
Abstract
Clinicians are increasingly faced with challenges regarding the pharmacological treatment of obese pediatric patients. To provide guidance for these treatments, a better understanding of the impact of obesity on pharmacological processes in children is needed. Results on pharmacological studies in adults show however ambiguous patterns regarding the impact of obesity on ADME processes or on drug pharmacodynamics. Additionally, based on the limited research performed in obese pediatric patients, it becomes clear that findings from obese adults cannot be expected to always translate directly to similar findings in obese children. To improve knowledge on drug pharmacology in obese pediatric patients, studies should focus on quantifying the impact of maturation, obesity, and other relevant variables on primary pharmacological parameters and on disentangling systemic (renal and/or hepatic) and presystemic (gut and/or first-pass hepatic) clearance. For this, data is required from well-designed clinical trials that include patients with not only a wide range in age but also a range in excess body weight, upon oral and intravenous dosing. Population modelling approaches are ideally suitable for this purpose and can also be used to link the pharmacokinetics to pharmacodynamics and to derive drug dosing regimens. Generalizability of research findings can be achieved by including mechanistic aspects in the data analysis, for instance, using either extrapolation approaches in population modelling or by applying physiologically based modelling principles. It is imperative that more and smarter studies are performed in obese pediatric patients to provide safe and effective treatment for this special patient population.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- ADME
- Obesity
- Pediatrics
- Pharmacodynamics
- Pharmacokinetics
- Physiologically based modelling
- Population modelling
1 Introduction
Around the world, the body weight of both adult and pediatric populations is generally increasing (Abarca-Gómez et al. 2017), and as a result, clinicians are increasingly faced with pharmacological treatment challenges for overweight and obese patients. In children, a global definition of overweight or obesity is lacking, but most definitions are based on a comparison of body mass index (BMI; the ratio of body weight in kg and the square of the length in m) to age- and sex-specific values, although there is evidence that using BMI to define obesity may lead to underestimation of the problem (Reilly et al. 2018). The CDC, for instance, defines overweight and obesity in children and adolescents as BMI above the 85th or 95th percentile of age- and sex-specific values in their growth charts, respectively. In 2013 an extensive survey found 23.8% of boys and 22.6% of girls to be either overweight or obese in developed countries, while in developing countries these numbers were 12.9% and 13.4%, respectively (Ng et al. 2014).
Obesity leads to physiological changes to the extent that the American Medical Association has officially classified obesity as a disease in 2013. Both adipose tissue and lean body weight are increased in obesity, with the ratio of fat and lean body mass being higher than in non-obese individuals. It is now also widely believed that both obese adult and obese pediatric patients are in a chronic state of low-grade inflammation (Wellen and Hotamisligil 2003; Rainone et al. 2016), which may impact the expression of metabolic enzymes, drug transporters, and plasma proteins (Ulvestad et al. 2013; Blouin et al. 1987). These and other changes may impact processes underlying the pharmacokinetics and pharmacodynamics of drugs, resulting in increased variability in drug pharmacology. The impact of these changes needs to be understood, to be able to provide guidance on safe and effective drug dosing in obese (pediatric) patients.
This paper provides an overview of current knowledge on the impact of overweight and obesity on the pharmacokinetics and pharmacodynamics of drugs. Specific focus is on pediatric patients; however, as the amount of information in this special population is very limited, findings in the adult population are also reviewed to assess whether generalizations from this population can be derived. Subsequently, guidance will be provided on how to perform future studies on drug pharmacology in obese children.
2 Drug Pharmacology in Obese Adults
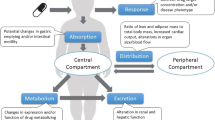
Drug exposure is dependent on processes related to absorption, distribution, metabolism, and excretion (ADME), which together results in pharmacokinetic profiles of drugs. The impact of obesity on ADME processes of different drugs has to a certain extent been studied in adults. However, findings from studies on different drugs, even when metabolized by the same pathway, are sometimes counterintuitive or may appear contradictory, making it difficult to derive generalizable dosing recommendations, even for adults.
Drug responses are dependent on both pharmacokinetics and pharmacodynamics, meaning that both drug exposure and the exposure-response relationships will result in desired and undesired drug effects. A small number of adult studies have shown that not just drug pharmacokinetics, but also drug pharmacodynamics may be altered with obesity.
