
Abstract
In angiosperms, the WOX family of transcription factors has important functions in meristem regulation and in control of the partitioning of developing embryos into functional domains. In this study, a putative WOX2 homologous gene was isolated from Picea abies, and its expression pattern during somatic embryo development was followed using real-time quantitative reverse transcription polymerase chain reaction (qRT-PCR). We used strategies of both absolute and relative quantification of gene expression, and benefits and disadvantages of the two methods are presented and discussed. During embryogenesis, PaWOX2 expression was highest at the earliest stages of development, but low levels were also detected in seedling tissues. No PaWOX2 expression was detected in a non-embryogenic cell culture, indicating that PaWOX2 plays a fundamental role during early somatic embryo development, and can be used as a possible marker for embryogenic potential. Additional results show that conifers, like angiosperms, contain a large number of WOX-related genes, many of them expressed during embryo development. In phylogenetic analysis based on the deduced homeodomain of retrieved pine and spruce EST sequences, no conifer WUS homolog was found. Neither did we find any homeodomain to cluster with WOX5. Interestingly, a clade including only conifer sequences derived from various tissues was resolved as sister to a Physcomitrella WOX-like gene, suggestive of the early origin of this gene family. Our results thus provide basic information for further studies of the evolution of this gene family and of their function in relation to meristem dynamics and specification of stem cells in gymnosperms.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Somatic embryogenesis is a developmental process, describing the in vitro production of bipolar structures capable of germination with a unified shoot and root induction phase. In conifers, plantlets were successfully produced by this method more than 20 years ago independently by Hakman and co-workers (Hakman 1985; Hakman et al. 1985; Hakman and von Arnold 1985) and Chalupa (1985), and is currently receiving considerable interest as a mass propagation method to be integrated with conventional breeding programs for conifers (Park 2002; Stasolla and Yeung 2003; Sutton 2002; Giri et al. 2004). Apart from its use in forest clonal propagation programs, somatic embryogenesis also serves as an invaluable experimental system in basic studies of conifer embryo development since it allow us do obtain large numbers of embryos of specific developmental stages the year around, and to manipulate their development in culture (Hakman et al. 1990). Embryogenic tissue cultures can also be used as target tissue for introducing new genes from which transgenic plants can be regenerated, although not as easy as many angiosperms. Even though plantlet production via somatic embryogenesis is now firmly established as an efficient technique for propagation, there are still problems to be solved concerning both initiation and maintenance of embryogenic cultures, as well as maturation and conversion of embryos into plantlets. Accordingly, a proper formation of functional root and shoot meristems in the embryos is essential. Although it is possible to discriminate embryogenic tissue cultures from non-embryogenic ones just by visual inspection (Hakman et al. 1985), molecular markers of embryogenic potential are useful. Contrary to non-embryogenic tissue cultures, embryogenic tissue cultures in conifers are translucent-to-white and do not turn green when cultured in light.
Embryogenesis in seed plants can be divided conceptually into two phases; an early morphogenic phase characterized by cell divisions and onset of cell differentiation, and a late maturation phase when the embryo accumulates major storage products and also prepares for desiccation, dormancy and germination. During morphogenesis, the primary body plan is established with regional specification of apical-basal and radial domains from which morphological structures derive. The process by which cells are specified in three dimensions has been termed pattern formation (see e.g., Jürgens 2001; Berleth and Chatfield 2002; Laux et al. 2004; Willemsen and Scheres 2004), and the first insights into pattern formation during plant embryogenesis came from analysis of Arabidopsis mutant seedlings with specific defects in root apical meristem (RAM) and shoot apical meristem (SAM) (see e.g., Laux et al. 2004; Weijers and Jürgens 2005). Among them, mutants of the gene WUSCHEL (WUS), which encodes a homeodomain transcription factor, failed to maintain a proper SAM (Laux et al. 1996). A WUS transcript was expressed already in the 16-cell embryo (Mayer et al. 1998); well before the shoot meristem can be recognized histologically. In later stages, WUS expression becomes restricted to a small sub-apical domain in the SAM, where it appears to play an essential role in maintaining stem cell identity of initial cells in the meristem through a feedback loop between the CLAVATA (CLV) genes (Groß-Hardt and Laux 2003).
Arabidopsis WUS is the founding member of the WUS-related WOX ( W US homeob ox ) gene family of transcription factors comprising at least 15 members in the Arabidopsis genome (Haecker et al. 2004). They all share a characteristic nucleotide sequence, called the homeobox (HB) that encodes a WUS-type homeodomain (HD), with a characteristic helix-turn-helix DNA-binding motif. Beside the HD, many WUS/WOX proteins share the WUS-box, located downstream of the HD. The function of the WUS-box is unknown (Haecker et al. 2004; Nardmann and Werr 2006). Otherwise, little sequence conservation is found. Like WUS, the other WOX genes seem to have important functions in meristem regulation. For example, expression of WOX5 shows that identity of the quiescent centre (QC) of the root meristem is initiated very early in the Arabidopsis embryo (Haecker et al. 2004), which is in agreement with the expression patterns of QHB and ZmWOX5B, rice and maize homologs of Arabidopsis WOX5, respectively (Kamiya et al. 2003; Nardmann et al. 2007). The plant hormone auxin, which is involved in root formation, has been shown to induce expression of WOX5 in both Arabidopsis (Gonzali et al. 2005; Dorantes-Acosta and Vielle-Calzada 2006) and Medicago truncatula (Imin et al. 2007). Based on recent evidence, WOX5 is suggested to have a similar function in the RAM in stem cell signalling as WUS has in the SAM (Kamiya et al. 2003; Sarkar et al. 2007). STIMPY/WOX9 (also called STIP), another member of the WOX family, has been proposed to act in the central zone of SAM where it maintains the stem-cell population by positively regulating the activity of WUS (Wu et al. 2005). STIMPY/WOX9 is also required for maintaining cell division in the embryo and the suspensor, and is thus essential for maintaining tissue growth during embryogenesis (Wu et al. 2007). Recently, Haecker et al. (2004) showed that several of the WOX gene family members are expressed very early during embryo development. The transcription factors, WOX2 and WOX8, are co-expressed in the egg cell and in the zygote, and are then specifically expressed in the apical and basal cell of the Arabidopsis two-celled embryo, respectively, suggesting that the two daughter cells of the zygote assume different transcriptional profiles. Moreover, WOX2 was shown to be functionally required for correct development of the apical domain of the Arabidopsis embryo. In Zea mays, ZmWOX2A, a homolog of Arabidopsis WOX2, is expressed in the zygote and, from the few-celled stage and onwards, transcripts are exclusively detected in the apical domain of the embryo proper. Later on, the transcriptional activity of the gene becomes confined to the adaxial face of the embryo proper, where the SAM will emerge (Nardmann et al. 2007). Several WOX genes, including WOX2, apparently constitute markers of cell fate during early embryogenesis, suggesting that they might have important functions in early embryonic patterning (Haecker et al. 2004; Nardmann et al. 2007).
Given that many of the WOX family members appears to function in meristem regulation and that WOX2 is expressed very early during Arabidopsis and Zea mays embryo development, we wanted to find out if the expression of a putative WOX2 gene in P. abies followed a similar developmental pattern and if it could be used as an early embryogenic marker in our tissue culture system. In the present work we isolated and analyzed a putative PaWOX2 gene (accession number AM286747) for its expression pattern during somatic embryo development using real-time quantitative reverse transcription polymerase chain reaction (qRT-PCR). To handle the problems of finding suitable endogenous reference genes for relative quantifications, innate to tissues with very dynamic gene activity program, such as developing embryos, as well as problems associated with the use of standard curves and normalization against total RNA mass for absolute quantification (Gonçalves et al. 2005; Bustin 2000; Huggett et al. 2005), both quantification strategies were used in this study. Here, we present the expression dynamic of PaWOX2 and show that it is highly expressed early during somatic embryo development, but declines as embryos mature. Our data also indicate that a high PaWOX2 expression is linked to the proliferation rate of the embryogenic cell cultures, but not necessarily to their capacity of forming well-organized early embryos or regenerating mature ones. We also analyzed the PaWOX2 expression in a non-embryogenic cell line. No expression was detected, indicating that PaWOX2 plays a fundamental role during early somatic embryo development, and can be used as a marker for embryogenic potential. In a recent study of the Pinus taeda embryo transcriptome (Cairney et al. 2006), WOX-like sequences were identified, and in this work we present a phylogenetic distribution of WOX-related genes of both pine and spruce in relation to those of the flowering plants and to a moss sequence.
