by the fact that palladium addition into copper catalyst facilitate the reduction of copper species and change the electronic properties of metallic copper. Additionally, it leads to active center formation on the catalyst surface, on which the CO or/and CO2 are reduced to main product methanol. Additionally, it is well known in literature data that the formate species formed on the copper or support surface plays pivotal role in the methanol synthesis reaction. The formate species formed on the support surface or on the perimeter between metal (copper) or alloy (PdCu) and support would play important role in the methanol synthesis as those formed on supported copper catalysts in our case.
TPD-NH3 is a conventional method for characterizing acidity in catalytic systems. The acidity of all investigated catalysts was evaluated by the TPD-NH3 method and ammonia desorption profiles for copper and palladium doped copper supported catalysts are shown in Figs. 10 and 11.

TPD-NH3 profiles for 20 % Cu/ZnAl2O4, 20 % Cu/ZnAl2O4–1 % CeO2 and 20 % Cu/ZnAl2O4–5 % CeO2 catalysts calcined in air atmosphere for 4 h at 400 °C

TPD-NH3 profiles for 20 % Cu/ZnAl2O4, 2 % Pd–20 % Cu/ZnAl2O4, 2 % Pd–20 % Cu/ZnAl2O4–1 % CeO2 and 2 % Pd–20 % Cu/ZnAl2O4–5 % CeO2 catalysts calcined in air atmosphere for 4 h at 400 °C
The total amount of desorbed ammonia expressed as μmol NH3 \({\text{g}}_{{{\text{cat}}.}}^{ - 1}\) and distribution of acid centers for all studied systems in the temperature range 100–600 °C are given in Table 2. It can be easily observed from Figs. 10 and 11, and Table 2 that all catalytic systems exhibited three different acid centers.
Weak, medium and strong acid sites are visible on TPD curves. The mutual relationship between three various kind of acidic centers is given in Table 2. The obtained results reveal that the total surface acidity of prepared systems increases with increasing of Ce content in the catalytic systems. The same trend was observed after palladium promotion of copper supported catalyst. The difference was only observed in the case of 2 % Pd–20 % Cu/ZnAl2O4 catalyst for which a slight decrease of total acidity was observed compared with the monometallic copper catalyst. The highest total amount of desorbed NH3 exhibited 2 % Pd–20 % Cu/ZnAl2O4–5 % CeO2 catalyst (228 NH3 μmol \({\text{g}}_{{{\text{cat}}.}}^{ - 1}\)).
It is worth noticing that the palladium–copper catalysts showed also the lowest content of weak acid centers (see Table 2). In addition, a correlation can be found between the activity and acidity of all catalysts. The highest activity and selectivity in methanol synthesis reaction was observed for catalyst exhibited the highest total acidity. The 20 % Cu/ZnAl2O4, catalyst which showed the lowest value of total acidity, was the least active and selective to methanol formation. Additionally, the acidity result explain the differences in the activity observed for all systems in methanol synthesis.
Conclusion
In this study, we prepared unpromoted and promoted by CeO2 monometallic copper and bimetallic Pd–Cu supported catalysts by a wet aqueous impregnation method and tested in CO hydrogenation reaction. The physicochemical and catalytic properties of prepared catalytic systems were studied using various techniques TPR, XRD, TPD-NH3 and FTIR. The following conclusions can be drawn from the present investigations dealing with the mono- and bimetallic catalysts. The catalytic activity of mono and bimetallic catalysts depends on the surface acidity and its chemical composition.
Activity results obtained for the investigated catalysts in the methanol synthesis reaction confirmed the promotion effect of Pd and CeO2 on catalytic activity and selectivity. The improvement in activity observed for copper catalysts after CeO2 addition is associated with the increasing of oxygen mobility on CeO2 doped surface catalysts which can oxidize the metallic Cu and form Cu+ ions on the catalyst surface and by the acidity of promoted catalyst.
Another factor that plays an important role during the reaction is the presence of oxygen vacancies, which can adsorb the CO or CO2 species on the catalyst surface, which then may migrate to the adjacent H–Cu site to be reduced into methanol.
In the case of a copper–palladium doped catalyst, the increased activity and selectivity is explained by the Pd–Cu alloy formation. The resulting alloy changes the electronic properties of metallic copper on which the CO or/and CO2 are reduced to main product methanol. In addition, Pd promotes hydrogen activation due to the hydrogen spillover effect and increases the adsorption of CO during the reaction.