2.1 Absorption in Obese Adults
The most common route for drug administration is oral. Factors that may impact the absorption rate and/or the bioavailability of orally administered drugs in obese patients include an increase in gastric emptying and intestinal motility (Xing and Chen 2004; Cardoso-Júnior et al. 2007), increased permeability of the gut due to loss of tight junction function and resulting increases in paracellular transport, which was observed both in adults and in children (Rainone et al. 2016; Teixeira et al. 2012), decreased expression of CYP enzymes in the gut and liver, which was found to increase with BMI in adults (Ulvestad et al. 2013), and increased splanchnic blood flow, which may carry drugs away faster from the metabolic enzymes in the gut wall (Alexander et al. 1962–1963).
Interestingly, for trazodone (Greenblatt et al. 1987), cyclosporine (Flechner et al. 1989), dexfenfluramine (Cheymol et al. 1995), and moxifloxacin (Kees et al. 2011), obesity was found to not impact bioavailability, despite the aforementioned physiological changes in obese patients. For propranolol a trend toward higher bioavailability in obese patients compared to non-obese patients was found, based on similar systemic clearance values in both groups upon intravenous dosing but smaller oral apparent clearance values in the obese upon oral dosing (Bowman et al. 1986). Similar trends in oral apparent clearance have been observed for alprazolam and triazolam, which for triazolam even reached statistical significance (Abernethy et al. 1984). However, in the absence of data upon intravenous dosing, it cannot be established whether these differences indeed arise from differences in bioavailability, from differences in clearance, or a combination of both. The same limitation applies to a study on vortioxetine, in which based on similar areas under the concentration-time curve (AUC) and steady-state concentrations (Greenblatt et al. 2018a), the bioavailability for vortioxetine can be assumed to be similar between obese and non-obese patients, while another possible explanation for the presented observations is that bioavailability and clearance change with similar magnitudes. For midazolam, one study did not find a difference in the bioavailability between obese and non-obese patients (Greenblatt et al. 1984), while a later study did find an increased bioavailability for midazolam in morbidly obese patients (Brill et al. 2014a). These differences in findings can potentially be attributed to differences in study design and data analysis, but another possible explanation could be that the latter study included patients with much higher body weights, which could imply that differences in bioavailability for midazolam only become apparent with extreme obesity, which may or may not also be linked to the duration a patient has been obese.
For midazolam, the absorption rate was found to be reduced in obese patients (Brill et al. 2014a). Studies using time to maximum concentration (Tmax) as a proxy for absorption rate suggest decreases in absorption rate of paracetamol (Lee et al. 1981) and levothyroxine (Michalaki et al. 2011) and unaltered absorption rate values for morphine (Lloret-Linares et al. 2014). It should, however, be noted that Tmax is impacted by parameters other than absorption rate and can therefore not be regarded as a pure proxy of absorption rate.
2.2 Distribution Volume in Obese Adults
Drug distribution is impacted by system-specific as well as drug-specific properties. With respect to system-specific changes in obesity, increases in the physical size of the body will increase blood and tissue volumes, additionally cardiac output and tissue perfusion might change (Alexander et al. 1962–1963; Lemmens et al. 2006), and all may be expected to impact drug distribution rates and distribution volume. Moreover, lipophilicity, a drug-specific property, may theoretically lead to increased partitioning into fat tissue, thereby increasing distribution volume for these drugs in the obese. However, specific and non-specific drug binding to blood or tissue constituents, the presence of blood constituents competing for plasma protein binding, as well as the presence of drug transporter proteins on tissue membranes may either further increase or decrease tissue partitioning and as a result lipophilicity is generally not a good predictor of distribution volume (Jain et al. 2011; Knibbe et al. 2015).
For drugs that are hydrophilic or only weakly or moderately lipophilic, partitioning into fat tissue is generally limited. It seems obesity mostly leads to at best moderate increases in distribution volume, as was seen for methylxanthines, aminoglycosides, beta-blockers, ibuprofen, phenazone, certain benzodiazepines, ranitidine, and heparin (Morgan and Bray 1994; Davis et al. 1990; Cheymol et al. 1997; Smit et al. 2019). For the hydrophilic drug vancomycin, however, both moderate and large increases in distribution volume values have been reported in the obese (Adane et al. 2015; Blouin et al. 1982).
Due to the impact of aforementioned mechanisms that may impact drug partitioning into fat, the expected increase in distribution volume of lipophilic compounds in the obese is in fact found to be highly variable. For posaconazole (Greenblatt et al. 2018b), lipophilic benzodiazepines such as midazolam (Brill et al. 2014a), thiopental sodium, phenytoin, verapamil, and lidocaine (Morgan and Bray 1994), distribution volume was found to be greatly increased in the obese, while the distribution volume for propofol, digoxin, cyclosporine, and prednisolone was found to remain constant in the obese (Morgan and Bray 1994; van Kralingen et al. 2011a; Abernethy et al. 1981).
