
Abstract
Purpose
To evaluate how obesity affects the pharmacokinetics of human IgG following subcutaneous (SC) and intravenous (IV) administration to rats and the homeostasis of endogenous rat IgG.
Methods
Differences in body weight and size, body composition, and serum concentration of endogenous rat IgG in male Zucker obese (ZUC-FA/FA) and control (ZUC-LEAN) rats were measured from the age of 5 weeks up to 30 weeks. At the age of 23–24 weeks animals received a single IV or SC dose of human IgG (1 g/kg of total body weight), and serum pharmacokinetics was followed for 7 weeks. A mechanistic model linking obesity-related changes in pharmacokinetics with animal growth and changes in body composition was developed.
Results
Significant differences were observed in both endogenous and exogenous IgG pharmacokinetics between obese and control groups. The AUC for human IgG was lower in obese groups (57.6% of control after IV and 48.1% after SC dosing), and clearance was 1.75-fold higher in obese animals. The mechanistic population model successfully captured the data and included several major components: endogenous rat IgG homeostasis with age-dependent synthesis rate; competition of human IgG and endogenous rat IgG for FcRn binding and its effect on endogenous rat IgG concentrations following injection of a high dose of human IgG; and the effect of body size and composition (changing over time and dependent on the obesity status) on pharmacokinetic parameters.
Conclusions
We identified important obesity-induced changes in the pharmacokinetics of IgG. Results can potentially facilitate optimization of the dosing of IgG-based therapeutics in the obese population.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Obesity is a complex disorder with excessive adipose tissue triggered by positive energy imbalance [1]. Obesity may be caused by unhealthy eating, lack of physical activity, genetic predisposition, and can be induced by other diseases or medications [2]. According to the Centers for Disease Control and Prevention (CDC), in 2016, over 1.9 billion adults worldwide were overweight (25 ≤ body mass index (BMI) < 30) or obese (BMI ≥ 30) [3]. Patients with obesity have an increased risk of various comorbidities, such as cardiovascular diseases, diabetes, and certain cancers [4], and might receive multiple medications. The obese population has been under-represented in clinical trials, and medication dosing approaches for these individuals often have not been sufficiently studied [5].
The dosing strategy for protein therapeutics is often based on a body size descriptor in clinical trials. Body size-based dosing can minimize variability of pharmacokinetics and efficacy in patients with different physiological conditions [6]. More than 50% of approved monoclonal antibodies (mAbs) are dosed based on total body weight [7]. Most protein therapeutics are highly hydrophilic and preferentially distributed into aqueous compartments with minimal distribution into adipose tissue [8, 9]. Therefore, dosing an obese subject based on total body weight could lead to an overdose by generating disproportionally high drug concentration in systemic circulation and highly perfused tissues, leading to toxicity [10]. Other body size metrics used for dosing drugs include body surface area and lean, ideal, and adjusted body weights; however, a universal approach still needs to be developed. Many of these metrics do not accurately reflect a patient’s body composition [5, 11,12,13].
For anticancer biologics, conflicting results were reported when comparing (for over 30 biologics) body size-based dosing or fixed dosing to improve inter-subject variability of pharmacokinetic exposure in morbidly obese patients [14]. Fixed dosing is recommended for first-in-human studies, while the effect of body size requires assessment in Phase 3 studies [7, 14].
Intravenous immunoglobulin (IVIG) and subcutaneous immunoglobulins (SCIG) are a purified mixture of IgG extracted from the pooled plasma of >1000 blood donors, which contains more than 95% unmodified IgG [15]. Since 1981, various IVIG and SCIG products have been approved to treat immunodeficiency and autoimmune and infectious diseases [16]. A wide range of doses is used clinically, from 0.2 g/kg to as high as 2 g/kg [17]. Obesity-related changes in IgG pharmacokinetics have yet to be adequately studied, but several theoretical factors were proposed [18]. The use of ideal body weight or adjusted body weight for dosing of IgG had been proposed based on low perfusion of adipose tissue [19], reduced blood volume in obesity [20], reduced expression of the FcRn receptor in adipose tissue compared to other tissues [21], and limited distribution of IgG into adipose due to its relative polarity [22, 23]. Potentially lower overall expression of FcRn in obesity can be significant as a high dose of IVIG might saturate FcRn-mediated recycling and lead to a shorter half-life of IgG (in turn requiring a higher dose or more frequent administration).
Obesity is often associated with a higher level of inflammation, and some inflammatory factors are produced by the adipose tissue [18]. Enhanced inflammation status aggravates immune dysregulation, which might require higher IVIG doses in obese, immunodeficient patients [24]. High levels of activated macrophages were shown to accumulate in the adipose tissue [24], and catabolism of IgG could be enhanced in obesity as the primary catabolism of IgG is via the macrophage-mediated reticuloendothelial system [25]. Subcutaneous (SC) administration of IgG has become more prevalent in recent years [26]. SC absorption is highly dependent on the structure of the SC tissue. The thickness of SC tissue increases with BMI [27]. Blood flow rate, capillary density and lymph drainage in the SC tissue are much lower in obese subjects [27]. Therefore, SC absorption of IgG formulations could be affected in obesity.
The main goal of this work was to investigate how obesity affects the pharmacokinetics of human IgG following SC and IV administration to rats and the homeostasis of endogenous rat IgG. The second goal was to develop a mechanistic model linking obesity-related changes in pharmacokinetics with animal growth and changes in body composition.
Materials and Methods
Materials
Human Immune Globulin Intravenous (10% Liquid, Privigen®) was purchased from CSL Behring UK Ltd. Meloxicam, carprofen, and bupivacaine were obtained from the animal facility at Rutgers University. Jugular vein catheters were made from PE50 tubing (Braintree Scientific Inc, Braintree, MA, USA). DietGel 76A and HydroGel were purchased from ClearH2O Inc. (Portland, ME).