Materials and methods
Plant material and somatic embryo culture
Embryogenic cultures of Norway spruce (Picea abies [L.] Karst.) were initiated as previously described (Hakman et al. 1985, 1990) by culturing immature zygotic embryos isolated from seed cones collected from an open-pollinated tree in Uppsala August 9, 2004. The cultures were initiated and maintained either on a Westvaco (WV5) medium including vitamins (W0260, Duchefa, The Netherlands) supplied with sucrose (30 gl−1) and casein hydrolysate (500 mgl−1), or a half-strength Litvay (1/2LV) medium including vitamins (L0218, Duchefa, The Netherlands) supplied with sucrose (10 gl−1), casein hydrolysate (500 mgl−1) and glutamine (250 mgl−1). Both media contained NAA (2 mgl−1) and BA (1 mgl−1), and were solidified with Phytagel (0.3% w/v). Prior to the experiments the cultures were maintained for over two years by monthly subculture to the same medium. To develop somatic embryos further, pieces of tissue was spread on top of a filter paper on an embryo maturation medium (full-strength LV-salts) containing glutamine (250 mgl−1), casein hydrolysate (500 mgl−1), sucrose (10 gl−1), PEG (7.5% w/w), ABA (2 mgl−1) and Phytagel (0.2% w/v). All cell cultures were kept at 25°C in darkness. About 100–250 mg of tissue was collected and immediately frozen in liquid nitrogen for later gene expression analysis. Samples were stored at −80°C. Proliferating tissues (“calli”) of four cell lines with varying embryogenic potential, here designated as 04WV5 + h#3, 04WV5 + h#3*, 04WV5 + h#6 and 04LV1/2 + h#5, were collected 10 days after their subculture to fresh medium. Developing somatic embryos of cell line 04WV5 + h#3 were isolated with the aid of a dissection microscope. Due to their very small size, stage 1 and stage 2 embryos (for description of stages see Fig. 1) were isolated together 6 days after subculture to maturation medium. Stage 2 and stage 3 embryos were isolated together after two-to-four weeks. Thus, two different tissue pools, both containing stage 2 embryos, were created for later gene expression analysis. Mature embryos (stage 4) were isolated after four-to-eight weeks. All these procedures, including separate transfers to subculture medium, were repeated three times to give three biological replicates.

Somatic embryogenesis in Picea abies. (a–d) Different stages of somatic embryo development in cell line 04WV5 + h#3. (a) Stage 1 embryo on maintenance medium (bar = 1 mm; insert, bar = 0.2 mm). (b) Stage 2 embryos after transfer to maturation medium (bar = 1 mm; insert, bar = 0.2 mm). (c) Stage 3 embryos on maturation medium (bar = 0.5 mm; insert, bar = 0.2 mm). Cotyledons are indicated with arrowheads. (d) Stage 4 embryos (mature embryos) on maturation medium (bar = 5 mm). Cotyledons are indicated with arrowheads. (e–g) Light micrographs comparing cells in calli of cell lines 04WV5 + h#3 and 04WV5 + h#3*. (e) Stage 1 embryo in cell line 04WV5 + 5#3 (bar = 50 μm). (F) Cell aggregates in cell line 04WV5 + h#3* (bar = 100 μm). (g) Small cell aggregates in 04WV5 + h#3*. Note the small dense cells from which larger cells protrude (bar = 50 μm)
Non-embryogenic cell cultures were initiated from hypocotyls of one-week-old seedlings. They were cultured on WV5 medium supplemented with hormones and maintained under the same conditions as described above for the embryogenic cell lines. When proliferation had started, pieces of non-embryogenic calli were placed in light to see if they turned green. One of the non-embryogenic cell lines, 07WV5 + h NE, was used in this study. It was grown for two and a half months before RNA was extracted from calli, 5 days after subculture to fresh medium.
Seeds of P. abies were germinated at 25°C under a 16 h photoperiod in a growth chamber. Seedlings were harvested after two weeks and divided into roots, shoot apices, hypocotyls and cotyledons that were immediately frozen separately in liquid nitrogen for later RNA isolation. This procedure was repeated three times to give three biological replicates.
RNA extraction and cDNA synthesis
Total RNA was extracted from the plant samples by using the RNeasy Plant Mini Kit (Qiagene Science, Maryland, USA). To eliminate the residual genomic DNA present in the samples, RNA was treated with both RNAse-free DNase I (Qiagene GmbH, Hilden, Germany) and TURBO-DNA free ™ (Ambion, Austin, TX, USA) according to the manufacturers’ protocols. Total RNA concentration was quantified using the Quant-it™ RiboGreen® RNA Assay Kit (Molecular Probes, Eugene, OR, USA) and a fluorescence plate reader. Each sample was analyzed in triplicate. The integrity of the RNA was verified by electrophoresis on 1% formaldehyde agarose gels followed by ethidium bromide staining.
For cDNA synthesis, 1 μg (seedling tissue) or 2 μg (somatic embryos and calli) of total RNA was reverse transcribed with oligo (dT) and random hexamer primers using the iScript™ cDNA synthesis kit (Bio-Rad, Hercules, CA, USA) according to the manufacturer’s protocol. Each reaction was run in duplicate generating two independent cDNA samples for every RNA sample.
Database analysis and cloning of PaWOX2
To isolate a potential WOX2-ortolog from P. abies, the pine [Release 6.0 (July 19, 2005)] and spruce [Release 2.0 (June 21, 2006)] EST databases at The Gene Index Databases (http://compbio.dfci.harvard.edu/tgi/) were first screened for sequences homologous to Arabidopsis WOX2 (accession no. NM125325) using the TBLAST algorithm (Altschul et al. 1997) with default parameter settings and the conserved WUS/WOX homeodomain sequence as a query. Homology searches were also performed with the NCBI BLAST program (http://www.ncbi.nlm.nih.gov/BLAST/). Retrieved conifer sequences were then aligned together with all the AtWUS/WOX sequences published recently (Haecker et al. 2004) and phylogenetic reconstructions were performed using the CLC Free Workbench (http://www.clcbio.com) or the free Geneious Basic (http://www.geneious.com) software on both protein and nucleotide data from the entire coding region or from the homeodomain alone (see Supplementary material online, Fig. 1). Based on these results primers were designed from one of the Pinus taeda EST cDNA clones (accession no. DR693345).
cDNA from early staged somatic embryos was used as a template in a polymerase chain reaction (PCR) with primers PtWOX2a (5′-ACTTCTCCCTCCACAGCAGC-3′) and PtWOX2b (5′-TGATGAAAATGCCCCCCG-3′), and the resulting PCR product (290 bp) was sequenced. Rapid amplification of the 3′-cDNA and 5′-cDNA ends was then performed with primers specific for PaWOX2 using the SMART™ RACE cDNA amplification kit (Clonetech, Palo Alto, CA, USA) according to the manufacturer’s protocol. 3′-RACE was performed with the primer PaWOX2a (5′-TGAACAGAACGATGCAAGCTCAGG-3′) and 5′-RACE with the primer PaWOX2b (5′-CGCCTGAGCTTGCATCGTTCTGTTC-3′). Resulting PCR products were cloned using the TOPO-TA cloning kit (Invitrogen, Carlsbad, CA, USA) and sequenced.