By reducing the unbound drug fraction, plasma protein binding may limit the distribution of drugs into peripheral tissue, as only unbound drug is believed to be able to diffuse into tissue. Results regarding potential changes in concentrations of serum albumin and α1-acid glycoprotein in the obese are contradictory but generally indicated only relatively small changes (Benedek et al. 1983; Cheymol et al. 1987; Pai et al. 2007). Increases in triglycerides that may compete with drugs for plasma protein binding (Benedek et al. 1983) may increase the unbound fraction of some drugs. A decrease in unbound drug fraction has been reported for propranolol, while unbound fractions of phenytoin, alprazolam, cefazolin, daptomycin, and various benzodiazepines have been reported to remain unchanged (Abernethy et al. 1984; Greenblatt et al. 1984; Benedek et al. 1983; Cheymol et al. 1987; Pai et al. 2007; Brill et al. 2014b). It should be noted that decreased unbound drug fractions will proportionally reduce the total clearance of drugs with a low or intermediate extraction ratio, which result in unaltered unbound drug concentrations in the obese.
In addition to changes in distribution volume, it also has to be kept in mind that tissue penetration of drugs may be reduced in obese patients. For cefazolin, subcutaneous tissue concentrations were found to be considerably reduced in obese patients, while systemic exposure to this drug was similar to values in non-obese patients. Such reduced local exposure may reduce the apparent efficacy of prophylactic treatments of postoperative wound infections with this antibiotic (Brill et al. 2014b).
2.3 Metabolism in Obese Adults
Drug metabolism mainly occurs in the liver. Due to abnormal fat disposition and the low-grade chronic inflammation, nonalcoholic fatty liver disease, ranging from steatosis to nonalcoholic steatohepatitis (NASH), is common in obese individuals (Harnois et al. 2006; Machado et al. 2006). Potential other changes including changes in plasma protein binding, liver blood flow and perfusion, expression of transporters on hepatocytes, and metabolic enzymes in the hepatocytes may all further impact hepatic metabolic drug clearance. How these changes impact the hepatic metabolism of drugs depends to a large extent on the extraction ratio.
For drugs with a low or intermediate extraction ratio, plasma protein binding may be a limiting factor for hepatic metabolism; however, generally no large changes in protein binding are observed in obesity (Benedek et al. 1983; Cheymol et al. 1987; Pai et al. 2007) as mentioned before. For these drugs, the intrinsic clearance, which is impacted by the expression and activity of metabolic enzymes, is likely the most limiting factor for hepatic metabolism in the obese. Generally, clearance of CYP3A4 substrates is reduced with obesity, although statistical significance of findings is not always reached in trials (Brill et al. 2012). This trend is in line with observations of reduced CYP3A4 activity in adult and pediatric patients with nonalcoholic fatty liver disease or inflammation (Woolsey et al. 2015; Kolwankar et al. 2007; Brussee et al. 2018). For caffeine and theophylline, both substrates for CYP1A2, no changes in clearance were observed with obesity (Caraco et al. 1995; Jusko et al. 1979), although for the latter a trend toward increased clearance was observed after correcting for confounding factors. The clearance of substrates for other CYP enzymes, including CYP2C9, CYP2C19, CYP2D6, and CYP2E1, also appears to be increased to various extents in obese patients (Brill et al. 2012; Emery et al. 2003; van Rongen et al. 2016), although especially for CYP2C19 and CYP2D6 the impact of polymorphisms on inter-individual differences in clearance seems to be more pronounced than the impact of obesity.
Remarkably, a drug-drug interaction study seems to suggest that compared to non-obese patients, the relative impact of CYP3A inhibition by posaconazole on oral exposure to lurasidone is less pronounced in obese, but that the effect of the inhibition does persist longer (Greenblatt et al. 2018b). It has to be mentioned however that at least 66% of this difference was reported to be attributable to the reduced oral exposure to posaconazole in obese patients in combination with a prolonged washout of this drug in the obese. The authors attribute the difference in the exposure of posaconazole, which is mainly metabolized by UGT enzymes, to increased clearance and distribution volume in the obese. In addition to posaconazole, the glucuronidation of paracetamol, lorazepam, oxazepam, and garenoxacin has been reported to be increased with obesity (van Rongen et al. 2016; Abernethy et al. 1982, 1983; Van Wart et al. 2004), although the finding for garenoxacin may have been confounded by an underestimation of creatinine clearance in the obese. Interestingly, UGT2B15 has been shown to be present in adipose tissue, which has been proposed to cause the reduced plasma levels for testosterone in obese men (Tchernof et al. 1999); it is however unknown whether this is also responsible for increased glucuronidation of drug substrates in obese patients.