Animals
All animal experiments were conducted in accordance with a protocol approved by the Institutional Animal Care and Use Committee (IACUC) at Rutgers, The State University of New Jersey. Male Zucker rats (ZUC-LEAN (control) and ZUC-FA/FA (obese)) were purchased from Charles River (Wilmington, MA). Upon arrival, 5-week-old animals were housed in pairs in solid bottom cages on a 12/12 h dark/light cycle and provided with a standard rat diet (LabDiet 5012, PMI Nutrition International) and water ad libitum. Various measures of body size were performed throughout the study, as described below. In IV dosing groups, the right jugular vein was cannulated (under isoflurane anesthesia) using PE-50 tubing to allow IV infusion. After surgery, animals were housed individually in cages and allowed to recover for 72 h; SC meloxicam/carprofen and intradermal bupivacaine analgesia were provided.
Measurement of body size and body composition
Each animal’s total body weight (TBW) was recorded weekly from arrival up to week 15 and then once a week from the dosing week until the end of the study (a measurement gap was due to COVID-19-related restrictions). Body composition was measured using the EchoMRI™ body composition analyzer (EchoMRI LLC, Houston, TX), which allows to measure amount of fat, lean body weight, free water, and total water weight (http://www.echomri.com) in unanesthetized animals. An animal was restrained in a cylinder holder for a scan that took approximately 2 min. Fat mass was reported as a mass of all the adipose tissues (canola oil was used for instrument calibration per manufacturer’s instructions. Lean mass was reported as a mass of all the water-containing tissues, excluding bone, fat, and NMR silent components such as hair and claws. Other body weight (OBW) was calculated by subtracting the fat mass from TBW. Free water was reported as the water mass mainly in the bladder. Total water mass was defined as a sum of free water and the water contained in lean mass. Body composition was obtained for each animal biweekly from arrival to week 15, then on the dosing day and the last day of the study. Each animal's abdominal circumference and length were measured on the same days. On the dosing day, the animals were 23–24 weeks old; the total body weight was approximately 440 g in control rats and 720 g in obese rats.
Experimental Design
A few days before the planned dosing of human IgG, the animals were randomly assigned to two obese and two control groups (n = 5–7 each), depending on the route of administration, IV or SC (to allow for jugular vein cannulation and animal recovery in IV groups before the dosing). Each animal received a single dose of human IgG at a dose level of 1 g per kg of total body weight (10% Liquid, Privigen®, CSL Behring) by IV infusion (IV lean, IV obese) or SC administration (SC lean, SC obese).
IV infusion was performed using an infusion pump (Harvard Apparatus 70–3005 PhD Ultra) at 2 mL/h. SC injection was administered in the back of animals (under isoflurane anesthesia). Lean and obese animals were dosed at the age of 23 and 24 weeks, respectively (18–19 weeks after arrival – a delay from the originally planned 15 weeks was due to COVID-19-related restrictions). Before dosing, blood samples (~ 100 μL) were collected from the saphenous vein biweekly from arrival up to week 15 in all the animals to measure endogenous rat IgG. For IV groups, blood samples were collected one week before the dose, then 1, 5, 8, 24, and 72 h, and weekly up to 6 weeks post-dose. For SC groups, blood samples were collected one week before the dose, then 1, 5, 8, 12, 24, 48, 72, and 96 h, and weekly up to 6 weeks post-dose. The blood samples were allowed to clot for 30–60 min at room temperature and centrifuged (3400 g for 7 min) to separate serum. Aliquots were stored at -80°C until analysis by ELISA.
Bioanalytics
The concentration of human IgG and endogenous rat IgG in rat serum was measured using a human IgG SimpleStep® ELISA kit (ab195215) and a rat IgG SimpleStep® ELISA kit (ab189578; Abcam, Cambridge, MA), respectively. The assays were performed according to manufacturer instructions. The working ranges of the human IgG and the rat IgG assay were 0.00023–150 mg/mL and 0.3125–20 mg/mL. The calibration curves were fitted with a 4-parameter logistic equation in GraphPad Prism 8. The precision and accuracy were within ± 20%.
Data Analysis and Mechanistic Population Pharmacokinetic Modeling
All body size and composition measurements and rat and human IgG serum concentration data were presented as mean ± standard deviation (SD). Standard noncompartmental analysis was performed for concentration-time data using Phoenix WinNonlin 8.3 (Certara, Princeton, NJ). Welch’s t-test (p < 0.05) was used to compare data between lean and obese groups statistically.
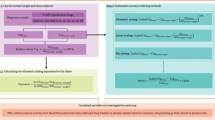
A mechanistic model was developed to capture the pharmacokinetics of endogenous rat IgG, and human IgG administered IV and SC in control and obese rats. The model combined several major components: 1) absorption and pre-systemic degradation of human IgG injected SC; 2) homeostasis of endogenous rat IgG through endosomal recycling and with synthesis rate being dependent on animal age; 3) competition of human IgG and endogenous rat IgG for FcRn binding in the endosomal compartment and its effect on endogenous rat IgG concentrations following injection of a high dose of human IgG; 4) and the effect of body size and composition (changing over time and dependent on the obesity status) on pharmacokinetic parameters. The model was developed based on human IgG and rat IgG serum concentration–time data and literature data from previous publications on the binding of rat FcRn and IgG from multiple species [28,29,30,31]. The schematic of the final model is presented in Fig. 1.

