
Abstract
l-Lactate is a commodity chemical used in various fields. Microorganisms have produced l-lactate via lactic fermentation using saccharides derived from crops as carbon sources. Recently, l-lactate production using microalgae, whose carbon source is carbon dioxide, has been spotlighted because the prices of the crops have increased. A red alga Cyanidioschyzon merolae produce l-lactate via lactic fermentation under dark anaerobic conditions. The l-lactate titer of C. merolae is higher than those of other microalgae but lower than those of heterotrophic bacteria. Therefore, an increase in the l-lactate titer is required in C. merolae. l-Lactate dehydrogenase (l-LDH) catalyzes the reduction of pyruvate to l-lactate during lactic fermentation. C. merolae possesses five isozymes of l-LDH. The results of previous transcriptome analysis suggested that l-LDHs are the key enzymes in the lactic fermentation of C. merolae. However, their biochemical characteristics, such as catalytic efficiency and tolerance for metabolites, have not been revealed. We compared the amino acid sequences of C. merolae l-LDHs (CmLDHs) and characterized one of the isozymes, CmLDH1. BLAST analysis revealed that the sequence similarities of CmLDH1 and the other isozymes were above 99%. The catalytic efficiency of CmLDH1 under its optimum conditions was higher than those of l-LDHs of other organisms. ATP decreased the affinity and turnover number of CmLDH1 for NADH. These findings contribute to understanding the characteristics of l-LDHs of microalgae and the regulatory mechanisms of lactic fermentation in C. merolae.
Key message
ATP inhibited Cyanidioschyzon merolae l-lactate dehydrogenase showing high catalytic efficiency for pyruvate reduction, possibly contributing to avoiding the overproduction of ATP via lactic fermentation at night.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Lactate/lactic acid is one of the commodity chemicals used for different fields such as foods, cosmetics, and medicines (Abdel-Rahman et al. 2013). Lactate has enantiomers, l-lactate and d-lactate. Both enantiomers are required for manufacturing bioplastic derived from lactate, namely polylactide (Tsuji 2005; Tsuji et al. 2006). Industrial lactate production uses lactic fermentation by microorganisms such as lactic acid bacteria, whose carbon sources are saccharides derived from crops (Ghaffar et al. 2014). However, the prices of the crops have increased because the prices are affected by population growth, soaring crude oil prices, and biofuel production (Bilgili et al. 2020). In recent years, when global warming accelerated, metabolite production from carbon dioxide using eukaryotic microalgae and cyanobacteria is spotlighted. Eukaryotic microalgae and cyanobacteria can produce lactate using carbon dioxide fixed via photosynthesis as the sole carbon source, minimizing the costs of carbon sources such as saccharides (Abdel-Rahman et al. 2013).
Cyanidioschyzon merolae is a unicellular red alga living in acid hot springs (pH 1–3 and 40–50 °C) and does not possess a cell wall (De Luca et al. 1978). The genome sequences of the nucleus, mitochondria, and chloroplast in C. merolae are completely elucidated (Ohta et al. 1998, 2003; Matsuzaki et al. 2004; Nozaki et al. 2007). Previous transcriptome analysis indicated that C. merolae performed anaerobic energy conversion, such as lactic fermentation, rather than aerobic respiration at night (Miyagishima et al. 2019). C. merolae produces l-lactate under dark anaerobic conditions (Yoshida et al. 2024). Among eukaryotic microalgae and cyanobacteria, a model cyanobacterium Synechocystis sp. PCC 6803 and Euglena gracilis also produce l-lactate (Angermayr and Hellingwerf 2013; Tomita et al. 2016). In Synechocystis sp. PCC 6803, genetic manipulation is necessary to produce l-lactate because wild-type does not produce l-lactate (Angermayr and Hellingwerf 2013). The l-lactate titer (3.2 g/L) and productivity (16.0–19.4 mg/L/h) of C. merolae are higher than those of the Synechocystis sp. PCC 6803 mutant (1.8 g/L and 2.7 mg/L/h, respectively) (Yoshida et al. 2024; Angermayr and Hellingwerf 2013). l-Lactate production in Euglena gracilis is not efficient because its l-lactate titer is occasionally below 10 mg/L (Tomita et al. 2016). Thus, C. merolae is a candidate for a host of l-lactate production from carbon dioxide. However, the l-lactate titer and productivity of C. merolae are lower than those of heterotrophic bacteria (Abdel-Rahman et al. 2013), and a further increase in the l-lactate titer of C. merolae is required.