For drugs with a high extraction ratio, hepatic blood flow and perfusion are the main rate limiting factors for hepatic metabolism. Fatty liver disease has been suggested to reduce sinusoidal perfusion (Farrell et al. 2008), which could reduce hepatic clearance, while an increased cardiac output in obese patients might overall direct more blood to the liver, which could increase hepatic clearance for drug with a high extraction ratio. For propofol, clearance was consistently found to be increased in obese adults (van Kralingen et al. 2011a; Dong et al. 2016; Diepstraten et al. 2013; Cortínez et al. 2010). Findings for sufentanil and paclitaxel also indicate a trend toward increased clearance in obese patients (Schwartz et al. 1991; Sparreboom et al. 2007). A tendency toward increased hepatic blood flow in obese patients is further supported by findings for midazolam and fentanyl, which are mainly metabolized by CYP3A4 metabolism and have an intermediate to high extraction ratio. Clearance for these drugs has been reported to be the same or even increased in obese versus non-obese individuals (Greenblatt et al. 1984; Brill et al. 2014a; Shibutani et al. 2004), which could be interpreted as that reduced CYP3A4 activity in obese individuals is compensated by increases in hepatic blood flow and perfusion. Findings for the clearance of morphine do however not fit this narrative. Morphine is mainly cleared through glucuronidation and has an intermediate to high extraction ratio; both factors are believed to yield increased clearance with obesity, yet in a clinical study the clearance of morphine in obese patients was found to be similar to the clearance in non-obese patients (de Hoogd et al. 2017); this could suggest a role for obesity-related changes in transporter expression or activity.
Influx or efflux transporters on hepatocytes may, respectively, increase or decrease the presentation of drugs to metabolic enzymes in the hepatocytes and can thereby impact hepatic metabolic clearance. The impact of hepatic drug transporters on drug clearance has hitherto remained largely understudied in most patient populations. Rodent studies do suggest that expression or functionality of hepatic transporters may be changed in models for NASH (Dzierlenga et al. 2015; Fisher et al. 2009). Human studies also found expression of transporters in obese patients to be altered, even to the extent that it can in some cases impact drug clearance (Ulvestad et al. 2013). The directionality of these transporters and the differences in their increased or decreased expression and functionality may result in enhanced or reduced hepatic metabolic clearance for various drugs in obese patients.
2.4 Excretion Clearance in Obese Adults
The kidneys are the primary drug excretion organs. As for the liver, kidney blood flow and perfusion may be altered in obesity, and as only unbound drug can be filtered in the glomeruli, potential changes in plasma protein binding can also impact excretion clearance of drugs. The biggest impact of altered excretion clearance in the obese and overweight is, however, likely the result of reported increases in glomerular filtration rate (GFR), which is believed to result from changed kidney function (Ribstein et al. 1995; Chagnac et al. 2008; Park et al. 2012). This may explain the increases in renal clearance observed in obese patient for drugs that are exclusively renally cleared including vancomycin, gentamycin, amikacin, and tobramycin (Adane et al. 2015; Blouin et al. 1982; Brill et al. 2012; Bauer et al. 1983, 1998). Interestingly, the excretion of morphine metabolites was found to be decreased in obese patients (de Hoogd et al. 2017). Although in animals these metabolites were found to be mainly excreted by the kidneys, one possible explanation provided by the authors is that in humans biliary excretion of morphine metabolites might be more relevant than renal excretion.
In addition to GFR, active tubular secretion facilitated by drug transporters is also believed to be increased in obese patients, which may further add to the increased renal excretion of drugs like oseltamivir and its active metabolite, procainamide, ciprofloxacin, and cisplatin in the obese (Sparreboom et al. 2007; Chairat et al. 2016; Christoff et al. 1983; Allard et al. 1993). Recently a more prominent role for obesity-related induction of OCT2 has been suggested to play a role in the increase in metformin clearance (van Rongen et al. 2018a) and gentamicin clearance (Smit et al. 2019), but not tobramycin clearance (Smit et al. 2019).
Less is known about tubular reabsorption of drugs in the obese. One study, for instance, found the clearance of lithium to be increased with obesity, which was attributed to impaired reuptake (Reiss et al. 1994). Another study, however, found the excretion of lithium to be reduced in those obese with an increased filtration fraction, as this increased filtration fraction increases the oncotic pressure, which in turn increases reabsorption (Chagnac et al. 2008).