Schematic of the mechanistic model used to describe the pharmacokinetics of IVIG following intravenous and subcutaneous administration to lean and obese rats. Model parameters are described in the Methods and Table II (hIgG - human IgG, rIgG - rat IgG).
The systemic disposition of IgG (human and rat) was described using a classical endosomal recycling model [32] with central (Cp and Vp) and endosomal (Ce and Ve) compartments, where subscripts “h” and “r” were used to designate human and rat. Human IgG was directly injected into the central distribution compartment for IV groups. For SC groups, human IgG was dosed into the absorption compartment (Abs), where it could be degraded (kdeg) or absorbed (ka) into the central distribution compartment (Eq. 1).
Homeostasis of endogenous rat IgG was maintained by endogenous production (kin), FcRn-mediated IgG recycling (kup, kret, and FcRn binding processes), and lysosomal catabolism (kdeg2). It was observed that the production of endogenous IgG increased with age and gradually reached maximum levels at adulthood in humans [33]. We observed in rats that the concentration of endogenous IgG increased with age (Fig. 4). A maturation model (Eq. 2) was used to describe the age-related increase of endogenous IgG production, where kin,base, kin,max, T50, and γ represented baseline (at birth) production rate, maximum production rate, the age with 50% of the maximum rate, and steepness of the curve, similar to the modeling of metabolism maturation in pediatric population [34]. For modeling purposes, the time scale was defined in days and time zero set when the first sample for endogenous IgG was taken (5 weeks of age – upon animal arrival); therefore, age was incorporated as time plus 35 days in Eq. 2.
We also observed that following administration of human IgG, endogenous IgG increased to a higher level in IV groups than in rats assigned to SC groups, which might be related to jugular vein surgery (and potential surgery-related inflammation processes) that was performed in animals assigned to IV groups. To capture this phenomenon, an additional IgG production rate constant stkin was added for all IV animals after the day of surgery; and ST was a switch variable that was set to 1 for this purpose and was equal to zero before surgery for IV groups and all the time for SC groups. During the final model run, the value of rate constants kin,base and kin,max were fixed to the values estimated in previous runs to facilitate convergence and calculation of precision for parameters.
Human and rat endogenous IgG were transported from the central compartment to the endosomal compartment with a rate constant (kup) - Eqs. 3 and 4. In the endosomal compartment, human IgG and rat IgG competed for binding to rat FcRn receptor– Eqs. 5–8. Administration of human IgG can affect the homeostasis of endogenous rat IgG by saturating FcRn binding and leading to more unbound rat IgG being degraded by lysosomal catabolism [35].
Total FcRn receptor concentration (Rtot) in the endosomal compartment was assumed to be constant, similar to previous models [29, 32]. IgG-FcRn complex (where Fh and Fr are the amounts of each complex, respectively) was recycled back to the central compartment (kret), and unbound IgG was degraded (kdeg2) via lysosomal catabolism. Full binding equations with species-specific rate constants for association (kon) and dissociation (koff) were used (which resolved the challenge of solving the equilibrium equation for two ligands). However, available data did not allow for estimating all binding parameters. The difference in binding affinity has been reported previously (KDh and KDr) [31]. The relationships between equilibrium dissociation constant KD, association rate constant kon, and dissociation rate constant koff were shown in Eqs. 9 and 10 for rat IgG and human IgG, respectively. In the final model, to facilitate performance, rat and human association rate constants (kon,r and kon,h) were constrained to be the same [31, 36], and the value was fixed to the previous model runs. Furthermore, a value for human KD was assumed to be 3.66-fold higher than rat KD based on previous studies [31, 36], and only a KDr was estimated.
In this study, we followed the changes in endogenous IgG and injected human IgG for many weeks to evaluate the effect of animal growth and the development of obesity on IgG biodisposition. Different body size and composition metrics were evaluated as time-varying covariates on various model parameters to allow for the simultaneous description of data in obese and lean animals. Relationships included in the final model provided the best description of the data and are described below.
Previously, we reported that incorporating the growth of rats into the pharmacokinetic model (by setting the volume of distribution to be proportional to total body weight) facilitated the description of rituximab pharmacokinetics [37]. Similarly, in this study, logistic growth function (implemented in GraphPad Prism v8) was used to describe individual increase of TBW and OBW of each animal (Eqs. 11 and 12), where TBW0, OBW0 and TBWmax, OBWmax were initial (5-week-old) and final (end of the study) individual weights and kgr, and kgr2 are growth rate constants (and these estimated individual parameters were included as covariates in NONMEM). In humans, FcRn receptors were shown to have a lower expression level in adipose tissue than in other tissues [38]. Vp and Ve were set to be proportional to TBW and OBW, respectively, with volume coefficients A_Vp and A_Ve (Eqs. 9 and 10). SC absorption could be potentially slower in the obese population because blood flow rate and capillary density in their SC tissue are lower [27]. The absorption rate constant ka was set to be proportional to the fraction of OBW (OBW/TBW) with the absorption rate coefficient (A_ka) (Eq. 15).
Obese patients often have chronic inflammation, and high levels of activated macrophages accumulate in the adipose tissue [24]. Catabolism of IgG is possibly increased in obese patients as the primary catabolism of IgG is via the macrophage-mediated reticuloendothelial system [25]. Therefore, kdeg2 was set to be proportional to a fat fraction with endosomal catabolism rate coefficient A_kdeg2 (Eq. 16). The rate constant for endosomal recycling (kret) was set to be proportional to an inverse of fat fraction with recycling rate coefficient A_kret to reduce the FcRn recycling in obese animals (Eq. 17).