l-Lactate dehydrogenase (l-LDH; EC 1.1.1.27) catalyzes the final step in lactic fermentation: pyruvate + NADH → l-lactate + NAD+. l-LDH is a paralog of malate dehydrogenase (MDH), and their substrate specificities are determined by five amino acid residues (Yin and Kirsch 2007). l-LDHs have been well characterized in bacteria (particularly lactic acid bacteria) and higher plants (Matoba et al. 2014; Gaspar et al. 2007; Jonas et al. 1972; Barman 1969; Dennis and Kaplan 1960; Götz and Schleifer 1975; Yoshida 1965; Oba et al 1977; Betsche 1981). Bacterial l-LDHs are allosteric enzymes, and fructose-1,6-bisphosphate (FBP) is necessary for their catalytic activities. On the other hand, there are non-allosteric l-LDHs in vertebrate cells (Matoba et al. 2014). Some organisms (Sporolactobacillus inulinus YBS 1-5, Bacillus coagulans, Enterococcus faecalis, Enterococcus mundtii 15-1A, Fusarium granearum) possess two isozymes of l-LDH (Wu et al. 2019; Sun et al. 2016; Jönsson et al. 2009; Matoba et al. 2014; Chen et al. 2019). On the other hand, C. merolae has five isozymes of l-LDH (Matsuzaki et al. 2004; Nozaki et al. 2007; Ohta et al. 1998, 2003). C. merolae does not possess other l-lactate-generating enzymes such as lactaldehyde dehydrogenase and malolactic enzyme (KEGG database URL: https://www.kegg.jp/pathway/map=cme00620&keyword=pyruvate). The expression level of a gene encoding l-LDH increases from day to night in C. merolae (Miyagishima et al. 2019), suggesting that l-LDH is the key enzyme in lactic fermentation in C. merolae. Previous analysis indicated that the amount of l-LDHs in C. merolae remains almost the same under photoautotrophic and dark anaerobic conditions (Yoshida et al. 2024). This suggests that the biochemical regulation of C. merolae l-LDHs (CmLDHs) by temperature, pH, and effectors enables them to convert pyruvate to l-lactate under dark anaerobic conditions. We presume that understanding the regulation leads to a further increase in the l-lactate titer of C. merolae.
In this study, we compared the amino acid sequences of five CmLDHs (CmLDH1–5) and biochemically analyzed one of the isozymes, CmLDH1.
Materials and methods
Preparation of a vector used for the expression of CmLDH1 in Escherichia coli
The sequence of the gene encoding CmLDH1 (CMA145C) was acquired from the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (https://www.genome.jp/kegg/kegg_ja.html). The sequence was synthesized by Eurofins Genomics Japan (Tokyo, Japan), and the synthesized sequence was introduced into the BamHI-XhoI site of vector pGEX6P-1 (G.E. Healthcare Japan, Tokyo, Japan). The vector was transformed into competent cells of Escherichia coli BL21 (DE3) (BioDynamics Laboratory Inc., Tokyo, Japan). After the transformation of the E. coli, the E. coli cells were cultured in an LB medium (2.4 L) at 30 °C with shaking (150 rpm). During the cultivation, the expression of the recombinant CmLDH1 was induced by 5 µM isopropyl β-d-1-thiogalactopyranoside (Wako Chemicals, Osaka, Japan) overnight.
Affinity purification of a glutathione-S-transferase (GST) -tagged CmLDH1
The E. coli cells in 600 mL culture were suspended in 10 mL phosphate-buffered saline/tween (PBS-T) (0.137 M NaCl, 0.27 mM KCl, 8.1 mM Na2HPO4.12H2O, 1.47 mM KH2PO4, and 0.001% Tween 20). The cells were sonicated twelve times for 15 s at 20% intensity using model VC-750 (EYELA, Tokyo, Japan). After centrifugation at 14,200 × g for 15 min at 4 °C, 800 µL of Glutathione Sepharose 4B resin (G.E. Healthcare Japan, Tokyo, Japan) was added to the supernatant. The sample was gently shaken on ice for 60 min. After that, 10 mM MgSO4·7H2O and 5 mM ATP were added to the sample, and the mixture was shaken for 30 min at 37 °C. The mixture was centrifugated at 5800 × g for 2 min at 4 °C to remove the supernatant. The resin was washed with 3 mL PBS-T five times and 700 µL of PBST five times. The GST-CmLDH1 was eluted by 500 µL glutathione-S-transferase (GST) elution buffer [50 mM Tris-HCl (pH 9.6) and 10 mM reduced glutathione] five times. Then, the GST-CmLDH1 was concentrated in a Vivaspin 500 MWCO 30000 device (Sartorius, Göttingen, Germany). The concentration of purified GST-CmLDH1 was measured by a Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, Rockford, IL, USA). Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) was conducted using 8% gels, and the gels were stained by QuickBlue stain reagent (BioDynamics Inc., Tokyo, Japan).
Enzyme assay
The reaction catalyzed by CmLDH1 proceeded in 1 mL assay solution [100 mM sodium acetate (pH 4.0–5.5), Tris-HCl (pH 7.0–8.0), or the phosphate-citrate buffer (pH 4.0–8.0), different concentrations of sodium pyruvate, NADH, and CmLDH1]. After incubating the assay solution without sodium pyruvate and NADH at different temperature for 5 min, sodium pyruvate and NADH was added to the assay solution to initiate the enzymatic reaction. During the reaction, the decrease in the NADH concentration, namely the change of the absorbance at 340 nm, was monitored for 1 min using a Hitachi U-3900H spectrophotometer (Hitachi High-Tech., Tokyo, Japan). The enzymatic activity of 1 unit was defined as the amount of enzyme that converts 1 μmol of substrate per minute. The Vmax (the maximum reaction velocity) and S0.5 (the substrate concentration at 1/2 Vmax) of CmLDH1 were calculated by curve fitting of the hill equation (Dixon and Webb 1979) (below) using the KaleidaGraph ver. 4.5 software.
The kcat (turnover number) were calculated from Vmax.