2.5 Pharmacodynamics in Obese Adults
For a limited number of drugs, differences in pharmacodynamics between obese and non-obese patients have been studied. A recent Chinese study, for instance, found for propofol that the EC50 (i.e., the concentration at which half the maximum effect is obtained) was reduced in morbidly obese patients compared to non-obese controls. Such an increase in drug potency suggests that a similar effect can be obtained at lower plasma concentrations (Dong et al. 2016). A previous study performed in Europe did, however, not find differences in the efficacy or potency of propofol between morbidly obese and non-obese patients (van Kralingen et al. 2011a).
For atracurium, a study reported no differences in the distribution volume and clearance between obese and non-obese patients. As atracurium in this study was dosed based on body weight, the concentrations in obese patients were systematically higher than in non-obese patients, yet no difference was observed in the time of recovery from neuromuscular blockade, from which the authors concluded that the efficacy of this drug may be reduced in obese patients (Varin et al. 1990). Another study found, however, that when atracurium is dosed based on ideal body weight, predictable efficacy profiles in terms of train-of-four (TOF) ratios, intubation conditions, and need for antagonism with neostigmine can be expected between individual morbidly obese patients (van Kralingen et al. 2011b). Unfortunately plasma concentrations were not obtained, but it is expected that dosing based on ideal body weight in the morbidly obese yields similar exposure than non-obese patients, which would suggest that there are in fact no differences in efficacy between these populations. From these studies it can therefore not be concluded if there truly is a difference in the relationship between concentration and effect of atracurium for obese and non-obese adults.
Reduced insulin sensitivity and type 2 diabetes are commonly encountered in obese patients, which is likely the result of the low-grade chronic inflammation in these patients. The reduced insulin sensitivity will also impact the sensitivity to exogenous insulin used in the pharmacological treatment of these patients.
For infectious diseases it is often believed that similar target exposures between patient subpopulations will yield similar efficacy, as the microorganisms that a drug is treating do generally not differ between these populations. However, as the immune response and immune cell differentiation and activity are dysregulated in obesity, anti-infective drugs may appear to be less effective in the obese as the hosts’ immune system is less efficient in clearing an infection (Huttunen and Syrjänen 2013). Similarly, the efficacy of other treatments that are based on interactions with the hosts’ immune response may be expected to be altered in obesity. In this respect immunogenicity in obese patients has been shown to be increased for the flu vaccine, while for hepatitis B no difference in the immune response between obese and non-obese patients has been observed (Xiong et al. 2017).
Reduced tissue penetration of antimicrobial drugs in obese patients, which has, for instance, been shown for cefazolin as mentioned before (Brill et al. 2014b), will cause a shift toward reduced efficacy when assessing the relationships between systemic drug exposure (e.g., blood concentrations) and observed efficacy. Although this presents itself as an apparent difference in drug pharmacodynamics, this should not be interpreted as such, because the underlying mechanism that is driving the observed differences is drug distribution to the site of action, which is a pharmacokinetics process.
3 Drug Pharmacology in Obese Pediatric Patients
Table 1 provides an overview of the changes in drug pharmacological parameters associated with obesity in the adult population, for the various drugs discussed above. In obese adults, multiple physiological changes occur, including increase in body size and volume of fat tissue and increase in cardiac output and altered organ perfusion; altered enzyme and transporter expression and functionality, intestinal, hepatic, and renal dysfunction; and altered plasma protein binding. The dynamics of these changes may differ depending on the underlying mechanism, and the impact of each change on the pharmacokinetics and/or pharmacodynamics of a drug may in some cases depend on drug properties. Additionally, some of the changes may have opposing effects on pharmacological parameters.
In children, growth and maturation are additional dynamic factors that may impact the pharmacokinetics and/or pharmacodynamics of drug. In obese children these changes occur in conjunction with the physiological changes induced by obesity. Moreover, the off-label and/or unlicensed use of drugs in the pediatric population is still relatively high, and as a result optimized drug dosing recommendations may not be available for a large number of drugs even in non-obese pediatric patients. For those drugs for which dedicated pediatric studies have been performed, body weight is often found as a predictive descriptor of the impact of changes in both body size and maturation on pharmacokinetic parameters. Whether the pediatric dosing recommendations based on body weight that are derived from such studies should be maintained in obese pediatric patients is essential to know in order to develop rational dosing guidelines for the growing number of obese pediatric patients, because inconsistent dosing strategies are currently being adopted in clinical practice for these patients (Gade et al. 2018).
Unfortunately, the number of studies that assess the relationship between size or age descriptors and pharmacological parameters in obese pediatric patients is much more limited than in adults. In this case it may be tempting to extrapolate findings from obese adult populations to children. However, also for this, findings between different drugs and between obese adults and obese children are sometimes counterintuitive or may appear contradictory, making it difficult to derived generalizable dosing recommendations or approaches for extrapolations of findings between adults and children.