The initial conditions for human IgG-related compartments (Cp,h(0), Abs(0), Ce,h(0), and Fh(0)) were set equal to 0. For rat IgG-related compartments, the initial conditions (Cp,r(0), Ce,r(0), and Fr(0)) were expressed using the system parameters (assuming the system was at equilibrium and using values at time zero for kin, kret, kdeg2, Vp and Ve) as showed in Eqs. 18–20.
A mixed-effect modeling approach was used in this work, and model parameters were estimated by simultaneously fitting individual serum human and rat IgG concentration-time data for all groups using first‐order conditional estimation with interaction (FOCE-I) implemented in NONMEM 7.4.3 (ICON plc, Hanover, MD, USA). The final structural model was chosen based on the precision of parameter estimates and goodness-of-fit criteria, including objective function value, visual inspection of fitted curves, and diagnostic plots. In addition, a visual predictive check (VPC) was performed by simulating 1000 data sets using the final parameter estimates. The observed concentration data were then compared with the simulated concentrations. An exponential model was used to describe inter-subject variability (IIV). Initially, IIV was included for all system parameters and was retained in the final model based on the precision of parameter estimates, shrinkage, and goodness-of-fit criteria. A proportional residual error model was used.
Covariate analysis was conducted to evaluate the effect of various body size metrics on different model parameters. Covariates were selected based on physiological plausibility and observed trends in the data. Initially, the effect of body composition on the volume of compartments was tested using all rat IgG data and only IV data for human IgG. Significant covariates were retained, and human IgG SC data were added to evaluate body composition’s effect on SC absorption. The model was further improved by including body composition metrics as covariates on rate constants. At each step, covariate selection was driven by a significant decrease (p < 0.05) in the objective function value, visual inspection of fitted curves, and diagnostic plots. In addition, stepwise covariate modeling (SCM) was conducted to assess a potential effect of fat percentage, TBW, abdominal circumference, and length on the dosing day on T50, kup, kin,max, and stkin. Stepwise covariate search was performed using forward selection at p = 0.05 and backward elimination at p = 0.01. None of these covariate relationships was found to be significant.
Following establishing the final model, simulations were performed to illustrate the effect of obesity on human IgG kinetics and rat IgG homeostasis under different dosing scenarios (0.5–2 g/kg commonly used for clinical dosing of human IgG). Mean body composition data for each group were used, and stkin factor in IV groups was not included.
Results
Rat body weight and body composition profiles from the age of 5 weeks until the end of the study are shown in Fig. 2. While data are shown separately for each group, animals were randomly assigned to IV and SC administration routes approximately 1 week before the dosing. As illustrated in Fig. 2, there was no significant difference in body metrics between the two obese groups and no difference between the two lean groups throughout the study period. TBW was significantly different between obese and control animals at the age of 5 weeks (Welch’s t-test, p < 0.05) (Fig. 2a). Lean body weight and OBW were comparable between obese and lean rats, approximately 400 g and 420 g at the end of the study (Fig. 2b and d). Total and free water mass were also comparable between obese and lean rats (Fig. 2e and f). Lean and obese animals had significantly different fat mass and abdominal circumference starting from the age of 5 weeks (Fig. 2c and g). The fat percentage at the time of dosing of human IgG was 41 ± 3.8% in obese rats and 7.2 ± 1.56% in control rats. The length was comparable initially and became slightly larger in obese animals than in lean animals at the age of 15 weeks (Fig. 2H). Observed differences in some of body size and composition metrics were associated with alterations in pharmacokinetics, as described below.

Time course of various metrics of body size and composition of lean and obese Zucker rats from 5 week of age till the end of study. Data are shown as mean ± SD (n = 5–8). Obese and control rats were randomly assigned to IV and SC dose groups (for human IgG administration) a few days before the dosing (lean and obese animals were dosed at the age of 23 and 24 weeks, respectively).
Serum rat IgG profiles in lean and obese rats starting from the age of 5 weeks are shown in Fig. 3. Initially, rat IgG serum concentration was higher in lean rats than in obese rats (Welch’s t-test, p < 0.05). Rat IgG serum concentration increased with animal age and reached an apparent plateau at approximately the age of 22-to-23 weeks. No significant difference existed between rats assigned to IV and SC groups before human IgG dosing. After IV infusion of 1 g /kg of human IgG, a rapid drop in rat IgG concentration was observed in both obese and control animals during the first few days (probably due to a saturation of FcRn-mediated recycling); the concentrations of rat IgG returned to pre-infusion levels at approximately 14 days post-dose. Furthermore, in some animals from the IV groups, the concentration of endogenous IgG continued to rise to significantly higher values than pre-infusion. Serum rat IgG profiles in obese and lean animals showed no significant drop following SC injection of human IgG.

Observed serum pharmacokinetic profiles of endogenous rat IgG in obese and lean (control) rats before and after IV and SC single-dose administration of human IgG 1 g/kg. Data are shown as mean ± SD (n = 4–7). Time zero corresponds to 5 weeks of age. Control and obese animals were dosed with human IgG one week apart: day 126 for control groups and day 133 for obese groups. Rats were randomly assigned to IV and SC dose groups a few days before the dosing.
Human IgG pharmacokinetics profiles in the serum of lean and obese rats following 1 g/kg dose are shown in Fig. 4. Pharmacokinetic parameters calculated using a noncompartmental approach are shown in Table I. During the first few days post-dose, no difference was observed for IV groups in the human IgG serum concentrations between obese and lean animals. Then the serum profiles diverged as obese animals exhibited a faster concentration decline than lean animals (Fig. 4a). In SC animals, higher human IgG Cmax was observed in lean animals. Then the serum concentrations declined at an approximately similar rate, and human IgG concentration was consistently higher in lean animals resulting in a twofold higher AUC (Fig. 4b). SC bioavailability was similar in obese and lean groups, approximately 19%.