Phylogenetic analysis
After cloning and sequencing of the P. abies WOX2 homolog, all the deduced homeodomains of 65 aa from the retrieved conifer sequences were assembled together with those of the Arabidopsis WUS/WOX proteins and selected high-scoring (BLAST; E > 10−5) sequences from other species. One sequence (accession no. BU052399) of the moss Physcomitrella patens retrieved from the Physcomitrella database PHYSCObase (http://moss.nibb.ac.jp/) was also included. After a preliminary alignment and analysis of 84 sequences, we eliminated 16 sequences that were either too distantly related to WUS/WOX (e.g., conifer HD-leucine zipper) or were shorter than 65 aa. Some representatives of the different sublineages that were almost identical were also removed. Our final data set (see Supplementary material online, Fig. 2) included 58 sequences; from Arabidopsis thaliana (15), Petunia x hybrida (1), Antirrhinum majus (1), Zea mays (9), Oryza sativa (7), Populus trichocarpa (10) Picea abies (1), P. sitchensis (2), Pinus palustris (1), P. pinaster (2), P. taeda (8) and Physcomitrella patens (1). The homeodomain sequences were aligned using ClustalX (Thompson et al. 1997), and phylogenetic reconstructions were performed with the Neighbor Joining (NJ) method in PAUP 4.0 (Swofford 2000) or within the CLC Free Workbench software (http://www.clcbio.com). Estimates of confidence were done by bootstrapping (1000 replicates). To evaluate the result further a Parsimony tree was made by a heuristic search. A consensus tree was computed by the 50%-majority rule and a final tree was generated. The Physcomitrella homolog was selected as outgroup based on the preliminary phylogenetic analysis.

Early staged embryos of Picea abies. Plastic embedded sections illustrate the similar morphology of early staged zygotic (a) and somatic (b) embryos. Note the characteristic organization of embryos with small meristematic cells in the embryo proper (E) subtended by long and highly vacuolated suspensor cells (S) (bar = 0.1 μm)
Reference gene selection and primer design
Three housekeeping genes were selected as endogenous reference genes in relative quantification: eukaryotic translation initiation factor 4A (eIF4A), glyceraldehyde-3-phosphate-dehydrogenase (GAPDH), and ubiquitin-conjugating enzyme 1 (UBC1). The nucleotide sequence for P. abies UBC1 was obtained from GenBank (accession no. AY639585) while P. abies GAPDH and P. glauca elF4A were identified by querying the spruce EST database with Pinus sylvestris (GAPDH) and Nicotiana tabacum (elF4A) nucleotide sequences, respectively, using BLASTN. Selected spruce sequences, including PaWOX2, were then used to query the Arabidopsis protein database using BLASTX (Table 1). Primers were designed using Primer3 software (Rozen and Skaletsky 2000) with optimal Tm at 60°C and GC content of 40–60%. The primers were tested to ensure amplification of single discrete bands with no primer-dimers using the same optimized conditions as described below for real-time qRT-PCR. Invitrogen synthesized all primers and all sequencing was done by MWG-Biotech (Ebersberg, Germany).
Absolute and relative real-time qRT-PCR
PCR reactions were performed with a MiniOpticon Real-Time PCR Detection System (Bio-Rad). Reactions were done in a 25 μl volume containing 12.5 μl iQ SYBR Green Supermix (Bio-Rad), 0.3 μM (PaWOX2, UBC1) or 0.4 μM (GAPDH, elF4A) of both primers and 2 μl of cDNA template. The thermal cycling conditions were as follows: 3 min at 95°C, followed by 40 cycles of 10 s at 95°C, 10 s at 60°C, and 10 s at 72°C. Data collection was performed during each extension phase and a melting curve was obtained immediately after amplification. Expression levels were determined as the number of cycles needed for the amplification to reach a threshold fixed in the exponential phase of the PCR reaction (Ct) (Walker 2002).
Absolute quantification of PaWOX2 copy number in each cDNA sample was determined using a standard curve and normalized against μg total RNA. The standard curve was generated with the purified PCR product obtained with primer set of PaWOX2 (Table 1), and was serially diluted from 106 to 101 copies with each step differing by 10-fold and quantified with a fluorescence plate reader using Quant-it™ PicoGreen® dsDNA Assay Kit (Molecular Probes). The corresponding copy number was calculated as previously described (Whelan et al. 2003). Inter- and intra-run variation was determined according to the equation: ±% Molecules = [(E + 1)SD − 1] × 100%, where E is the PCR efficiency (E = 10[−1/slope]) and SD is the standard deviation of Ct obtained from replicate amplifications of the standard curve (Rutledge and Côté 2003).
The relative expression ratio of PaWOX2 was computed based on a variant of the comparative Ct (2−ΔΔCt) method, which combines gene quantification and normalization into a single calculation, incorporating the PCR efficiencies of the target and endogenous reference genes to correct for differences between the assays (Pfaffl 2001). Three housekeeping genes (UBC1, GAPDH, and elF4A) were considered for use in normalization of relative PaWOX2 expression. To determine their stability, Ct values were converted into relative quantities and imported into geNorm v3.4 software (Vandesompele et al. 2002). For each gene, PCR efficiencies were calculated using a calibration curve derived from a pooled cDNA mixture (a 5-fold dilution series with 5 measuring points). The pooled cDNA sample was obtained from the various developmental stages of somatic embryos and the different cell lines of calli, using the same RNA extraction and cDNA synthesis protocols as described above.
All samples were run in quadruplicate (n = 4, two replicates per cDNA sample) and three assays were performed using three independent samples of each tissue collected. Standard and calibration curves were run in triplicates. Negative (distilled water) and no-template (total RNA) controls were included in each run.
After normalization, average values and standard errors of gene copy numbers and relative expression ratios were calculated using data from triplicate assays. Statistical significant differences were evaluated by one-way ANOVA, followed by post hoc Tukey´s HSD comparisons, using STATISTICA v7.1 (StatSoft, Inc.). Differences of P < 0.05 were regarded as significant.
Microscopy
Somatic embryo development was followed with an Olympus SZX12 stereomicroscope equipped with an Olympus DP10 camera and pictures were taken regularly for documentation. Micrographs were also taken with a Zeiss Axiovert 10 inverted microscope equipped with a Leica DFC 320 camera. Pictures were further processed using Adobe® Photoshop® CS.
Results
Cell line characterization and embryo development
Four embryogenic cell lines were selected for this study based on their uniform growth behaviour and consistent regeneration capacity. Pieces of “callus” were regularly transferred to a maturation medium and development of somatic embryos was followed. Four different embryo stages, depicted in Fig. 1, were defined (stage 1–4) and collected. Briefly, stage 1 embryos (Fig. 1a insert and Fig. 1e) that were continuously formed on maintenance medium consisted of an embryo region composed of small cytoplasmic cells subtended by long, highly vacuolated suspensor-like cells. Stage 2 embryos (Fig. 1b insert) were formed after transfer of callus to maturation medium. They were opaque and club-like with a smooth surface. Stage 3 embryos (Fig. 1c insert) bore cotyledons but were still quite small. Stage 4 embryos (Fig. 1d) were fully mature. For comparison, sections of an early staged zygotic (Fig. 2a) and a somatic embryo (Fig. 2b) are presented in Fig. 2. A more thorough comparison of developing Picea abies zygotic and somatic embryos can be found e.g., in Hakman (1993).
The four cell lines had different capacities to form mature embryos (stage 4) on maturation medium, and they also differed in how fast the embryos continued to develop after transfer to that medium as well as in the manner they did so. Judged by how many well-formed embryos were produced, cell line 04WV5 + h#3 was superior to the others. The different cell lines also differed in their proliferation rate, both on the maintenance medium as well as initially on the maturation medium after transfer, before the onset of embryo maturation. With this in mind, cell line 04WV5 + h#6 produced many embryos of the younger stages (stage 1–3), but in comparison to cell line 04WV5 + h#3, not as many of them developed to complete maturity. The embryos sometimes also bore cotyledons in several whorls. Cell line 04LV1/2 + h#5 was both very slow growing and produced only a few mature embryos. The growth behaviour of the different cell lines is summarized in Table 2.
In the beginning of the monthly subculture of cell line 04WV5 + h#3, a very small piece of callus with a greatly altered appearance was detected. It was removed and cultured separately and produced after subdivision a “new” cell line, here named 04WV5 + h#3*, characterized as being white and very fast growing. It did not, however, show much signs of differentiation or regeneration capacity when transferred to the maturation medium. Instead the callus generally turned brown with no further growth of embryos (Table 2). However, a very few mature embryos were occasionally formed at the start of this cell line. Microscopic inspection of proliferating calli of the two cell lines 04WV5 + h#3 and 04WV5 + h#3* showed that cell line 04WV5 + h#3 contained well-organized stage 1 somatic embryos (Fig. 1e) as well as smaller cell aggregates and single cells of various size and shape while cell line 04WV5 + h#3* was much more uniform in composition with many equal sized cells (Fig. 1f). Even though well-organized stage 1 embryos were not observed, aggregates of smaller cells together with more vacuolated ones could be seen (Fig. 1g). The two cell lines thus represent the same genotype albeit with very different growth characters.