3.1 Similarities and Differences in Drug Clearance Between Obese Adults and Children
As drug clearance is the main driver of drug exposure at steady state and therefore of required maintenance doses, we focus on this parameter in this section.
When analyzing the pharmacokinetics of propofol in obese and non-obese adults, adolescents, and children simultaneously, a relationship was found between total body weight and clearance, with decreased values in young adults, adolescents, and children (Diepstraten et al. 2013). In this case obese adolescents with the same body weight as obese adults had lower clearance values than these adults, but their clearance values could be higher than those of non-obese adults with lower body weights.
For busulfan, the covariate relationship that defined the impact of maturation on clearance in non-obese individuals across a wide pediatric age range was based on body weight. It was found that this weight-based relationship could also be used to define clearance in obese pediatric individuals (Bartelink et al. 2012). This means that for two children of the same age, total clearance will be higher in the obese child compared to the non-obese child and, alternatively, that total clearance is the same in an obese child, compared to a non-obese child of the same weight (that is inevitably older than the obese child). Others have found that busulfan clearance per kilogram body weight is lower in children with a BMI higher than the 85th percentile of their age, compared to children with a lower BMI (Browning et al. 2011). Although the results of these studies cannot be directly compared due to the different parameterizations of the impact of weight and age on clearance, the results could point in the same direction. This is because to obtain total clearance, the clearance per kilogram is multiplied by more kilograms in obese children compared to non-obese children. It cannot, however, be excluded that in terms of total values, there are differences between obese and non-obese children for the clearance of busulfan. In adults it has, for instance, also been observed that for busulfan, the apparent oral clearance per kilogram body weight is lower in obese and even lower in morbidly obese patients compared to non-obese and underweight patients, while in terms of total apparent oral clearance values, this translated into elevated values in obese adults and even further elevated values in morbidly obese adults (Gibbs et al. 1999). For absolute clearance values obtained upon intravenous administration of busulfan in adults, values expressed per kilogram were also found to decrease with severity of obesity, but it was not reported how this translated into total absolute clearance values (Nguyen et al. 2006).
Findings on the pharmacokinetics of metformin in overweight and obese adolescents were in line with the findings above for propofol and busulfan, in that clearance increases with increasing age, which was parameterized in this study as weight-for-age-and-length (van Rongen et al. 2018a). On top of this first function, a second function was identified describing the linear increase in metformin clearance with excess body weight in this adolescent population (van Rongen et al. 2018a), which is also in line with findings for propofol and busulfan. Compared to values reported in literature, the clearance values in the obese adolescents were found to be comparable to values in non-obese adults and showed a tendency to be lower compared to clearance in obese adults. Most interestingly, however, a study on metformin in obese adults showed that lean body weight was the best size descriptor for inter-individual differences in clearance (Bardin et al. 2012), which suggest a much more limited impact of excess body weight on the clearance of metformin in the obese adult population, compared to the obese adolescent population.
Correlations between a reduced metabolic clearance by CYP3A isoenzymes and both decreasing age in children and increasing inflammation and organ failure have been reported (Woolsey et al. 2015; Kolwankar et al. 2007; Brussee et al. 2018). In adults, obesity was found to generally reduce the apparent oral clearance of CYP3A substrates (Brill et al. 2012), while studies in morbidly obese adults with midazolam, which is predominantly metabolized by CYP3A, showed systemic clearance not to be impacted by obesity, potentially due to compensations by increasing blood flow (Brill et al. 2014a). Interestingly, and quite unexpectedly, a dedicated pharmacokinetic study for midazolam in overweight and obese adolescents showed the systemic clearance in these adolescents to be substantially higher than the systemic clearance in the morbidly obese adults (van Rongen et al. 2018b). In fact, the clearance values in the obese adolescents were more in line with clearance values obtained in the same morbidly obese patients described above, 1 year after bariatric surgery and after considerable weight loss (Brill et al. 2015). The clearance values of obese adults after weight loss were found to be higher than their clearance values before weight loss surgery. Potentially these findings can be explained by recovery of CYP3A activity in the liver after substantial weight loss. The differences between obese adolescents and obese adults can possibly be explained by the hypothesis that in obesity suppression of the CYP3A activity increases with the duration of the disease and therefore takes time to fully manifest, while the increase in cardiac output and hepatic blood flow is more closely linked to the increase in excess body weight. This was further confirmed by the finding that within the population of obese adolescents, midazolam clearance was found to increase with weight, which may in its turn be related to an increased liver blood flow. As described above, an increasing impact of obesity with increased duration of this disease has also been suggested as a possible explanation for the fact that bioavailability of midazolam in the morbidly obese adults was increased compared to non-obese adults, while in obese patients that have not reached the state of morbid obesity, the bioavailability is not (yet) impacted (Greenblatt et al. 1984; Brill et al. 2014a).