Observed serum pharmacokinetic profiles of human IgG in obese and lean (control) rats following IV and SC single-dose administration of 1 g/kg. Data are shown as mean ± SD (n = 4–7). For better visualization, the time scale was shifted in this figure to show time zero as time of injection (during the experiment lean and obese animals were dosed one week apart: day 126 for control groups and day 133 for obese groups).
A mechanistic model was successfully developed to describe serum pharmacokinetics of human IgG and changes in serum endogenous IgG concentrations after IV and SC administration of 1 g/kg human IgG in lean and obese rats (Fig. 1). Observed data were described well by the model. Population and individual fitted profiles were shown in Fig. 5a (human IgG) and 5B (rat IgG). VPC plots based on final model estimates were in agreement with the observed data (Fig. 6). All system parameters were estimated with sufficient precision (Table II); however, some parameters were fixed to published or previously estimated values. Between-subject variability was successfully estimated for several parameters, except for stkin, which might be related to a small number of animals. TBW and OBW growth of all animals were successfully described by a logistic growth function (Fig. 7).


Individual observed and population model fitted serum concentration-time profiles of human IgG (Panel a) following IV and SC dosing (1 g/kg TBW) and endogenous rat IgG (Panel b) in Zucker lean and obese rats. Symbols are observed data, solid red lines are individual model fits and dashed blue lines are population model fits. Time zero corresponds to 5 weeks of age. Lean (control) and obese animals were dosed with human IgG one week apart: day 126 for control groups and day 133 for obese groups.

Visual predictive check using final parameter estimates based on 1000 subjects per group. Simulated human IgG (panel a) and rat IgG (panel b) concentration–time profiles in Zucker obese and lean control rats. The solid line represents the median prediction, the dashed lines represent the 5th and 95th prediction interval, and circles are observed data.

Different combinations of TBW, OBW, lean mass, and abdominal circumference were added to Vp and Ve for comparison in the base model for data of IV groups. TBW on Vp and OBW on Ve were chosen based on objective function value (Supplementary Table 1). Then percentage of OBW or lean mass percentages were added to ka for comparison in the model with data from all groups. The base model that included TBW as a covariate on Vp, OBW as a covariate to Ve, and OBW/TBW as a covariate to ka provided the lowest objection function value and the best fit. To describe significantly faster lysosomal catabolism of human IgG in obese groups, adding FW/TBW to kdeg2 decreased the objection function. Faster lysosomal degradation of human IgG in obese animals did not generate enough separation for human IgG serum profiles between obese and lean groups. Therefore, TBW/FW was added to kret to further reduce the FcRn recycling rate in obese animals. The final model generated the lowest objection value, good fits, and parameter estimates with sufficient precision.
Discussion
Human IgG (in the form of IVIG and SCIG) is an important treatment modality for various immune and infectious diseases, and a wide range of doses is used for different indications. The effect of obesity on the biodistribution of antibodies has not been studied sufficiently, and the best body size metric for optimization of dosing is currently unknown. This animal study provides important mechanistic insights into the effects of obesity on the pharmacokinetics of both endogenous rat IgG and exogenous human IgG (after IV and SC administration).
The study showed that endogenous rat IgG concentrations increased with age in both strains of Zucker rats (obese and lean) and reached an apparent plateau at approximately 22 weeks (Fig. 3). These data were captured using a maturation function for the synthesis rate for rat IgG (kin - Eq. 2). We previously reported a similar age-dependent trend in Sprague Dawley rats when endogenous rat IgG increased from 0.67 µM at 7 weeks to 2.7 µM at 20 weeks [37]. This observation is important because animals of different ages are used in pharmacokinetic studies, and endogenous IgG concentration is a central parameter in FcRn-mediated endosomal recycling models (due to the competition between IgG-based therapeutics and endogenous IgG for FcRn binding). This parameter is often fixed to previously published values and is not directly measured during pharmacokinetic studies [28, 32, 39]. Furthermore, we observed that rat IgG concentration in Zucker lean animals was statistically significantly higher than in Zucker obese animals from the beginning of the study.
During the first few days following IV infusion of human IgG (1 g/kg of TBW), a rapid drop was observed in rat IgG concentration in both obese and lean animals, likely resulting from saturation of FcRn binding sites by exogenous IgG. There was no significant decrease in rat IgG following the SC dose of human IgG (as systemic concentrations of human IgG were at least tenfold lower than after IV dosing). The effect of high dose of human IgG on the clearance of endogenous IgG was reported before in mice [35], and several mechanistic models simulating this process in humans [40] and rodents [29, 32, 35] were published.
Rat IgG rebounded to pre-infusion (or higher) levels at approximately 14 days post-IV dose. While there is no consensus, data from wild-type and FcRn knockout mice support the hypothesis that no immunoregulatory feedback mechanism stimulates IgG synthesis after partial depletion [41]. Although a sterile surgery technique was used in this study to introduce jugular vein catheters needed for delivering the IV infusion, the procedure itself might have stimulated rat IgG synthesis; and this was reflected in the model by introducing an additional synthesis rate constant (stkin in Eq. 2). This higher than pre-dose rebound concentration phenomenon will need further evaluation in future studies.
Obesity substantially affected the pharmacokinetics of human IgG after IV and SC delivery, with lower concentrations in obese animals, faster elimination, and lower Cmax after SC delivery (Fig. 4 and Table I). These changes are consistent with previous findings of a relatively lower FcRn expression in adipose tissue [38]; and therefore, relatively less FcRn-mediated protection for IgG in obese subjects. Obesity is associated with changes in other organs, not only an increase in adipose tissues. For example, in humans, obesity is associated with increased weight of the intestines, liver, spleen, kidneys, and heart, but not the lungs [42]. To capture these differences between lean and obese rats, various body size metrics were used as covariates on different model compartments (Fig. 1).