Cultivation of C. merolae and measurement of CmLDH activity in the cell extracts
Cyanidioschyzon merolae NIES-3377 (from the National Institute for Environmental Studies) was cultivated in 70 mL of Modified Allen’s medium containing 20 mM (NH4)2SO4 (pH 2.5) at 40 °C (Minoda et al. 2004). During the cultivation, the cultures were bubbled with 1% (v/v) CO2 in the air under a white light (25 μmol/m2/s photons). After 3 days of the cultivation, the cell density (OD730) was measured by a Shimadzu UV-2400 spectrophotometer (Shimadzu, Kyoto, Japan). C. merolae cells were recultivated for 3 days from OD730 = 0.4. Measurement of CmLDH activity in cell extracts of C. merolae were performed as described previously (Yoshida et al. 2024). After 3 days of the cultivation, C. merolae cells [OD730 × culture volume (mL) = 100] were collected by centrifugation at 5800 × g for 2 min. The cells were resuspended in 1 mL of PBS-T [0.137 M NaCl, 2.7 mM KCl, 8.1 mM Na2HPO4⋅12H2O, 1.47 mM KH2PO4, 0.005% (w/v) Tween-20] and sonicated on ice by a model VC-750 sonicator (EYELA, Tokyo, Japan) at 20% intensity for 10 s. The sonication was repeated five times. The mixture was centrifugated at 17,400 × g for 5 min at 4 °C. The total protein concentration in the supernatant was measured by a Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, Rockford, IL, USA), and 200 μg of total proteins was used for enzyme assay.
Results
We compared the amino acid sequences of five CmLDH isozymes (CmLDH1–5) (Fig. 1). l-LDH is a paralog of MDH, and the five amino acid residues determine l-LDH or MDH (Yin and Kirsch 2007). CmLDH isozymes excluding CmLDH4 possessed the five amino acid residues that are widely conserved in l-LDHs (aa47: Valine, aa115: glutamine, aa119: glutamate, aa258: Alanine, aa262: Isoleucine) (Yin and Kirsch 2007) (Fig. 1). The N-terminal sequence of CmLDH4 was 80 residues shorter than those of the other CmLDHs (Fig. 1). Therefore, CmLDH4 did not possess one of the amino acid residues determining l-LDH or MDH (aa47) (Fig. 1). The BLAST analysis when CmLDH1 was set at a query sequence revealed that the sequence identities and similarities (positives) of CmLDH1 and the other isozymes were 99% and ≥ 99%, respectively (Table 1). CmLDH isozymes excluding CmLDH4 possessed identical amino acid sequences without five amino acids residues (aa19, 26, 144, 150, and 300) (Fig. 1). The five residues were not included in the substrate binding site defined in Homo sapience LDH (Pineda et al. 2007) and the NADH binding site defined in Bacillus stearothermophilus LDH (Wigley et al. 1992) (Fig. 1). CmLDH4 did not possess one of the amino acids residues composing the NADH binding site defined in B. stearothermophilus LDH (aa68) (Fig. 1). An amino acid sequence of CmLDH1 has been used to determine the localization of CmLDHs in the cells as representative CmLDH (Moriyama et al. 2015). Hence, we biochemically characterized CmLDH1 in this study.

Comparison of amino acid sequences of LDHs and E. coli MDH. Amino acid sequences of LDHs and E. coli MDH were aligned using CLC Sequence Viewer ver. 8.0. The eMDH, HLDH, and BLDH are E. coli MDH, Homo sapience LDH, and Bacillus stearothermophilus LDH, respectively. The order of amino acid residues of these enzymes is based on that of CmLDH1. The orange squares represent the amino acid residues that differ between CmLDHs (aa19, 26, 144, 150, and 300). The blue squares represent the amino acid residues distinguishing l-LDH and MDH (aa47, 115, 119, 258, and 262) (Yin and Kirsch 2007). The green, gray, and purple squares represent the substrate binding site defined in H. sapience LDH (aa121, 153, 184, 208, and 268) (Pineda et al. 2007), NADH binding site defined in B. stearothermophilus LDH (aa68, 110, 113, 114, 115, 151, 153 and 177) (Wigley et al. 1992), and FBP binding site defined in B. stearothermophilus LDH (aa186 and 201) (Wigley et al. 1992), respectively
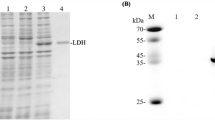
We purified and biochemically characterized a glutathione-S-transferase (GST)-tagged CmLDH1. The single band was localized between 75 and 50 kDa in the SDS-PAGE after purification of CmLDH1 (Fig. 2a). The position of the single band corresponded to the molecular weight of GST-CmLDH1 (63.9 kDa) (Fig. 2a). The purified CmLDH1 exhibited the highest activity under 57 °C and pH 4.5 (Fig. 2b). The CmLDH1 activity on different concentrations of pyruvate and NADH were measured for calculation of kinetic parameters of CmLDH1 under 57 °C and pH 4.5 (Fig. 3). The S0.5 (the substrate concentration at 1/2 Vmax), kcat (turnover number), and kcat/S0.5 (catalytic efficiency) of CmLDH1 for pyruvate were 0.13 mM, 314 s–1, and 2461 s−1 mM−1 under 57 °C and pH 4.5 (Table 2). The S0.5, kcat, and kcat/S0.5 of CmLDH1 for NADH were 0.011 mM, 324 s−1, and 29,473 s−1 mM−1 under 57 °C and pH 4.5 (Table 2). The pH of cytosol in C. merolae is pH 6.35 to 7.1 (Zenvirth et al. 1985). We also measured the CmLDH1 activity on different concentrations of pyruvate and NADH under 57 °C and pH 7.0 (Fig. 3). The S0.5, kcat, and kcat/S0.5 of CmLDH1 for pyruvate were 0.20 mM, 79 s−1, and 387 s−1 mM−1 under 57 °C and pH 7.0 (Table 2). The S0.5, kcat, and kcat/S0.5 of CmLDH1 for NADH were 0.0064 mM, 65 s−1, and 10,213 s−1 mM−1 under 57 °C and pH 7.0 (Table 2). CmLDH1 activity linearly decreased depending on incubation time at pH 4.5 and 7.0 (Fig. 4). The t1/2 (time where the residual activity was 50%) of CmLDH1 at pH 4.5 and 7.0 was calculated as 192 and 518 min, respectively (Fig. 4).