References
Tijm PJA, Waller FJ, Brown DM (2001) Methanol technology developments for the new millennium. Appl Catal A 221:275–282
Liu X-M, Lu GQ, Yan Z-F, Beltramini J (2003) Recent advances in catalysts for methanol synthesis via hydrogenation of CO and CO2. Ind Eng Chem Res 42:6518–6530
Waugh KC (1992) Methanol synthesis. Catal Today 15:51–75
Mierczynski P, Kaczorowski P, Maniecki TP, Bawolak-Olczak K, Maniukiewicz W (2013) The influence of Pd loading on the physicochemical properties of the Cu–Cr–Al methanol synthesis catalysts. Reac Kinet Mech Cat 109:13–27
Bradford MCJ, Konduru MV, Fuentes DX (2003) Preparation, characterization and application of Cr2O3/ZnO catalysts for methanol synthesis. Fuel Process Technol 83:11–25
Gunter M, Ressler T, Bems B, Buscher C, Genger T, Hinrichsen O, Muhler M, Schlögl R (2001) Implication of the microstructure of binary Cu/ZnO catalysts for their catalytic activity in methanol synthesis. Catal Lett 71:37–44
Grunwaldt JD, Molenbroek AM, Topsøe NY, Topsøe H, Clausen BS (2000) In situ investigations of structural changes in Cu/ZnO catalysts. J Catal 194:452–460
Yoshihara J, Campbell CT (1996) Methanol synthesis and reverse water–gas shift kinetics over Cu(110) model catalysts: structural sensitivity. J Catal 161:776–782
Nakamura J, Choi Y, Fujitani T (2003) On the issue of the active site and the role of ZnO in Cu/ZnO methanol synthesis catalysts. Top Catal 22(3–4):277–285
Li B, Jens K-J (2014) Low-temperature and low-pressure methanol synthesis in the liquid phase catalyzed by copper alkoxide systems. Ind Eng Chem Res 53(5):1735–1740
Li C, Sakata Y, Arai T, Domen K, Maruya K-I, Onish T (1989) Adsorption of carbon monoxide and carbon dioxide on cerium oxide studied by Fourier-transform infrared spectroscopy. Part 2.—Formation of formate species on partially reduced CeO2 at room temperature. J Chem Soc Faraday Trans I 85(6):1451–1461
Si R, Zhang Y-W, Li S-J, Lin B-X, Yan C-H (2004) Urea-based hydrothermally derived homogeneous nanostructured Ce1−x Zr x O2 (x = 0–0.8) solid solutions: a strong correlation between oxygen storage capacity and lattice strain. J Phys Chem B 108(33):12481–12488
Kapoor MP, Raj A, Matsumura Y (2001) Methanol decomposition over palladium supported mesoporous CeO2–ZrO2 mixed oxides. Microporous Mesoporous Mater 44–45:565–572
Galtayries A, Sporken R, Riga J, Blanchard G, Caudano R (1998) XPS comparative study of ceria/zirconia mixed oxides: powders and thin film characterisation. J Electron Spectrosc Relat Phenom 88–91:951–956
Yoo C-J, Lee D-W, Kim M-S, Moon DJ, Lee K-Y (2013) The synthesis of methanol from CO/CO2/H2 gas over Cu/Ce1−x Zr x O2 catalysts. J Mol Catal A 378:255–262
Ma Y, Ge Q, Li W, Xu H (2009) Methanol synthesis from sulfur-containing syngas over Pd/CeO2 catalyst. Appl Catal B 90:99–104
Shen W-J, Ichihashi Y, Andoa H, Okumuraa M, Haruta M, Matsumura Y (2001) Influence of palladium precursors on methanol synthesis from CO hydrogenation over Pd/CeO2 catalysts prepared by deposition–precipitation method. Appl Catal A 217:165–172
Maniecki TP, Stadnichenko AI, Maniukiewicz W, Bawolak K, Mierczynski P, Boronin AI, Jozwiak WK (2010) An active phase transformation on surface of Ni–Au/Al2O3 catalyst during partial oxidation of methane to synthesis gas. Kinet Catal 51:573–578
Maniecki TP, Bawolak-Olczak K, Mierczynski P, Maniukiewicz W, Jóźwiak WK (2009) Effect of the chemical composition of (MgO)(x)(Al2O3)(y) support on the catalytic performance of Ni and Ni–Au catalysts for the partial oxidation of methane. Chem Eng J 154(1–3):142–148
Maniecki TP, Bawolak K, Mierczynski P, Jozwiak WK (2008) Gold as promoter of nickel supported catalysts for semi combustion of methane. Pol J Chem 82:2389–2399