A mechanistic model was successfully developed to capture all observed data (Figs. 1, 5, 6, 7, and Table II). The final model included various body composition metrics as covariates on several model parameters, which allowed for a good simultaneous description of pharmacokinetic profiles in lean and obese animals. As a partial model qualification, some of the model parameter estimates were compared to previously published data [31, 36, 40, 41, 43,44,45,46]. Endogenous rat IgG production was studied in mice [41] and estimated as zero-order synthesis in several studies [28, 29, 32, 43]. In our study, rat endogenous IgG concentration increased with animal age, which allowed for capturing the maturation of IgG production rate (kin). The value of kin was estimated to increase from 0.0018 to 0.0156 µmol/day from birth to adulthood, which was similar to 0.013 µmol/day reported in mice [41] (production rate data in rats could not be identified in the literature). Total endosomal FcRn concentration (Rtot) was estimated to be 60.5 µM, similar to 33–50 µM estimated using PBPK models of rat data before [43, 44]. In-vitro kon,h and kon,r were reported as 0.01 and 0.08 µM−1·day−1 [31], lower than our estimated value of 0.229 µM−1·day−1, which might reflect the difference between in-vitro and in-vivo conditions. Different approaches are used in modeling literature regarding endosomal return and uptake rate constants (kret and kup), where they are either constrained to be equal [29, 32, 43] or estimated separately [45, 46]. In our study, kret and kup were estimated separately (and kret was also depended on body composition). The estimates from our model were consistent with previous studies: 0.5–1 day−1 for kret and 0.18–1.96 day−1 for kup [40, 45, 46]. Binding affinity in-vitro between rat FcRn and rat and human IgG was previously reported (KDr = 4.1–8.2 nM and KDh = 15–30 nM at 37°C) [31, 36]. To facilitate model performance, we only estimated KDr (and the estimate was 4.75 nM), and KDh was constrained to be 3.66-fold higher (17.8 nM effective value).
Multiple models and covariates combinations were tested during the development process. A model with TBW as covariates to both Vp and Ve was compared with other models (with multiple combinations of TBW, OBW, lean body weight, and abdominal circumference as covariates to Vp and Ve). The model with TBW on Vp and OBW on Ve better described the data from obese and lean cohorts. Different covariates were also explored for the SC absorption process (including lean body weight and OBW percentages). The model with the percentage of OBW as a covariate on ka yielded the best model performance and was consistent with the observed data and reported changes in SC tissue in obesity [27]. A more complex saturable absorption model (previously reported for describing dose-dependent absorption of rituximab in rats [47]) did not improve model fits, which might be related to the fact that only a single dose level was tested in this study. Dissociation rate constants for human and rat IgG and rat FcRn (koffr and koffh) were reported to be comparable in-vitro [31, 43]. However, a model with koffr and koffh constrained to be equal did not provide a satisfactory fit (Supplementary Table 1).
One limitation of this study is that only a single dose level was tested (1 g/kg). To overcome this limitation, simulations were performed using the final model estimates to generate human and rat IgG profiles under three human IgG different dose level scenarios for comparison (Fig. 8). All three dose levels resulted in a similar general trend in the data; however, the extent of the decrease in rat IgG depended on the dose. An interesting finding from the simulation (and observed phenomenon in some rats in this study) is an apparent slight drop in endogenous IgG in the first few weeks (at approximately 6-to-7 weeks of age). It may be related to a relatively slow increase in IgG production rate in comparison to faster growth of the body size. These results should be further evaluated in future studies.

Model simulated human IgG and rat IgG profiles following IV and SC administration of 0.5, 1, and 2 g/kg human IgG. Mean body composition data for each group was used and stkin factor in IV groups was not included. For rat IgG figures, time zero corresponds to 5 weeks of age. Control and obese animals were dosed with human IgG one week apart: day 126 for control groups and day 133 for obese groups. For human IgG, the time scale was shifted in this figure to show time zero as time of injection for better visualization.
In the clinic, human IgG is administered intravenously or subcutaneously in cycles at different dose levels (depending on the indication) [17]. Although there is no consensus, a trough concentration in the range of 500-to-1000 mg/dL has been suggested for maintaining efficacy, and often dosing is adjusted based on patients’ response [48,49,50,51,52,53]. A series of simulations was conducted to illustrate how the difference in IgG pharmacokinetics between obese and lean individuals might affect the dose level and/or frequency of administration requirements (Fig. 9). Although the simulation is performed in rats, a trough level of 33 µM (500 mg/dL) was used as a hypothetical therapeutic threshold. Simulations showed that IV administration of 1 g/kg once weekly was sufficient to maintain human IgG concentration above the threshold in lean rats. However, increasing the IV dose to 2.4 g/kg once weekly or administering 1 g/kg every 4 days would be required in obese rats. Following SC dosing, the threshold was not achieved in either lean nor obese rats after a single SC dose, and a multiple-dose schedule was required to achieve the goal. Similar to IV dosing, a higher SC dose level or higher dosing frequency was required in obese rats than in lean rats to reach the set steady-state trough threshold. Model simulations should be verified in future studies.

Simulations illustrating the need to use a higher dose level or a higher dosing frequency for human IgG in obese rats compared to lean rats in order to achieve a trough concentration of 33 µM (500 mg/dL). Mean body composition data for each group was used and stkin factor in IV groups was not included. Time zero corresponds to the age and body composition at the time of human IgG dosing in this study.