Table 1 also summarizes the findings regarding the impact of obesity on drug clearance in the pediatric population for the drugs discussed in this section. The examples above illustrate that generalizable conclusions on how to translate findings regarding the impact of obesity from adults to children are not obvious. Findings for propofol are in line with the expectation that, due to maturation, clearance in children is lower than clearance in adults and indeed show that clearance in obese children is lower than clearance on obese adults. Additionally, total clearance increased with excess body weight, meaning that obese adolescents with body weights higher than non-obese adults can have higher propofol clearance values than the non-obese adults. Similar trends are seen in both adult and pediatric patients for busulfan in which obesity or excess body weight increases the clearance. Findings for metformin are in line with propofol and busulfan in adolescents, as excess body weight increases clearance in this population as well, but clearance values in adolescents do not appear to increase far beyond values in non-obese adults, nor do clearance values in obese adults increase far beyond the values in non-obese adults, suggesting that the impact of excess body weight on the clearance of metformin diminishes when patients reach adulthood. In adults there is – unexpectedly – also no difference in the clearance of midazolam between obese and non-obese individuals, potentially because the reduced CYP3A activity that may be anticipated in the obese is compensated by increased blood flow. Yet in adolescents, midazolam clearance is in fact increasing with obesity. If these observations for midazolam can indeed be attributed to the duration of obesity as influencer on CYP3A activity, as hypothesized, the generalizability of findings in any study, whether in the adult or pediatric population, is limited without taking disease duration into account.
In summary, the highlighted examples show that there may or may not be contradictions in terms of the absence or presence of differences in drug clearance between obese and non-obese individuals of the adult or pediatric population. Moreover, the directionality of the impact of obesity on drug clearance cannot be presumed to follow the same pattern in adults and children.
4 How to Study Drug Pharmacology in Obese Children
Although research efforts are increasing, generally the pharmacokinetics and pharmacodynamics of drugs in children remain understudied. Studies in obese pediatric patients are even more limited. As generic scaling cannot be derived based on body weight (as proxy for maturation) nor on results described in current literature, further research efforts are required on drug pharmacology in obese children. For this research several considerations should be taken into account.
4.1 Focus on Primary Pharmacological Parameters with Population Modelling
Pharmacokinetic studies are ideally focused on quantifying primary pharmacokinetic parameters like absorption rate, bioavailability, clearance, and distribution volume, rather than descriptive metrics like Cmax, tmax, and observed AUC. This has the advantage that findings can be more directly linked to physiological mechanisms underlying drug pharmacokinetics, which will increase our understanding of the impact of obesity-related physiological changes, which may in turn aid in the definition of generalizable conclusions.
In pediatric patients, the population approach, also known as non-linear mixed effects modelling, is the preferred method to quantify primary pharmacological parameters (De Cock et al. 2011). In this approach, observed data on pharmacological outcome measures of all individuals in a study are analyzed simultaneously while still taking into account that different observations come from different individuals. This method can handle dense, sparse, and unbalanced data that are obtained during routine clinical practice or in multiple studies with different designs. Since this method can separate inter-individual variability from residual unexplained variability, it is ideally suitable for the establishment of covariate relationships that define correlations between patient or treatment characteristics and inter-individual variability in model parameters. Defined covariate relationships (related to obesity or other patient-related variables) on primary parameters can be directly used as the basis for drug dosing recommendations. Clearance is, for instance, the only determinant for steady-state concentration and drives through concentrations and is therefore important for the establishment of maintenance doses and dosing intervals. Distribution volume, on the other hand, drives peak concentrations and time-to-steady-state and is therefore important for peak concentrations and loading doses and the dosing of drugs that are driven by peak concentrations.