Temperature and pH dependence of CmLDH1 activity. a Result of SDS-PAGE after purification of CmLDH1. b Effects of temperature (top) and pH (bottom) on CmLDH1 activity. Regarding the measurement of temperature dependence of CmLDH1 activity, pH was fixed at pH 4.5. Regarding the measurement of pH dependence of CmLDH1 activity, the temperature was fixed at 57 °C. The sodium pyruvate and NADH concentrations were 1 mM and 0.15 mM, respectively. The amount of CmLDH1 was 3 pmol. Data exhibit average ± standard deviation obtained from three independent experiments

Saturation curves of CmLDH1 for pyruvate and NADH. a Saturation curves of CmLDH1 for pyruvate. The experiments were performed under 57 °C and pH 4.5 or 7.0. The concentration of NADH was 0.15 mM. The amount of CmLDH1 was 3 pmol. b Saturation curves of CmLDH1 for NADH. The experiments were performed under 57 °C and pH 4.5 or 7.0. The concentration of sodium pyruvate was 2 mM. The amount of CmLDH1 was 0.5 pmol. All data in Fig. 3 exhibit average ± standard deviation from three independent experiments

pH stability of CmLDH1. CmLDH1 activities are represented by residual activities, and the activity without incubation at pH 4.5 or 7.0 was 100%. The blue and orange makers indicate residual activities after incubation at pH 4.5 and 7.0, respectively. The temperature was set at 57 °C. The sodium pyruvate and NADH concentrations were 2 mM and 0.15 mM, respectively. The amount of CmLDH1 was 3 pmol. The t1/2 (time where the residual activity was 50%) was calculated by a linear equation obtained from all the values. Data exhibit average ± standard deviation obtained from three independent experiments
We examined the effect of the five metabolites, effectors of l-LDHs from other organisms, on CmLDH1 (Fig. 5) (Oba et al. 1977; Betsche 1981; Götz and Schleifer 1975; Gaspar et al. 2007; Feldman-Salit et al. 2013; Matoba et al. 2014; Steinbüchel and Schlegel 1983; Davies and Davies 1972). Under 57 °C and pH 4.5, the five metabolites decreased CmLDH1 activity (Fig. 5a). Under 57 °C and pH 7.0, ATP and ADP (particularly ATP) decreased CmLDH1 activity (Fig. 5b). ATP also decreased CmLDH1 activity under 30–50 °C and CmLDH activity in cell extracts of C. merolae (Fig. 6). ATP increased the S0.5 of CmLDH1 for NADH and decreased the kcat and kcat/S0.5 of CmLDH1 for pyruvate and NADH (Table 2). Under 57 °C and pH 7.0, CmLDH1 activity did not change and decreased in the presence of 1 mM and 5 mM AMP, respectively (Fig. 5b). Under 57 °C and pH 7.0, CmLDH1 activity did not change and increased in the presence of 1 mM and 5 mM FBP, respectively (Fig. 5b). Under 57 °C and pH 7.0, CmLDH1 activity did not change and decreased in the presence of 1 mM and 5 mM phosphoenolpyruvate (PEP), respectively (Fig. 5b).