In conclusion, important obesity-induced differences in the pharmacokinetics of endogenous and exogenous IgG were observed, and a mechanism-based model describing the observed data was developed. The model combined several major components: endogenous rat IgG homeostasis with age-dependent synthesis rate; competition of human IgG and endogenous rat IgG for FcRn binding and its effect on endogenous rat IgG concentrations following injection of a high dose of human IgG; and the effect of body size and composition (changing over time and dependent on the obesity status) on pharmacokinetic parameters. These results can potentially facilitate optimization of the dosing of IgG-based therapeutics in the obese population.
References
Bays HE. Adiposopathy is “sick fat” a cardiovascular disease? J Am Coll Cardiol. 2011;57(25):2461–73.
Bouchard C. Defining the genetic architecture of the predisposition to obesity: a challenging but not insurmountable task. Am J Clin Nutr. 2010;91(1):5–6.
World Health Organization. Obesity and overweight, Geneva: WHO; 2020. Available from: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight. Accessed 11 Nov 2022.
Nguyen DM, El-Serag HB. The epidemiology of obesity. Gastroenterol Clin North Am. 2010;39(1):1–7.
Barras M, Legg A. Drug dosing in obese adults. Aust Prescr. 2017;40(5):189–93.
Bai S, Jorga K, Xin Y, Jin D, Zheng Y, Damico-Beyer LA, et al. A guide to rational dosing of monoclonal antibodies. Clin Pharmacokinet. 2012;51(2):119–35.
Zhao L, Ren TH, Wang DD. Clinical pharmacology considerations in biologics development. Acta Pharmacol Sin. 2012;33(11):1339–47.
Wang W, Wang EQ, Balthasar JP. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin Pharmacol Ther. 2008;84(5):548–58.
Bensch F, Smeenk MM, van Es SC, de Jong JR, Schroder CP, Oosting SF, et al. Comparative biodistribution analysis across four different (89)Zr-monoclonal antibody tracers-The first step towards an imaging warehouse. Theranostics. 2018;8(16):4295–304.
Hanley MJ, Abernethy DR, Greenblatt DJ. Effect of obesity on the pharmacokinetics of drugs in humans. Clin Pharmacokinet. 2010;49(2):71–87.
Jeffrey F, Barletta PD, FCCM. Drug Dosing in Special Populations: Obesity and Geriatrics. CCSAP 2020 Book 2 • Issues in Critical Care Practice. 2020.
MacDonald JJ, Moore J, Davey V, Pickering S, Dunne T. The weight debate. J Intensive Care Soc. 2015;16(3):234–8.
Morgan DJ, Bray KM. Lean body mass as a predictor of drug dosage. Implications for drug therapy. Clin Pharmacokinet. 1994;26(4):292-307.
Zhang S, Shi R, Li C, Parivar K, Wang DD. Fixed dosing versus body size-based dosing of therapeutic peptides and proteins in adults. J Clin Pharmacol. 2012;52(1):18–28.
Rutter A, Luger TA. High-dose intravenous immunoglobulins: an approach to treat severe immune-mediated and autoimmune diseases of the skin. J Am Acad Dermatol. 2001;44(6):1010–24.
Jolles S, Sewell WA, Misbah SA. Clinical uses of intravenous immunoglobulin. Clin Exp Immunol. 2005;142(1):1–11.
Lexicomp. Immune globulin (Intravenous, subcutaneous, and intramuscular): Drug information. UpToDate. https://www.wolterskluwer.com/en/solutions/lexicomp. Accessed 18 Dec 2021.
Hodkinson JP. Considerations for dosing immunoglobulin in obese patients. Clin Exp Immunol. 2017;188(3):353–62.
Han PY, Duffull SB, Kirkpatrick CM, Green B. Dosing in obesity: a simple solution to a big problem. Clin Pharmacol Ther. 2007;82(5):505–8.
Leykin Y, Miotto L, Pellis T. Pharmacokinetic considerations in the obese. Best Pract Res Clin Anaesthesiol. 2011;25(1):27–36.
Borvak J, Richardson J, Medesan C, Antohe F, Radu C, Simionescu M, et al. Functional expression of the MHC class I-related receptor, FcRn, in endothelial cells of mice. Int Immunol. 1998;10(9):1289–98.
Poduslo JF, Curran GL, Berg CT. Macromolecular permeability across the blood-nerve and blood-brain barriers. Proc Natl Acad Sci U S A. 1994;91(12):5705–9.
Ubogu EE. The molecular and biophysical characterization of the human blood-nerve barrier: current concepts. J Vasc Res. 2013;50(4):289–303.
Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112(12):1796–808.
Wohlrab J. Pharmacokinetic characteristics of therapeutic antibodies. J Dtsch Dermatol Ges. 2015;13(6):530–4.
Kobrynski L. Subcutaneous immunoglobulin therapy: a new option for patients with primary immunodeficiency diseases. Biologics. 2012;6:277–87.
Richter WF, Bhansali SG, Morris ME. Mechanistic determinants of biotherapeutics absorption following SC administration. AAPS J. 2012;14(3):559–70.
Xiao JJ. Pharmacokinetic models for FcRn-mediated IgG disposition. J Biomed Biotechnol. 2012;2012:282989.
Xiao JJ, Krzyzanski W, Wang YM, Li H, Rose MJ, Ma M, et al. Pharmacokinetics of anti-hepcidin monoclonal antibody Ab 12B9m and hepcidin in cynomolgus monkeys. AAPS J. 2010;12(4):646–57.
Li T, Balthasar JP. Application of physiologically based pharmacokinetic modeling to predict the effects of FcRn inhibitors in mice, rats, and monkeys. J Pharm Sci. 2019;108(1):701–13.