In addition to these cell lines, a non-embryogenic cell line, 07WV5 + h NE, was created. This cell line turned green when exposed to light while maintained on proliferation medium and did not produce any embryos when transferred to maturation medium.
Isolation and characterization of PaWOX2
To investigate the possibility of WOX-genes having important functions during conifer somatic embryo patterning, we set out to isolate a WOX2-homolog from the proliferating cell line 04WV5 + h#3. Conifer EST-databases were searched for homologous genes and a number of WOX-like sequences were identified, most of them from embryo libraries (Table 3). Primers were design based on the DNA sequence from one Pinus taeda EST cDNA clone (accession no. DR693345), recently described as a WOX2 homolog by Cairney et al. (2006). A full-length cDNA was prepared from P. abies and designated as PaWOX2 (accession no. AM286747).
The deduced protein (see Supplementary material online, Fig. 1a) contained the regular WUS-homeodomain subtype with 65 amino acids (66 aa in WUS; Mayer et al. 1998; Haecker et al. 2004) instead of the 60 aa found in most animal HDs (Gehring 1993). Although the sequence similarity of the HD varies among species, specific positions in helix I (Leu 16) and helix III (Trp 48, Phe 49 and Asn 51) are almost always conserved (Banarjee-Basu and Baxevanis 2001). The positions were conserved in PaWOX2 as well as in the other plant WOX HDs (in plant HDs: Trp 54, Phe 55, Asn 57, see Supplementary material online, Fig. 2). The high conservation of these residues in the third helix has major implications for DNA binding and overall stability of the tertiary structure of the homeodomain (Banarjee-Basu and Baxevanis 2001). The protein also contained the so-called “WUS-box” (TLELFPLH) which has unknown function, but present in many WUS/WOX-family members (Haecker et al. 2004). The alignment of PaWOX2 protein with other conifer homologs is shown in Fig. 1a, Supplementary material online.
Phylogenetic analysis of conifer WOX homologs
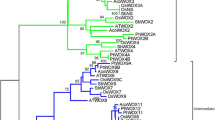
To investigate the evolutionary relationship between PaWOX2 and other retrieved conifer sequences with the angiosperm WUS/WOX homologs, we used the amino acid sequence of the HDs to create phylogenetic trees. In total we included 58 sequences from representatives of both eudicotyledons and monocotyledons together with those from the conifers (Supplementary material online, Fig. 2). For some conifer members, their amino acid sequences were almost identical, not only over the HD but for the entire deduced protein (Supplementary material online, Fig. 1a). If they also showed high degree of identity over the whole DNA sequence, we then included only one or a few members of that group in the phylogenetic analysis. The omission or inclusion of gene family members had no discernable affect on the overall tree topology. For clarity of trees, nearly identical sequences from other plant species were removed as well.
Both a distance method and a tree-searching method creating Neighbor Joining (NJ) trees and Parsimony trees, respectively, were used to perform this analysis. A resulting NJ tree after bootstrapping (1000 replicates) is shown in Fig. 3. The tree is resolved into several clusters. One cluster contains the majority of the WUS/WOX sequences (WUS, WOX1, WOX2, WOX4, WOX3 and WOX6), including P. abies PaWOX2 (PicabWOX2) and its Pinus ortholog (DR693345, PintaWOX2). A sister group consisting of two conifer sequences, DR691632 (PintaWOX4/6) and BX250513 (PinpiWOX4/6), derived from an embryo library and a xylem library, respectively, was found outside this clade. Interestingly, apart from the two conifer WOX2 sequences, the encoding sequence of DR691632 also contains the WUS-box (TLELFPLH), while a potential degenerated sequence (SGESVLW) is found in BX250513 (Table 3). No conifer sequence, however, was found to cluster with the WUS proteins, all of which contain an extra tyrosine residue within their homeodomain in the loop between helix I and helix II (Supplementary material online, Fig. 2). In the tree presented in Fig. 3, WOX5/7 can be seen to form a separate cluster. No conifer representatives were found within this group. Another interesting feature is that a clade including only conifer sequences, TC57841 (PinusWOX13) and TC14114 (PiceaWOX13), both derived from various tissues of Pine and Picea, respectively, are resolved as sister to the Physcomitrella gene (Fig. 3). Another conifer homolog is found within the WOX13 cluster together with the other angiosperms, while several others conifer proteins cluster with WOX8/9, although their relationship is not clear. None of these sequences contained the WUS-box. Interestingly, all of these Arabidopsis WOX proteins also have a less conserved WUS-box (Haecker et al. 2004).

Phylogenetic analysis of plant WUS/WOX proteins based on their HDs. (Neighbor-joining tree of the data set presented in Fig. 2, Supplementary material online. AM286747, PicabWOX2; AJ012310, ArathWUS; AY251394, ArathWOX1; AY251392, ArathWOX2; AY251397, ArathWOX3; AY251396, ArathWOX4; AY251398, ArathWOX5; AY251399, ArathWOX6; NM_120659, ArathWOX7; AY251400, ArathWOX8; AY251401, ArathWOX9; NM_101923, ArathWOX10; AY251402, ArathWOX11; AY251403, ArathWOX12; AY251404, ArathWOX13; NM_101922, ArathWOX14; AF481951, PethyWUS; AY162209, AntmaWUS; AM234744, ZeamaWUS1; AM234767, ZeamaWOX2A; AJ536578, ZeamaWOX3; AM234768, ZeamaWOX4; AM234769, ZeamaWOX5; AM234772, ZeamaWOX9B; AM234773, ZeamaWOX9C; AM234774, ZeamaWOX11/12A; AM234776, ZeamaWOX13A; AB218894, OrysaWUS; AM234749, OrysaWOX2; AM234748, OrysaWOX3; AM234752, OrysaWOX9; AM234753, OrysaWOX9C; AM234754, OrysaWOX11/12; AM234755, OrysaWOX13; AM234747, PoptrWUS; AM234756, PoptrWOX1A; AM234758, PoptrWOX2; AM234759, PoptrWOX4; AM234766, PoptrWOX5/7B; AM234760, PoptrWOX6; AM234761, PoptrWOX9; AM234763, PoptrWOX11/12A; AM234765, PoptrWOX5/7A; AM234762, PoptrWOX13; DR693345, PintaWOX2; DR691632, PintaWOX4/6; TC78309, PintaWOX8/9E; DR692518, PintaWOX8/9; DR685804, PintaWOX8/9F; AW981538, PintaWOX8/9C; BX250513, PinpiWOX4/6; TC61212, PintaWOX8/9A; DR691319, PintaWOX8/9D; TC57841, PinusWOX13; DR687694, PintaWOX8/9B; TC34052, PicsiWOX13A; TC14114, PiceaWOX13; BU052399, PhymiWOX)
Verification of absolute and relative qRT-PCR
A standard curve was generated for the target gene PaWOX2 and effective PCR amplification kinetics was shown by high PCR efficiency per cycle (Table 4). Assay sensitivities were confirmed by detection limits down to 10 ssDNA molecules and linear quantification ranges between 101 and 106 molecules. Intra- and inter-assay variations (expressed as % molecules) of 12.2 ± 3.2 to 7.82, respectively, were determined over the entire quantitative range, showing high precision and reproducibility of the applied assay. Calibration curves with five orders of magnitude were generated for the target gene PaWOX2 and the endogenous housekeeping genes GAPDH, eIF4A, and UBC1 considered for use in relative quantification. The calibration curves gave correlation coefficients (r) greater than 0.99 and PCR efficiencies greater than 87% for all genes (Table 1). Note that the PCR efficiency for PaWOX2 was higher when calculated from the calibration curve used for relative quantification (1.88, Table 1) compared to the standard curve used for absolute quantification (1.86, Table 4). The reaction efficiencies of the housekeeping genes were used to transform the Ct values into relative quantities for analysis of gene expression stability in the geNorm software. UBC1 and GAPDH were the most stable genes overall whereas the third housekeeping gene, eIF4A, had a higher level of variability in expression compared to both UBC1 and GAPDH (data not shown). As a result, the geometric mean of UBC1 and GAPDH expression ratios was selected as a normalization factor in the analysis of all assays and samples.