Effects of metabolites on CmLDH1 activity. a CmLDH1 activities in the presence of different metabolites under 57 °C and pH 4.5. The concentration of sodium pyruvate was 0.13 mM (S0.5 at pH 4.5). The concentration of NADH was 0.05 mM because the absorbance change in the presence of inhibitors was not detected when the concentrations of both substrates were S0.5. The amount of CmLDH1 was 1 pmol. b CmLDH1 activities in the presence of different metabolites under 57 °C and pH 7.0. The sodium pyruvate and NADH concentrations were 0.20 mM (S0.5 at pH 7.0) and 0.05 mM, respectively. The amount of CmLDH1 was 1 pmol. CmLDH1 activity in Fig. 5 was represented by relative activity when the activity in the absence of metabolites was 100%. All data in Fig. 5 exhibit average ± standard deviation from three independent experiments. Asterisks exhibit significant differences between CmLDH1 activities in the presence and absence of metabolites (Welch’s t-test: *P < 0.05, **P < 0.005). All metabolites used in this experiment as effectors are sodium salt. FBP: Fructose-1,6-bisphosphate, PEP Phosphoenolpyruvate

Effect of ATP on CmLDH activities. a CmLDH1 activities in the presence of 1 mM ATP at different temperatures. The pH was fixed at pH 7.0. The sodium pyruvate and NADH concentrations were 0.20 mM and 0.05 mM, respectively. The amount of CmLDH1 was 1 pmol. b CmLDH activities in cell extracts of C. merolae in the presence and absence of 1 mM ATP. The sodium pyruvate and NADH concentrations were 0.20 mM and 0.05 mM, respectively. The amount of total proteins was 200 μg. CmLDH activity in Fig. 6 was represented by relative activity when the activity without ATP was 100%. All data in Fig. 6 exhibit average ± standard deviation obtained from three independent experiments. Asterisks exhibit significant differences between CmLDH activities in the presence and absence of ATP (Welch’s t-test: **P < 0.005)
Discussion
In this study, we compared the amino acid sequences of five CmLDH isozymes and examined the biochemical properties of CmLDH1, such as catalytic efficiency and tolerance to effectors.
CmLDHs excluding CmLDH4 possessed almost identical amino acid sequences (Fig. 1 and Table 1). Although the N-terminal sequence of CmLDH4 was shorter than those of the other CmLDHs, the upstream sequence of CmLDH4 was similar to the N-terminal sequence of the other CmLDHs (Fig. 1). In Cyanidiophyceae, including C. merolae, gene duplications are observed in subtelomeric regions, and the composition of the duplicated genes varies depending on the lineages (Cho et al. 2023). In C. merolae genome, all genes encoding CmLDHs are located in the subtelomeric regions (Nozaki et al. 2007). These results suggest that genes encoding CmLDHs were generated by gene duplication in the subtelomeres. Among CmLDHs, only CmLDH4 did not possess amino acid residues equivalent to positions 47 and 68 of CmLDH1 (Fig. 1), suggesting that CmLDH4 cannot catalyze pyruvate reduction.
The catalytic efficiency of CmLDH1 for both substrates at pH 4.5 was higher than those of l-LDHs from other organisms (4 species) (Table 3). The catalytic efficiency of CmLDH1 for pyruvate at pH 7.0 was higher than those of l-LDHs from Cryptosporidium parvum, Limosilactobacillus fermentum, and Sporolactobacillus inulinus and similar to that of Enterococcus Mundtii (pH 7.5, 3 mM FBP) (Table 3). The catalytic efficiency of CmLDH1 for NADH at pH 7.0 was higher than those of l-LDHs from Cryptosporidium parvum and Limosilactobacillus fermentum and similar to that of Enterococcus Mundtii (pH 7.5, 3 mM FBP) (Table 3). These comparisons suggest that CmLDH1 is a high-activity l-LDH. Although absolute concentrations (molar concentrations) of pyruvate and NADH in C. merolae have been not reported, those of yeast have been reported as those of unicellular eukaryotes (pyruvate: 9.4 mM, NADH: 0.11 mM) (Park et al. 2016). These concentrations of pyruvate and NADH were markedly higher than the S0.5 of CmLDH1 (pyruvate: 0.13–0.20 mM, NADH: 0.0064–0.011 mM) (Table 2). This result suggests that CmLDH1 shows high activity similar to Vmax in the cells. Absolute quantification of intracellular metabolites of C. merolae is also necessary to determine the CmLDH1 activity in the cells accurately in the future. Previous microarray analysis revealed that the expression levels of genes encoding CmLDH and glycolysis enzymes rather than the tricarboxylic acid cycle enzymes increase at night (Miyagishima et al. 2019), suggesting that lactic fermentation is one of the main energy conversions at night in C. merolae. The high catalytic activity of CmLDH1 might enable C. merolae to perform efficient lactate fermentation at night. The stability of CmLDH1 was higher at pH 7.0 than at pH 4.5 (Fig. 4). Unlike l-lactate production at neutral pH in C. merolae, that at acidic pH leads to a decrease in intracellular pH and reaches a plateau at an early period (Yoshida et al. 2024). This might be due to the low stability of CmLDHs at acidic pH.
CmLDH1 activity was inhibited by ATP, ADP, and AMP (particularly ATP) in vitro (Fig. 5). These metabolites inhibit l-LDHs from sweet potato roots, Lactuca sativa L, and Staphylococcus epidermidis (Oba et al. 1977; Betsche 1981; Götz and Schleifer 1975). In C. merolae, the concentration of ATP is similar to that of ADP and higher than that of AMP (Miyagishima et al. 2019). Also, the absolute concentration of ATP in yeast (1.9 mM) (Park et al. 2016) is higher than the ATP concentration where ATP inhibited both CmLDHs in cell extracts of C. merolae and purified CmLDH1 (1 mM) (Figs. 5 and 6). These results suggest that among the adenine nucleotides, ATP mainly acts as an inhibitor of CmLDH1 in vivo. In L. sativa LDH, ATP decreases the affinity for NADH and acts as a competitive inhibitor for NADH (Betsche 1981). In CmLDH1, ATP decreased not only the affinity but also the kcat for NADH (Table 2). This suggests that ATP acts as a mixed inhibitor for NADH and does not bind to the NADH binding site in CmLDH1 (Fig. 1). C. merolae keeps the adenylate energy charge (balance of adenine nucleotides) almost constant throughout the day/night cycle (Miyagishima et al. 2019). Therefore, we presume that ATP generated via lactic fermentation strongly inhibits CmLDHs to avoid the overproduction of ATP at night.