Vaughn DE, Bjorkman PJ. High-affinity binding of the neonatal Fc receptor to its IgG ligand requires receptor immobilization. Biochemistry. 1997;36(31):9374–80.
Hansen RJ, Balthasar JP. Pharmacokinetic/pharmacodynamic modeling of the effects of intravenous immunoglobulin on the disposition of antiplatelet antibodies in a rat model of immune thrombocytopenia. J Pharm Sci. 2003;92(6):1206–15.
Blanco E, Perez-Andres M, Arriba-Mendez S, Contreras-Sanfeliciano T, Criado I, Pelak O, et al. Age-associated distribution of normal B-cell and plasma cell subsets in peripheral blood. J Allergy Clin Immunol. 2018;141(6):2208–19 e16.
Back HM, Lee JB, Han N, Goo S, Jung E, Kim J, et al. Application of size and maturation functions to population pharmacokinetic modeling of pediatric patients. Pharmaceutics. 2019;11(6).
Bleeker WK, Teeling JL, Hack CE. Accelerated autoantibody clearance by intravenous immunoglobulin therapy: studies in experimental models to determine the magnitude and time course of the effect. Blood. 2001;98(10):3136–42.
Abdiche YN, Yeung YA, Chaparro-Riggers J, Barman I, Strop P, Chin SM, et al. The neonatal Fc receptor (FcRn) binds independently to both sites of the IgG homodimer with identical affinity. MAbs. 2015;7(2):331–43.
Chapy H, Kagan L. Evaluation of the effects of animal growth and previous exposure on the pharmacokinetics of rituximab in rats. J Pharm Sci. 2018;107(7):1987–94.
Fan YY, Farrokhi V, Caiazzo T, Wang M, O'Hara DM, Neubert H. Human FcRn tissue expression profile and half-life in PBMCs. Biomolecules. 2019;9(8):373.
al-Bander HA, Martin VI, Kaysen GA. Plasma IgG pool is not defended from urinary loss in nephrotic syndrome. Am J Physiol. 1992;262(3 Pt 2):F333–7.
Kim J, Hayton WL, Robinson JM, Anderson CL. Kinetics of FcRn-mediated recycling of IgG and albumin in human: pathophysiology and therapeutic implications using a simplified mechanism-based model. Clin Immunol. 2007;122(2):146–55.
Junghans RP. IgG biosynthesis: no “immunoregulatory feedback.” Blood. 1997;90(10):3815–8.
Mandal R, Loeffler AG, Salamat S, Fritsch MK. Organ weight changes associated with body mass index determined from a medical autopsy population. Am J Forensic Med Pathol. 2012;33(4):382–9.
Urva SR, Yang VC, Balthasar JP. Physiologically based pharmacokinetic model for T84.66: a monoclonal anti-CEA antibody. J Pharm Sci. 2010;99(3):1582–600.
Shah DK, Betts AM. Towards a platform PBPK model to characterize the plasma and tissue disposition of monoclonal antibodies in preclinical species and human. J Pharmacokinet Pharmacodyn. 2012;39(1):67–86.
Ferl GZ, Wu AM, DiStefano JJ 3rd. A predictive model of therapeutic monoclonal antibody dynamics and regulation by the neonatal Fc receptor (FcRn). Ann Biomed Eng. 2005;33(11):1640–52.
Garg A, Balthasar JP. Physiologically-based pharmacokinetic (PBPK) model to predict IgG tissue kinetics in wild-type and FcRn-knockout mice. J Pharmacokinet Pharmacodyn. 2007;34(5):687–709.
Kagan L, Mager DE. Mechanisms of subcutaneous absorption of rituximab in rats. Drug Metab Dispos. 2013;41(1):248–55.
Rojavin M, Sidhu J, Pfister M, Hubsch A. Subcutaneous immunoglobulin loading regimens for previously untreated patients with primary antibody deficiency. Clin Exp Immunol. 2014;178(Suppl 1):146–8.
Ochs HD, Fischer SH, Wedgwood RJ, Wara DW, Cowan MJ, Ammann AJ, et al. Comparison of high-dose and low-dose intravenous immunoglobulin therapy in patients with primary immunodeficiency diseases. Am J Med. 1984;76(3A):78–82.
Orange JS, Hossny EM, Weiler CR, Ballow M, Berger M, Bonilla FA, et al. Use of intravenous immunoglobulin in human disease: a review of evidence by members of the Primary Immunodeficiency Committee of the American Academy of Allergy, Asthma and Immunology. J Allergy Clin Immunol. 2006;117(4 Suppl):S525-53.
Orange JS, Grossman WJ, Navickis RJ, Wilkes MM. Impact of trough IgG on pneumonia incidence in primary immunodeficiency: a meta-analysis of clinical studies. Clin Immunol. 2010;137(1):21–30.
Shehata N, Palda V, Bowen T, Haddad E, Issekutz TB, Mazer B, et al. The use of immunoglobulin therapy for patients with primary immune deficiency: an evidence-based practice guideline. Transfus Med Rev. 2010;24(Suppl 1):S28–50.
Perez EE, Orange JS, Bonilla F, Chinen J, Chinn IK, Dorsey M, et al. Update on the use of immunoglobulin in human disease: a review of evidence. J Allergy Clin Immunol. 2017;139(3S):S1–46.
Funding
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number R01GM124046 to Leonid Kagan. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Authors do not report any conflict of interest for this work.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Gao, X., Sheng, YH., Yu, S. et al. Mechanisms of Obesity-Induced Changes in Pharmacokinetics of IgG in Rats. Pharm Res 40, 1223–1238 (2023). https://doi.org/10.1007/s11095-023-03496-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-023-03496-y