PaWOX2 is expressed specifically during the earliest stages of somatic embryo development
Both absolute and relative quantification revealed that PaWOX2 was expressed specifically during the earliest stages of somatic embryo development (Fig. 4a). Expression level was highest in samples taken six days after transfer to the maturation medium, containing both stage 1 and stage 2 embryos together, compared to the callus growing on maintenance medium. It was 1.9- and 1.4-fold higher for absolute and relative expression, respectively. The callus samples taken from the maintenance medium always contained aggregates of small rapidly dividing cells and more elongated non-dividing ones in addition to the stage 1 somatic embryos. After transfer to the maturation medium with its high concentration of PEG, the tissues continued to proliferate initially but the number of highly vacuolated cells tended to decline, and after six days both stage 1 and stage 2 somatic embryos were present. In samples that included both stage 2 and stage 3 embryos, both the absolute and relative expression level decreased to 4.5- and 3.6-fold, respectively, below the initial expression level in the callus. In mature embryos (stage 4) the absolute and relative expression decreased 94- and 113-fold, respectively, compared with the callus, reaching as low as 407 copies per μg total RNA. In absolute quantification, statistical analysis showed a significant difference in PaWOX2 expression between the callus (containing stage 1 embryos) and the samples containing stage 1 and stage 2 embryos. However, this difference was not significant in relative quantification. The variation between assays was higher with relative quantification compared to absolute quantification, especially in regards to the earlier stages of somatic embryo development.

Absolute and relative expression of PaWOX2 during somatic embryo development. Absolute and relative expression of PaWOX2 normalized against μg total RNA and the two housekeeping genes UBC1 and GAPDH, respectively, during somatic embryo development in cell line 04WV5 + h#3 (a) and in calli from cell lines 04WV5 + h#3, 04WV5 + h#6, 04LV1/2 + h#5, and 04WV5 + h#3* (b). Each bar is the mean ± SE of triplicate assays. In (a), means are statistically significant different (P < 0.05), except between stage 2–3 and stage 4 in both absolute and relative quantification and between stage 1 (in callus) and stage 1–2 in relative quantification. In (b), means are statistically significant different (P < 0.05), except between 04WV5 + h#3 and 04LV1/2 + h#5 in absolute quantification and between 04LV1/2 + h#5 and 04WV5 + h#3* in relative quantification
PaWOX2 expression in different cell lines
Both absolute and relative expression level of PaWOX2 was compared in calli of four different embryogenic cell lines (Fig. 4b). The cell lines varied both in their growth characteristics and in their capacity to form mature embryos (Table 2). In both absolute and relative quantification the PaWOX2 expression was 2.9- and 1.9-fold, respectively, higher in calli of cell line 04WV5 + h#6 compared to cell line 04WV5 + h#3, and it was 1.2- and 4-fold, respectively, lower in 04LV1/2 + h#5 when compared to 04WV5 + h#3. This difference in PaWOX2 expression was significant in relative quantification but not in absolute quantification. PaWOX2 expression was also analyzed in the fast growing calli of the cell line 04WV5 + h#3* (Fig. 4b), that had no visible organized embryos (Fig. 1f–g) and did not form mature somatic embryos on maturation medium (Table 2). Absolute and relative quantification yielded widely different results. In absolute qRT-PCR the PaWOX2 expression was 4.7-fold higher in callus of cell line 04WV5 + h#3* compared to that of cell line 04WV5 + h#3, but it was 2.2-fold lower with relative qRT-PCR. Statistical analysis showed a significant difference in PaWOX2 expression between 04WV5 + h#3 and 04WV5 + h#3* in both absolute and relative quantification. In the non-embryogenic cell line 07WV5 + h NE, no PaWOX2 expression could be detected. Cell activity in this cell line was confirmed by UBC1 expression.
PaWOX2 is expressed in the hypocotyl, apical shoot, and cotyledon of seedlings
PaWOX2 expression could be detected in hypocotyl, apical shoot, and cotyledon from seedlings, but not in the root (Fig. 5). The results show that PaWOX2 expression was similar in both hypocotyl and apical shoot and comparable to PaWOX2 expression in stage 4 of somatic embryo development. However, the PaWOX2 expression was significantly higher in cotyledon compared to both hypocotyl and apical shoot.

Absolute expression of PaWOX2 in root, hypocotyl, apical shoot, and cotyledon from seedlings normalized against μg total RNA, and analysis of PaWOX2 expression in the same tissues using gel electrophoresis (insert). Each bar is the mean ± SE of triplicate assays. Means are statistically significant different (P < 0.05), except between hypocotyl and apical shoot
Discussion
Embryo developmental program
Due to their inaccessibility within the seed and that developing seed embryos of species like Picea abies are only available during a very short period of the year, if at all, somatic embryogenesis offers an attractive model system for fundamental studies of conifer embryology. Somatic embryos follow a developmental pathway that is very similar to their zygotic counterparts (Figs. 1 and 2), and they also show an extensive suspensor-like system similar to the zygotic embryos (Fig. 2) (Hakman 1993). Somatic embryos of conifers thus clearly demonstrate that the pattern formation of an embryo is not dependent upon any fixed spatial relationship to maternal tissues or structures (Berleth and Chatfield 2002). Knowledge of the regulatory pathway leading to well-formed mature embryos with a high conversion rate into plantlets is also of fundamental importance for the applied aspects of this area. Different cell lines have quite variable potential of forming embryos. They also differ in how well the embryos mature and germinate into plantlets and in other growth characteristics, such as growth rate (Table 2 and Fig. 1). Reliable expression markers associated with the developmental program of somatic embryos should therefore be of great interest for applications in generating large number of plants for reforestation programs.
Currently, most of our knowledge about the genes and developmental programs operating during embryo pattern formation comes from mutagenesis screens aimed at identifying embryo-defective mutants in model species like Arabidopsis (see e.g., Jürgens 2001; Berleth and Chatfield 2002; Laux et al. 2004; Willemsen and Scheres 2004). The expression dynamic of the newly described WUS RELATED HOMEOBOX (WOX) gene family members WOX2, WOX9, and WOX8 suggest their involvement in partitioning the early embryo into apical, central, and basal domains, respectively, from the zygote to eight-cell stage of embryogenesis. After the eight-cell stage, WOX2 marks shoot-specific pattern elements and WOX5 mark the quiescent centre (QC) in the basal part of the embryo (Haecker et al. 2004; Kamiya et al. 2003; Nardmann et al. 2007). Compared to Arabidopsis embryos that have a very regular pattern of division during development (see e.g., Berleth and Chatfield 2002), changes in embryo morphology of conifers are less predictable and pattern formation is quite subtle (Spurr 1949). Nevertheless, several putative homologs of angiosperm genes implicated in embryo development are also present in the large pine EST collection, including members of the WOX family of genes (Cairney et al. 2006).