CmLDH1 activity was affected by FBP and PEP in vitro (Fig. 5). FBP inhibited and slightly activated CmLDH1 activity at pH 4.5 and 7.0, respectively (Fig. 5). The pH of cytosol in C. merolae is neutral pH (Zenvirth et al. 1985), suggesting that FBP activates CmLDH activity in vivo. The activation of l-LDHs by FBP has been confirmed in bacteria (Lactococcus lactis, Lactobacillus plantarum, Streptococcus pyogenes, Enterococcus faecalis, Enterococcus mundtii, B. stearothermophilus) (Gaspar et al. 2007; Feldman-Salit et al. 2013; Matoba et al. 2014; Flores and Ellington 2005). The activities of l-LDHs from L. lactis, L. plantarum, S. pyogenes, and E. faecalis increase 1000, 1.05, 83, and 7.8-fold in the presence of 3 mM FBP (Gaspar et al. 2007; Feldman-Salit et al. 2013). B. stearothermophilus LDH activity increases 15-fold in the presence of 5 mM FBP (Flores and Ellington 2005). Although CmLDH1 activity increased 1.2-fold in the presence of 5 mM FBP (Fig. 5b), the absolute concentration of FBP in yeast (4 mM) is below 5 mM (Park et al. 2016). These results suggest that FBP is not essential for the catalytic activity of CmLDH1. CmLDHs did not possess histidine at position 201 composing the FBP binding site defined in B. stearothermophilus LDH (Fig. 1). This might be why CmLDH1 activity hardly depended on FBP. CmLDH1 activity did not change and decreased in the presence of 1 mM and 5 mM PEP at pH 7.0, respectively (Fig. 5b). The inhibition of l-LDHs by PEP has been confirmed in Cupriavidus necator, Ipomoea batatas, and Solanum tuberosum (Steinbüchel and Schlegel 1983; Oba et al. 1977; Davies and Davies 1972). The absolute concentration of PEP in yeast (0.029 mM) is below 1 mM (Park et al. 2016), suggesting that PEP hardly affects CmLDH1 activity in vivo.
This study revealed the biochemical properties of CmLDH1. Our findings contribute to understanding the biochemical characteristics of l-LDHs in microalgae and the regulatory mechanism of lactic fermentation in C. merolae. CmLDH1 was inhibited by ATP (Figs. 5 and 6). Therefore, the relief of the inhibition by novel culture methods and genetic manipulation of C. merolae might lead to an increase in l-lactate production of C. merolae.
Data availability
Not applicable.
References
Abdel-Rahman MA, Tashiro Y, Sonomoto K (2013) Recent advances in lactic acid production by microbial fermentation processes. Biotechnol Adv 31:877–902. https://doi.org/10.1016/j.biotechadv.2013.04.002
Angermayr SA, Hellingwerf KJ (2013) On the use of metabolic control analysis in the optimization of cyanobacterial biosolar cell factories. J Phys Chem B 117:11169–11175. https://doi.org/10.1021/jp4013152
Barman TE (1969) Enzyme handbook, vol 1. Springer, Berlin
Betsche T (1981) l-Lactate dehydrogenase from leaves of higher plants. Kinetics and regulation of the enzyme from lettuce (Lactuca sativa L). Biochem J 195:615–622. https://doi.org/10.1042/bj1950615
Bilgili F, Koçak E, Kuşkaya S, Bulut Ü (2020) Estimation of the co-movements between biofuel production and food prices: a wavelet-based analysis. Energy. https://doi.org/10.1016/j.energy.2020.118777
Chen W, Wei L, Zhang Y, Shi D, Ren W, Zhang Z, Wang J, Shao W, Liu X, Chen C, Gao Q (2019) Involvement of the two l-lactate dehydrogenase in development and pathogenicity in Fusarium graminearum. Curr Genet 65:591–605. https://doi.org/10.1007/s00294-018-0909-6
Cho CH, Park SI, Huang TY, Lee Y, Ciniglia C, Yadavalli HC, Yang SW, Bhattacharya D, Yoon HS (2023) Genome-wide signatures of adaptation to extreme environments in red algae. Nat Commun 14:10. https://doi.org/10.1038/s41467-022-35566-x
Cook WJ, Senkovich O, Hernandez A, Speed H, Chattopadhyay D (2015) Biochemical and structural characterization of Cryptosporidium parvum lactate dehydrogenase. Int J Biol Macromol 74:608–619. https://doi.org/10.1016/j.ijbiomac.2014.12.019
Davies DD, Davies S (1972) Purification and properties of l(+)-lactate dehydrogenase from potato tubers. Biochem J 129:831–839. https://doi.org/10.1042/bj1290831
De Luca P, Taddei R, Varano L (1978) ‘Cyanidioschyzon merolae’: a new alga of thermal acidic environments. Webbia 33:37–44
Dennis D, Kaplan NO (1960) d- and l-lactic acid dehydrogenases in Lactobacillus plantarum. J Biol Chem 235:810–818
Dixon M, Webb EC (1979) Enzymes. Longman, London, pp 400–402