Maniecki TP, Mierczynski P, Maniukiewicz W, Bawolak K, Gebauer D, Jozwiak WK (2008) Methanol synthesis from mixture of CO, CO2 and H2 under atmospheric pressure over Au, Ag–Cu/FeAlO3 supported catalysts. Pol J Chem 82(2008):2379–2388
Mierczynski P, Maniecki TP, Chalupka K, Maniukiewicz W, Jozwiak WK (2011) Cu/Zn x Al y O z supported catalysts (ZnO:Al2O3 = 1, 2, 4) for methanol synthesis. Catal Today 176:21–27
Mierczynski P, Vasilev K, Mierczynska A, Maniukiewicz W, Maniecki TP (2013) The effect of ZnAl2O4 on the performance of Cu/Zn x Al y O x+1.5y supported catalysts in steam reforming of methanol. Top Catal 56:1015–1025
Maniecki TP, Mierczynski P, Bawolak-Olczak K, Jozwiak WK (2009) Methanol synthesis from CO2 and H2 mixture over 60 % Cu/support (FeAlO3, ZnAl2O4) catalysts. Pol J Chem 83:1653–1662
Maniecki TP, Mierczynski P, Jozwiak WK (2010) Copper-supported catalysts in methanol synthesis and water gas shift reaction. Kinet Catal 51:843–848
Mierczynski P, Vasilev K, Mierczynska A, Maniukiewicz W, Maniecki TP (2014) Highly selective Pd–Cu/ZnAl2O4 catalyst for hydrogen production. Appl Catal A 479:26–34
Mierczynski P, Ciesielski R, Kedziora A, Zaborowski M, Maniukiewicz W, Nowosielska M, Szynkowska MI, Maniecki TP (2014) Novel Pd–Cu/ZnAl2O4–ZrO2 catalysts for methanol synthesis. Catal Lett 144:723–735
Mierczynski P, Kaczorowski P, Ura A, Maniukiewicz W, Zaborowski M, Ciesielski R, Kedziora A, Maniecki TP (2014) Promoted ternary CuO–ZrO2–Al2O3 catalysts for methanol synthesis. Cent Eur J Chem 12(2):206–212
Mierczynski P, Maniukiewicz W, Maniecki TP (2013) Comparative studies of Pd, Ru, Ni, Cu/ZnAl2O4 catalysts for the water gas shift reaction. Cent Eur J Chem 11(6):912–919
Cubeiro ML, Fierro JLG (1998) Selective production of hydrogen by partial oxidation of methanol over ZnO-supported palladium catalysts. J Catal 179:150–162
Nilsson M, Jansson K, Jozsa P, Pettersson LJ (2009) Catalytic properties of Pd supported on ZnO/ZnAl2O4/Al2O3 mixtures in dimethyl ether autothermal reforming. Appl Catal B 86:18–26
Wang F, Lu G (2010) Hydrogen feed gas purification over bimetallic Cu–Pd catalysts—effects of copper precursors on CO oxidation. Int J Hydrog Energy 35:7253–7260
Canner WC, Falconer JL (1995) Spillover in heterogeneous catalysis. Chem Rev 95:759–788
Kugai J, Miller JT, Guo N, Song C (2011) Oxygen-enhanced water gas shift on ceria-supported Pd–Cu and Pt–Cu bimetallic catalysts. J Catal 277:46–53
Fierro G, Lojacono M, Inversi M, Porta P, Lavecchia R, Cioci F (1994) A study of anomalous temperature-programmed reduction profiles of Cu2O, CuO, and CuO–ZnO catalysts. J Catal 178:709–721
Abdus Subhana Md, Ahmed T, Awal R, Makioka R, Nakata H, Pakkanen TT, Suvanto M, Moon Kim B (2014) Synthesis, structure, luminescence and photophysical properties of nano CuO·ZnO·ZnAl2O4 multi metal oxide. J Lumin 146:123–127
Zou L, Xiang X, Wei M, Yang L, Li F, Evans DG (2008) A facile and green synthesis route to mesoporous spinel-type Zn–Al complex oxide. Ind Eng Chem Res 47:1495–1500
Kugai J, Fox EB, Song C (2013) Role of CeO2 support for Pd–Cu bimetallic catalysts for oxygen-enhanced water gas shift. Appl Catal A 456:204–214
Meshesha BT, Barrabés N, Llorca J, Dafinov A, Medina F, Fottinger K (2013) PdCu alloy nanoparticles on alumina as selective catalysts for trichloroethylene hydrodechlorination to ethylene. Appl Catal A 453:130–141
Acknowledgments
Partially financed from Grant number 0680/B/H03/2011/40 is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Mierczynski, P., Maniukiewicz, W., Zaborowski, M. et al. The effect of palladium and CeO2 on the catalytic and physicochemical properties of copper catalysts in methanol synthesis. Reac Kinet Mech Cat 114, 211–228 (2015). https://doi.org/10.1007/s11144-014-0785-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11144-014-0785-0