The PaWOX2 gene encodes a WUS/WOX-like HD protein
Homeodomain-containing proteins are thought to act as transcription factor that regulate the expression of genes in a temporal, spatial, and tissue-specific fashion (Gehring 1987; Wolberger 1996). The homebox sequences are highly conserved during evolution whereas the flanking sequences are not (Gehring 1987; Kappen et al. 1993; Banerjee and Baxevanis 2001). Within the Arabidopsis homeobox (HB) family of transcription factors, domain shuffling and divergence in protein evolution have generated several subfamilies, some with plant-specific combinations of modules (Riechmann et al. 2000). Among them, the WUS/WOX-like transcription factors are plant-specific and comprise at least fifteen different members in Arabidopsis (Haecker et al. 2004). They have an atypical HD, unique to the plant kingdom, that is characterized by the insertion of two (WUS) or one (WOX) extra amino acids in the loop between helix 1 and helix 2 and four extra amino acids in the turn between helix 2 and helix 3 (Supplementary material online, Fig. 2). The role of extra amino acid residues in the turn is not known, but it is suggested they may be involved in the recognition of their target sequence(s) (Kamiya et al. 2003). Binding studies of WUS (Lohmann et al. 2001) and QBH, the rice WOX5 homolog (Kamiya et al. 2003), suggests they both interact with same/similar nucleotide sequences, although the proteins differ in their primary structure. Yeast two-hybrid studies have also indicated the region to be involved in homo- and/or heterodimer formation within the subgroup of HD-proteins (Nagasaki et al. 2005). The WUS-box is another domain that is also present in PaWOX2 (Table 3). This domain with unknown function, located downstream of the HD and present in most but not all WOX proteins, is more or less conserved (Haecker et al. 2004). In addition to the two WOX2 homologs that were retrieved from the pine EST-database, the WUS-box motif was only found in one other conifer sequence (accession no. DR691632, PintaWOX4/6) whose HD sequence also clustered closer to that of WOX2 in the phylogenetic analysis (Table 3 and Fig. 3). We did not find SLELXL motif in any of the retrieved sequences. This sequence is present in the C-terminal end of WUS proteins and is similar to repressor sequences, such as the EAR-like-motif (Nagasaki et al. 2005). It should be added, though, that many of the retrieved conifer sequences were incomplete and could, therefore, still contain any of these motifs.
WOX family in conifers
Compilation of WUS/WOX related family members of pine and spruce obtained from the public databases reveal that many members have very similar or almost identical sequence. Consequently, only one member was used for phylogenetic tree construction. Two tree-building methods were used and both supported the phylogenetic tree topology illustrated in Fig. 3. Comparison of the WUS/WOX-HD sequences gives evidence for at least three evolutionary lineages: one leading to WUS and most of the WOX proteins, including WOX2; one leading to WOX8, 9, 11 and 12; and one to WOX10, 13 and 14. Both PaWOX2 and the two pine orthologs obtained from the pine EST database are of embryo origin. They are almost identical to each other and are also closely related to the other angiosperm WOX2 proteins, suggesting they all share a similar fundamental function during embryo development. We also found that a large number of the other conifer clones encoding WOX-like proteins are from embryo libraries (Table 3) and that the majority of them group with Arabidopsis WOX8/9 proteins (Fig. 3). Others, that include clones of more diverse tissue origin, are found in the cluster with WOX10, 13 and 14. The WOX sequence from Physcomitrella is also found in this group. The substantial length separating the diverging points and the presence of a moss sequence in this group suggests it as ancestral to the others and that it may have existed for a long time, before the separation of moss and seed plants. Our results are in agreement with those recently presented at PlantTAPDB (http://www.cosmoss.org/bm/plantapdb) where phylogenetic analyses indicated that the WUS family of transcription factors exhibits a rigorous land-plant specific taxonomic profile, and that WUS evolved from an ancestral homeobox gene that was present already after the water-to-land transition (Richardt et al. 2007).
Apart from embryos, many of the coniferous WOX-like clones are from root libraries. Still none of them cluster with WOX5, a transcription factor thought to have similar function in the root meristem as WUS in the shoot meristem. Neither did we find any sequences to cluster with WUS, which leads to an interesting question of which, if any, of the conifer WOX homologs have similar function(s) in meristem regulation. Of course, both WUS and WOX5 homologs may be present in conifer genomes but have not yet been identified. Recently, Nardmann et al. (2007) showed that in Zea mays, a monocot containing two WOX5 paralogs (ZmWOX5A and ZmWOX5B), ZmWOX5B not only marks a single row of cells in the root QC but also the base of provascular bundles specified immediately above the QC. They also showed that the ZmWOX5A paralog has acquired a completely different expression pattern, with expression in early endosperm and in the embryo after early leaf stage 1, 4–6 cell layers below the root QC. Thus, ZmWOX5 is expressed differently to WOX5 in Arabidopsis and QHB in rice, indicating a possible difference in function. Although no conifer WUS homolog could be identified in the public databases, genes homologous to both the receptor kinase CLAVATA1 (CLV1) and receptor-like protein CLAVATA2 (CLV2) are present in the assembled pine embryo-derived EST-sequences (Cairney et al. 2006). In addition, sequences with CLE-motif similar to that of CLAVATA3/ESR-RELATED are present in the spruce EST database, indicating that the major players of the CLV/WUS negative feedback loop are present in conifer. In angiosperms the role of WUS appears to be evolutionary conserved and an interaction of conserved domain(s) in its C-terminal region, which include the WUS-box, with other proteins appears to be essential for its function (Kieffer et al. 2006). All conifer sequences with a WUS-box were found to cluster with WUS and the major WOX proteins of Arabidopsis in the phylogenetic tree. Non-embryogenic tissues expressing WOX include differentiating xylem and bark, including cambium and phloem (Table 3). It is, thus, possible that some WOX genes are expressed in cambial tissue, perhaps with a function of repressing differentiation of cells within the dividing zone.
PaWOX2 is transiently expressed during somatic embryo development
Since WOX2 expression was detected in very young embryos of Arabidopsis, we wanted to see if this was also the case for Picea abies somatic embryos, and if so, we then wanted to know if WOX2 could be used as a marker for embryogenic potential of our tissue cultures. The transcript level of PaWOX2 was highest in early stage somatic embryos and, declined as the embryos matured (Fig. 4a), which follows the expression pattern of both Arabidopsis seed embryos (Haecker et al. 2004) as well as that of microspore-derived embryos (MDEs) and developing seeds of Brassica napus (Malik et al. 2007). In their search of marker genes for microspore embryos, Malik et al. (2007) not only identified the B. napus ortholog BnWOX2 but also BnWOX9 as suitable. Transcripts of both these genes were also detected in vegetative tissues, which is in line with our result showing that P. abies seedlings express PaWOX2 in hypocotyls, apical shoots, and cotyledons (Fig. 5). Results from Arabidopsis also show WOX2 expression in cotyledons (http://www.weigelworld.org/resources/microarray/AtGenExpress/) (Schmid et al. 2005). In addition, a recent study suggests a role for WOX2, together with WOX8 (STPL), in promoting cotyledon separation, which further corroborates our findings of PaWOX2 expression in cotyledons (Wu et al. 2007).
According to Malik et al. (2007), both BnWOX2 and BnWOX9 are good markers to predict embryogenic potential of Brassica cultivars. Our results with the different embryogenic cell lines, showing different capacities to form mature embryos, were not that unambiguous. When using absolute quantification, the highest expression level of PaWOX2 was found in the cell line with the poorest regeneration capacity but which also had the highest growth rate on the maintenance medium (04WV5 + h#3*, Table 2). Therefore, we started a new cell line from a hypocotyl of a one-week-old seedling. The cell line was considered as non-embryogenic, based on its lack of plant regeneration capacity and its capacity to turn green in light. No PaWOX2 expression could be detected in the non-embryogenic cell line, which indicates that PaWOX2 can be used as a marker of embryogenic potential. These results also suggests that PaWOX2 is expressed in the small cell aggregates observed in calli from cell line 04WV5 + h#3* (Fig. 1f–g), and not only in well-defined embryos. Assuming a role for PaWOX2 during very early embryo development, results from both Arabidopsis and Zea mays showing expression of WOX2 and ZmWOX2A in the egg and the zygote, respectively, support our findings (Haecker et al. 2004; Wu et al. 2007). Thus, the higher growth rate of the cell line 04WV5 + h#3*, which includes many of these small cell aggregates would also lead to higher absolute expression of PaWOX2. This implies that PaWOX2 plays a fundamental role in early somatic embryo development, possibly with a function related to regulating cell division and/or differentiation in the developing embryos.