Feldman-Salit A, Hering S, Messiha HL, Veith N, Cojocaru V, Sieg A, Westerhoff HV, Kreikemeyer B, Wade RC, Fiedler T (2013) Regulation of the activity of lactate dehydrogenases from four lactic acid bacteria. J Biol Chem 288:21295–21306. https://doi.org/10.1074/jbc.M113.458265
Flores H, Ellington AD (2005) A modified consensus approach to mutagenesis inverts the cofactor specificity of Bacillus stearothermophilus lactate dehydrogenase. Protein Eng Des Sel 18:369–377. https://doi.org/10.1093/protein/gzi043
Gaspar P, Neves AR, Shearman CA, Gasson MJ, Baptista AM, Turner DL, Soares CM, Santos H (2007) The lactate dehydrogenases encoded by the ldh and ldhB genes in Lactococcus lactis exhibit distinct regulation and catalytic properties - comparative modeling to probe the molecular basis. FEBS J 274:5924–5936. https://doi.org/10.1111/j.1742-4658.2007.06115.x
Ghaffar T, Irshad M, Anwar Z, Aqil T, Zulifqar Z, Tariq A, Kamran M, Ehsan N, Mehmood S (2014) Recent trends in lactic acid biotechnology: a brief review on production to purification. J Radiat Res Appl Sci 7:222–229. https://doi.org/10.1016/j.jrras.2014.03.002
Götz F, Schleifer KH (1975) Purification and properties of a fructose-1,6-diphosphate activated l-lactate dehydrogenase from Staphylococcus epidermidis. Arch Microbiol 105:303–312. https://doi.org/10.1007/BF00447150
Jonas HA, Anders RF, Jago GR (1972) Factors affecting the activity of the lactate dehydrognease of Streptococcus cremoris. J Bacteriol 111:397–403. https://doi.org/10.1128/jb.111.2.397-403.1972
Jönsson M, Saleihan Z, Nes IF, Holo H (2009) Construction and characterization of three lactate dehydrogenase-negative Enterococcus faecalis V583 mutants. Appl Environ Microbiol 75:4901–4903. https://doi.org/10.1128/AEM.00344-09
Lu H, Bai Y, Fan T, Zhao Y, Zheng X, Cai Y (2018) Identification of a l-lactate dehydrogenase with 3,4-dihydroxyphenylpyruvic reduction activity for l-danshensu production. Process Biochem 72:119–123. https://doi.org/10.1016/j.procbio.2018.06.011
Matoba Y, Miyasako M, Matsuo K, Oda K, Noda M, Higashikawa F, Kumagai T, Sugiyama M (2014) An alternative allosteric regulation mechanism of an acidophilic l-lactate dehydrogenase from Enterococcus mundtii 15-1A. FEBS Open Bio 4:834–847. https://doi.org/10.1016/j.fob.2014.08.006
Matsuzaki M, Misumi O, Shin-I T, Maruyama S, Takahara M, Miyagishima SY, Mori T, Nishida K, Yagisawa F, Nishida K, Yoshida Y, Nishimura Y, Nakao S, Kobayashi T, Momoyama Y, Higashiyama T, Minuteoda A, Sano M, Nomoto H, Oishi K, Hayashi H, Ohta F, Nishizaka S, Haga S, Miura S, Morishita T, Kabeya Y, Terasawa K, Suzuki Y, Ishii Y, Asakawa S, Takano H, Ohta N, Kuroiwa H, Tanaka K, Shimizu N, Sugano S, Sato N, Nozaki H, Ogasawara N, Kohara Y, Kuroiwa T (2004) Genome sequence of the ultrasmall unicellular red alga Cyanidioschyzon merolae 10D. Nature 428:653–657
Minoda A, Sakagami R, Yagisawa F, Kuroiwa T, Tanaka K (2004) Improvement of culture conditions and evidence for nuclear transformation by homologous recombination in a red alga, Cyanidioschyzon merolae 10D. Plant Cell Physiol 45:667–671. https://doi.org/10.1093/pcp/pch087
Miyagishima SY, Era A, Hasunuma T, Matsuda M, Hirooka S, Sumiya N, Kondo A, Fujiwara T (2019) Day/night separation of oxygenic energy metabolism and nuclear DNA replication in the unicellular red alga Cyanidioschyzon merolae. Mbio 10:e00833-e919. https://doi.org/10.1128/mbio.00833-19
Moriyama T, Mori N, Sato N (2015) Activation of oxidative carbon metabolism by nutritional enrichment by photosynthesis and exogenous organic compounds in the red alga Cyanidioschyzon merolae: evidence for heterotrophic growth. Springerplus 4:559. https://doi.org/10.1186/s40064-015-1365-0
Nozaki H, Takano H, Misumi O, Terasawa K, Matsuzaki M, Maruyama S, Nishida K, Yagisawa F, Yoshida Y, Fujiwara T, Takio S, Tamura K, Chung SJ, Nakamura S, Kuroiwa H, Tanaka K, Sato N, Kuroiwa T (2007) A 100%-complete sequence reveals unusually simple genomic features in the hot-spring red alga Cyanidioschyzon merolae. BMC Biol 5:28. https://doi.org/10.1186/1741-7007-5-28
Oba K, Murakami S, Uritani I (1977) Partial purification and characterization of l-lactate dehydrogenase isozymes from sweet potato roots. J Biochem 81:1193–1201
Ohta N, Sato N, Kuroiwa T (1998) Structure and organization of the mitochondrial genome of the unicellular red alga Cyanidioschyzon merolae deduced from the complete nucleotide sequence. Nucleic Acids Res 26:5190–5198. https://doi.org/10.1093/nar/26.22.5190
Ohta N, Matsuzaki M, Misumi O, Miyagishima SY, Nozaki H, Tanaka K, Shin-I T, Kohara Y, Kuroiwa T (2003) Complete sequence and analysis of the plastid genome of the unicellular red alga Cyanidioschyzon merolae. DNA Res 10:67–77. https://doi.org/10.1093/dnares/10.2.67
Park JO, Rubin SA, Xu YF, Amador-Noguez D, Fan J, Shlomi T, Rabinowitz JD (2016) Metabolite concentrations, fluxes, and free energies imply efficient enzyme usage. Nat Chem Biol 12:482–489. https://doi.org/10.1038/nchembio.2077
Pineda JR, Callender R, Schwartz SD (2007) Ligand binding and protein dynamics in lactate dehydrogenase. Biophys J 93:1474–1483. https://doi.org/10.1529/biophysj.107.106146
Steinbüchel A, Schlegel HG (1983) NAD-linked l(+)-lactate dehydrogenase from the strict aerobe Alcaligenes eutrophus. 1. Purification and properties. Eur J Biochem 130:321–328. https://doi.org/10.1111/j.1432-1033.1983.tb07155.x
Sun L, Zhang C, Lyu P, Wang Y, Wang L, Yu B (2016) Contributory roles of two l-lactate dehydrogenases for l-lactic acid production in thermotolerant Bacillus coagulans. Sci Rep 6:37916. https://doi.org/10.1038/srep37916
Tomita Y, Yoshioka K, Iijima H, Nakashima A, Iwata O, Suzuki K, Hasunuma T, Kondo A, Hirai MY, Osanai T (2016) Succinate and lactate production from Euglena gracilis during dark, anaerobic conditions. Front Microbiol 7:2050. https://doi.org/10.3389/fmicb.2016.02050
Tsuji H (2005) Poly(lactide) stereocomplexes: formation, structure, properties, degradation, and applications. Macromol Biosci 5:569–597. https://doi.org/10.1002/mabi.200500062
Tsuji H, Takai H, Saha SK (2006) Isothermal and non-isothermal crystallization behavior of poly(l-lactic acid): effects of stereocomplex as nucleating agent. Polymer 47:3826–3837. https://doi.org/10.1016/j.polymer.2006.03.074
Wigley DB, Gamblin SJ, Turkenburg JP, Dodson EJ, Piontek K, Muirhead H, Holbrook JJ (1992) Structure of a ternary complex of an allosteric lactate dehydrogenase from Bacillus stearothermophilus at 2.5 a resolution. J Mol Biol 223:317–335. https://doi.org/10.1016/0022-2836(92)90733-Z
Wu B, Yu Q, Zheng S, Pedroso MM, Guddat LW, He B, Schenk G (2019) Relative catalytic efficiencies and transcript levels of three d- and two l-lactate dehydrogenases for optically pure d-lactate production in Sporolactobacillus inulinus. MicrobiologyOpen 8:e00704. https://doi.org/10.1002/mbo3.704
Yin Y, Kirsch JF (2007) Identification of functional paralog shift mutations: conversion of Escherichia coli malate dehydrogenase to a lactate dehydrogenase. Proc Natl Acad Sci USA 104:17353–17357. https://doi.org/10.1073/pnas.0708265104
Yoshida A (1965) Enzymatic properties of lactate dehydrogenase of Bacillus subtilis. Biochem Biophys Acta 99:66–77. https://doi.org/10.1016/s0926-6593(65)80008-x
Yoshida C, Akiyama Y, Iwazumi K, Osanai T, Ito S (2024) l-Lactate production from carbon dioxide in the red alga Cyanidioschyzon merolae. Algal Res 80:103526. https://doi.org/10.1016/j.algal.2024.103526
Zenvirth D, Volokita M, Kaplan A (1985) Photosynthesis and inorganic carbon accumulation in the acidophilic alga Cyanidioschyzon merolae. Plant Physiol 77:237–239. https://doi.org/10.1104/pp.77.1.237
Funding
Open Access funding provided by Meiji University. The following grants to TO supported this work: JSPS KAKENHI Grant-in-Aid for Scientific Research (B) (grant number 20H02905), JST-ALCA of the Japan Science and Technology Agency (grant number JPMJAL1306), Asahi Glass Foundation, and Sugar Industry Association.
Author information
Authors and Affiliations
Contributions
M.Y. designed the study, performed the experiments, analyzed the data, and wrote the manuscript. T.O. designed the study and wrote the manuscript. S.I. designed the study, analyzed the data, and wrote the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yamamoto, M., Osanai, T. & Ito, S. l-Lactate dehydrogenase from Cyanidioschyzon merolae shows high catalytic efficiency for pyruvate reduction and is inhibited by ATP. Plant Mol Biol 114, 98 (2024). https://doi.org/10.1007/s11103-024-01495-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11103-024-01495-0