Comparison of absolute and relative qRT-PCR
Because of problem with suitable reference genes for relative qRT-PCR we used both absolute and relative quantification methods to determine the expression pattern of PaWOX2 in developing somatic embryos. While the use of endogenous reference genes in relative quantification avoids the problems and limitations of absolute quantification, such as variations in RT and PCR efficiencies due to difference in quantity and quality of RNA (Bustin and Nolan 2004; Huggett et al. 2005), selection of proper endogenous reference genes, expressed at a nearly constant level in all tissue samples being investigated, is required. According to Thellin et al. (1999), Vandesompele et al. (2002) and Dheda et al. (2005), at least two or three housekeeping genes should be used as endogenous reference genes because the use of a single gene for normalization could lead to relative large errors. Furthermore, it is essential to validate potential reference genes to establish whether they are appropriate for a specific experimental purpose. GAPDH and UBC1 were selected, after analyzing gene stability in geNorm, as endogenous reference genes in all assays. In Arabidopsis both GADPH and UBC1 transcript levels are fairly stable over a wide range of developmental and environmental conditions (Czechowski et al. 2005), and GAPDH has also been demonstrated as an appropriate reference gene for real-time qRT-PCR in bovine preimplantation embryos (Goossens et al. 2005) and during development of grape berries (Reid et al. 2006). In contrast, developing seed embryos of Pinus pinaster varied in their expression of GAPDH (Gonçalves et al. 2005). Relative quantifications of PaWOX2 expression showed, in this study, high variation between assays of the earlier stages of somatic embryos. This is probably due to the variability in expression of the endogenous reference genes, statistically affecting study conclusions. It is also likely that the PCR efficiencies for GAPDH, UBC1, elF4A, and PaWOX2 were overestimated due to tissue-specific factors and RT components in the calibration curves (Suslov and Steindler 2005; Tichopad et al. 2003, 2004). This explains the difference in PCR efficiency of PaWOX2 between the calibration and standard curve. Our results support the conclusion of Gonçalves et al. (2005) that absolute quantification, when normalized against high quality, accurately measured total RNA mass, is the preferred method to use when determining gene expression in developing embryos due to the difficulty of finding stable housekeeping genes. However, this does not apply when analyzing gene expression in calli with different growth rates and embryogenic potential. The high expression of PaWOX2 in calli of cell line 04WV5 + h#3* in absolute quantification is likely due to the very high proliferation rate of that cell line (Table 2). A faster growth rate of cell line 04WV5 + h#6 compared to 04WV5 + h#3 may also explain why the fold difference was much higher in absolute quantification compared to relative quantification. Normalizing to total RNA primarily measures rRNA, which makes up ∼80% of the fraction, and there are thus problems comparing tissues that are proliferating at different rates, since their mRNA/rRNA ratios are likely to be different (Nolan et al. 2006). This is probably why the results are different between the two methods and why relative quantification is the preferred method to use when analyzing gene expression in different proliferating calli. To improve the experimental accuracy of both relative and, more importantly, absolute quantification we chose to run all samples in duplicate starting with the RT-reaction. Experimental accuracy is two times higher when a sample is split into aliquots before the RT-reaction compared to when it is split before qRT-PCR (Ståhlberg et al. 2004).
Conclusions
In conclusion, our results indicate that the WOX family of transcription factors is ancient, with some members of the conifer WOX13 clade being resolved as sister to a Physcomitrella gene. Based both on the structural similarities of the HD and the WUS-box, as well as the expression profile of WOX2 in Arabidopsis, Zea mays and Picea abies, it is tempting to consider that these genes represent orthologous gene function. This implies that WOX2 provides an ancient function associated with embryo development prior to the separation of angiosperms and gymnosperms. The highest expression level of PaWOX2 was found in a cell line with the highest proliferation rate, but with very poor embryo maturation capacity, while no expression was found in a non-embryogenic cell line. Together, this implies that PaWOX2 plays a fundamental role in early somatic embryo development, possibly with a function related to regulating cell division and/or differentiation in the embryos, and that it can be used as molecular marker of embryogenic potential, but not necessary for the regeneration capacity of cell lines. Our results also show that absolute qRT-PCR is the preferred method in determining gene expression in developing embryos, while relative qRT-PCR is the preferred method for calli with different growth rates.
Abbreviations
- ABA:
-
Abscisic acid
- BA:
-
6-Benzyladenine
- CLV :
-
CLAVATA
- HB:
-
Homeobox
- HD:
-
Homeodomain
- NAA:
-
1-Naphthalene acetic acid
- PEG:
-
Polyethylene glycol
- qRT-PCR:
-
Quantitative reverse transcription polymerase chain reaction
- QC:
-
Quiescent centre
- RAM:
-
Root apical meristem
- SAM:
-
Shoot apical meristem
- WOX :
-
WUS homeobox
- WUS :
-
WUSCHEL
References
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucl Acids Res 25:3389–3402
Banerjee-Basu S, Baxevanis AD (2001) Molecular evolution of the homeodomain family of transcription factors. Nucl Acid Res 29:3258–3269
Berleth T, Chatfield S (2002) Embryogenesis: pattern formation from a single cell. In: Somerville CR, Meyerowitz EM (eds) The arabidopsis book. American Society of Plant Biologists, Rockville, MD, doi:10.1199/tab.0009, http://www.aspb.org/publications/arabidopsis/
Bustin SA, Nolan T (2004) Pitfalls of quantitative real-time reverse-transcription polymerase chain reaction. J Biomol Tech 15:155–166
Bustin SA (2000) Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. J Mol Endocrinol 25:169–193
Cairney J, Zheng L, Cowels A, Hsiao J, Zismann V, Liu J, Ouyang S, Thibaud-Nissen F, Hamilton J, Childs K, Pullman GS, Zhang Y, Oh T, Buell CR (2006) Expressed sequence tags from loblolly pine embryos reveal similarities with angiosperm embryogenesis. Plant Mol Biol 62:485–501
Chalupa V (1985) Somatic embryogenesis and plant regeneration from cultured immature and mature embryos of Picea abies (L.) Karst. Commun Inst For Cech 14:57–63
Czechowski T, Stitt M, Altmann T, Udvardi MK, Scheible WR (2005) Genome-wide identification and testing of superior reference genes for transcript normalization in Arabidopsis. Plant Physiol 139:5–17
Dheda K, Hugget JF, Chang JS, Kim LU, Bustin SA, Johnson MA, Rook GAW, Zumla A (2005) The implications of using an inappropriate reference gene for real-time reverse transcription PCR data normalization. Anal Biochem 344:141–143
Dorantes-Acosta AE, Vielle-Calzada JP 2006 The male gametophytic mutant tepitzin1 indicates a requirement of the homeobox gene WOX5 for pollen tube growth in Arabidopsis. Sex Plant Reprod 19:163–173
Gehring WJ (1987) Homeo boxes in the study of development. Science 236:1245–1252
Gehring WJ (1993) Exploring the homeobox. Gene 135:215–221
Giri AV, Anandkumar N, Muthukumaran G, Pennathur G (2004) A novel medium for the enhanced cell growth and production of prodigiosin from Serratia marcescens isolated from soil. BMC Microbiol 4:11
Gonçalves S, Cairney J, Maroco J, Oliveira MM, Miguel C (2005) Evaluation of control transcripts in real-time RT-PCR expression analysis during maritime pine embryogenesis. Planta 222:556–563
Gonzali S, Novi G, Loreti E, Paolicchi F, Poggi A, Alpi A, Perata P (2005) A turanose-insensitive mutant suggests a role for WOX5 in auxin homeostasis in Arabidopsis thaliana. Plant J 44:633–645
Goossens K, Van Poucke M, Van Soom A, Vandesompele J, Van Zeveren A, Peelman LJ (2005) Selection of reference genes for quantitative real-time PCR in bovine preimplantation embryos. BMC Dev Biol 5:27
Groß-Hardt R, Laux T (2003) Stem cell regulation in the shoot meristem. J Cell Sci 116:1659–1666
Haecker A, Gross-Hardt R, Geiges B, Sarkar A, Breuninger H, Herrmann M, Laux T (2004) Expression dynamics of WOX genes mark cell fate decisions during early embryonic patterning in Arabidopsis thaliana. Development 131:657–668
Hakman I (1985) Tissue culture in conifers with emphasis on somatic embryogenesis in Norway spruce (Picea abies (L.) Karst.). Dissertation, Uppsala University. ISBN 91-554-1693-4
Hakman I (1993) Embryology in Norway spruce (Picea abies). An analysis of the composition of seed storage proteins and deposition of storage reserves during seed development and somatic embryogenesis. Physiol Plant 87:148–159
Hakman I, Fowke LC, von Arnold S, Eriksson T (1985) The development of somatic embryos in tissue cultures initiated from immature embryos of Picea abies (Norway spruce). Plant Sci 38:53–59
Hakman I, von Arnold S (1985) Plantlet regeneration through somatic embryogenesis in Picea abies (Norway spruce). J Plant Physiol 121:149–158
Hakman I, Stabel P, Engström P, Eriksson T (1990) Storage protein accumulation during zygotic and somatic embryo development in Picea abies (Norway spruce). Physiol Plant 80:441–445
Huggett J, Dheda K, Bustin S, Zumla A (2005) Real-time RT-PCR normalisation; strategies and considerations. Genes and Immunity 6:279–284
Imin N, Nizamidin M, Wu T, Rolfe BG (2007) Factors involved in root formation in Medicago truncatula. J Exp Bot 58:439–451
Jürgens G (2001) Apical-basal pattern formation in Arabidopsis embryogenesis. EMBO J 20:3609–3616
Kamiya N, Nagasaki H, Morikami A, Sato Y, Matsuoka M (2003) Isolation and characterization of a rice WUSCHEL-type homeobox gene that is specifically expressed in the central cells of a quiescent center in the root apical meristem. Plant J 35:429–441
Kappen C, Schughart K, Ruddle FH (1993) Early evolutionary origin of major homeodomain sequence classes. Genomics 18:54–70
Kieffer M, Stern Y, Cook H, Clerici E, Maulbetsch C, Laux T, Davies B (2006) Analysis of the transcription factor WUSCHEL and its functional homologue in Antirrhinum reveals a potential mechanism for their roles in meristem maintenance. Plant Cell 18:560–573
Laux T, Mayer KFX, Berger J, Jürgens G (1996) The WUSCHEL gene is required for shoot and floral meristem integrity in Arabidopsis. Development 122:87–96
Laux T, Würschum T, Breuninger H (2004) Genetic regulation of embryonic pattern formation. Plant Cell 16:190–202
Lohmann JU, Hong RL, Hobe M, Busch MA, Parcy F, Simon R, Weigel D (2001) A molecular link between stem cell regulation and floral patterning in Arabidopsis. Cell 105:793–803
Malik MR, Wang F, Dirpaul JM, Zhou N, Polowick PL, Ferrie AMR, Krochko JE (2007) Transcript Profiling and Identification of Molecular Markers for Early Microspor Embryogenesis in Brassica napus. Plant Physiol 144:134–154
Mayer KFX, Schoof H, Haecker A, Lenhard M, Jürgens G, Laux T (1998) Role of WUSCHEL in regulating stem cell fate in the Arabidopsis shoot meristem. Cell 95:805–815
Nagasaki H, Matsuoka M, Sato Y (2005) Members of TALE and WUS subfamilies of homeodomain proteins with potentially important functions in development form dimers within each subfamily in rice. Genes Genet Syst 80:261–267
Nardmann J, Werr W (2006) The shoot stem cell niche in angiosperms: expression patterns of WUS orthologues in rice and maize imply major modifications in the course of mono- and dicot evolution. Mol Biol Evol 23:2492–2504
Nardmann J, Zimmermann R, Durantini D, Kranz E, Werr W (2007) WOX gene phylogeny in Poaceae: a comparative approach addressing leaf and embryo development. Mol Biol Evol 24:2474–2484
Nolan T, Hands RE, Bustin SA (2006) Quantification of mRNA using real-time RT-PCR. Nat Protoc 1:1559–1582
Park YS (2002) Implementation of conifer somatic embryogenesis in clonal forestry: technical requirements and deployment considerations. Ann Forest Sci 59:651–656
Pfaffl MW (2001) A new mathematical model for relative quantification in real-time PCR. Nucl Acids Res 29:2002–2007
Reid KE, Olsson N, Schlosser J, Peng F, Lund ST (2006) An optimized grapevine RNA isolation procedure and statistical determination of reference genes for real-time RT-PCR during berry development. BMC Plant Biol 6:27
Richardt S, Lang D, Reski R, Frank W, Rensing SA (2007) PlanTAPDB, a phylogeny-based resource of plant transcription-associated proteins. Plant Physiol 143:1452–1466
Riechmann JL, Heard J, Martin G, Reuber L, Jiang C, Keddie J, Adam L, Pineda O, Ratcliffe OJ, Samaha RR, Creelman R, Pilgrim M, Broun P, Zhang JZ, Ghandehari D, Sherman BK, Yu G (2000) Arabidopsis transcription factors: genome-wide comparative analysis among eukaryotes. Science 290:2105–2110
Rozen S, Skaletsky HJ (2000) Primer3 on the WWW for general users and for biologist programmers. In: Krawetz S, Misener S (eds) Bioinformatics methods and protocols: methods in molecular biology. Humana Press, Totowa, NJ, pp 365–383
Rutledge RG, Côté C (2003) Mathematics of quantitative kinetic PCR and the application of standard curves. Nucl Acids Res 31:e93
Sarkar AK, Luijten M, Miyashima S, Lenhard M, Hashimoto T, Nakajima K, Scheres B, Heidstra R, Laux T (2007) Conserved factors regulate signalling in Arabidopsis thaliana shoot and root stem cell organizers. Nature 446:811–814
Schmid M, Davison TS, Henz SR, Pape UJ, Demar M, Vingron M, Schölkopf B, Weigel D, Lohmann JU (2005) A gene expression map of Arabidopsis thaliana development. Nature Gen 37:501–506
Spurr AR (1949) Histogenesis and organization of the embryo in Pinus strobus L. Amer J Bot 36:629–641
Stasolla C, Yeung EC (2003) Advances on embryogenesis in culture of coniferous species: improving somatic embryo quality. Plant Cell Tissue Organ Cult 74:15–35
Stahlberg A, Hakansson J, Xian X, Semb H, Kubista M (2004) Properties of the reverse transcription reaction in mRNA quantification. Clin Chem 50:509–515
Suslov O, Steindler DA (2005) PCR inhibition by reverse transcriptase leads to an overestimation of amplification efficiency. Nucleic Acids Res 33:e181
Sutton B (2002) Commercial delivery of genetic improvement to conifer plantations using somatic embryogenesis. Ann For Sci 59:657–661
Swofford DL (2000) PAUP*: phylogenetic analysis using parsimony (*and other methods), version 4. Sinauer Associates, Sunderland, MA
Thellin O, Zorzi W, Lakaye B, De Borman B, Coumans B, Hennen G, Grisar T, Igout A, Heinen E (1999) Housekeeping genes as internal standards: use and limits. J Biotechnol 75:291–295
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucl Acids Res 25:4876–4882
Tichopad A, Didier A, Pfaffl MW (2004) Inhibition of real-time RT-PCR quantification due to tissue-specific contaminants. Mol Cell Probes 18:45–50
Tichopad A, Dilger M, Schwarz G, Pfaffl MW (2003) Standardized determination of real-time PCR efficiency from a single reaction set-up. Nucleic Acids Res 31:e122
Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F (2002) Accurate normalizaton of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 3:research0034.1-0034.11
Walker NJ (2002) A technique whose time has come. Science 269:257–259
Weijers D, Jurgens G (2005) Auxin and embryo axis formation: the ends in sight? Curr Opin Plant Biol 8:32–37
Whelan JA, Russell NB, Whelan MA (2003) A method for the absolute quantification of cDNA using real-time PCR. J Immun Methods 278:261–269
Willemsen V, Scheres B (2004) Mechanisms of pattern formation in plant embryogenesis. Annu Rev Genet 38:587–614
Wolberger C (1996) Homeodomain interactions. Curr Opin Struct Biol 6:62–68
Wu X, Chory J, Weigel D (2007) Combinations of WOX activities regulate tissue proliferation during Arabidopsis embryonic development. Dev Biol 309:306–316
Wu X, Dabi T, Weigel D (2005) Requirement of homeobox gene STIMPY/WOX9 for Arabidopsis meristem growth and maintenance. Curr Biol 15:436–440
Acknowledgements
The support from the University of Kalmar is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.

{kind=link}
Rights and permissions
About this article
Cite this article
Palovaara, J., Hakman, I. Conifer WOX-related homeodomain transcription factors, developmental consideration and expression dynamic of WOX2 during Picea abies somatic embryogenesis. Plant Mol Biol 66, 533–549 (2008). https://doi.org/10.1007/s11103-008-9289-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11103-008-9289-5