4.1.1 Disentangle Presystemic and Systemic Processes Impacting Exposure
Both presystemic (gut and/or first-pass hepatic) and systemic (renal and/or hepatic) clearance impact the overall exposure, quantified as AUC, of orally administered drugs. The parameters quantifying these processes are bioavailability and clearance, respectively. When only data upon oral drug administration is available, bioavailability cannot be quantified and obtained values for clearance, as well as distribution volume is indicated as apparent parameter values or “parameter over F,” meaning that the true values of these parameters change proportionally with bioavailability. To enable a physiological interpretation of observed differences in AUC between obese and non-obese patients of orally administered drugs, it would be beneficial to study the pharmacokinetics of these drugs both after intravenous and oral administration. Ideally, this is performed in the same patient at the same time, to exclude the impact of inter-individual and inter-occasion differences. Different approaches are available to achieve this experimentally. In a semi-simultaneous approach proposed by Brill et al., patients receive an oral drug dose first, followed by an intravenous dose a few hours later, with blood sampling to measure drug concentrations following both administrations (Brill et al. 2014a). With microdosing designs truly simultaneous oral and intravenous administration can be achieved. By having therapeutic intravenous drug administration and simultaneous administration of an oral microdose of the isotopically labeled drug and measuring the concentration-time profiles of both labeled and unlabeled drug, systemic and presystemic clearance can be quantified with low experimental variability (Hohmann et al. 2015). Proof-of-principle studies have confirmed that microdose studies are also suitable throughout the pediatric population (Mooij et al. 2014).
4.1.2 Inclusion Criterion in Pediatric Clinical Trials
Body weight is often identified as most predictive descriptor for the impact of maturational changes on pharmacokinetic parameters in populations of children with age-appropriate body weights, but how such findings should be extrapolated to obese children is generally unknown. An important question in obese children is how weight related to obesity versus weight related to maturation or growth adds to changes in the pharmacokinetics or pharmacodynamics of drugs. Proposed approaches to disentangle the impact of maturation and obesity in obese pediatric patients are focused on defining sex, length, and age-appropriate body weights for normal weight children as descriptor of maturation and quantifying excess weight as descriptor of obesity (van Rongen et al. 2018a). To enable the disentanglement of age- and obesity-induced changes in body size in the data analysis process, patient inclusion of the clinical study should ascertain inclusion of not just a wide range in body weight per se but also in a wide range in deviations from appropriate body weights or excess body weight.
Also, given that disease duration has been suggested to drive to what extent the impact of obesity on processes underlying drug pharmacology manifests for some parameters, but not for others, it may be prudent to record disease duration in future studies on the impact of obesity on drug pharmacology. This may be true for both the adult and pediatric population, particularly when predictions based on obese adults to obese adolescents are foreseen.
Finally, it has to be noted that the pediatric population is already relatively small and as a result the absolute amount of obese pediatric patients is small. Consequently, inclusion of a sufficient number of obese pediatric patients in clinical trials may be challenging (Tamborlane et al. 2016). Therefore, instead of RCTs, alternative designs for studies in this special patient population should be explored. For this the Learning Healthcare System may offer relevant insights (http://www.learninghealthcareproject.org/section/background/learning-healthcare-system).
4.1.3 Linking Pharmacokinetics to Pharmacodynamics
In developing evidence-based drug dosing regimens, it should be remembered that the efficacy and safety of drugs are based on both the pharmacokinetics and pharmacodynamics of these drugs and it is possible for changes in one to (partially) counteract or enhance the change in the other. It may not be necessary to perform dedicated pediatric pharmacodynamics studies for all drugs. The pediatric decision tree from the FDA, for instance, defines when pediatric pharmacokinetic studies to develop dosing regimen that yield the same exposure in adults and children suffice (https://www.fda.gov/media/71277/download). However for a large number of drugs that do not meet the required criteria, age-appropriate target exposures are not known for children in general and, also in this case, even less is known about obese children. Regarding the obese pediatric subpopulation, priority should be given in this regard to pharmacodynamics studies on drugs for disease conditions that result from or are common with obesity, including, for instance, type 2 diabetes. Additionally, due to the low-grade systemic inflammation in obesity, pediatric pharmacodynamics studies on drugs that act on or with the immune system are imperative.
4.1.4 Generalization of Findings
Population modelling approaches are ideally suitable to study the impact of obesity in conjunction with maturation on the pharmacokinetics and pharmacodynamics of drugs. A drawback of this method is, however, that it would require dedicated studies for all relevant drugs in obese patients of all ages which would require an unrealistic amount of resources. Therefore, research efforts should also be directed toward gaining a deeper understanding of how obesity-related physiological changes in processes underlying drug pharmacokinetics and pharmacodynamics translate into changes in pharmacokinetic and pharmacodynamics parameters, both in adults and in children. Methods that combine population approaches with physiological insight would contribute to the generalizability of findings (Knibbe et al. 2011).
In pediatric patients with age-appropriate body weights, the between-drug extrapolation of covariate relationships for clearance for drugs sharing an elimination route has, for instance, been explored (Krekels et al. 2012a; De Cock et al. 2014; Calvier et al. 2018a). In this approach, pharmacological parameters are considered to reflect system-specific properties, drug-specific properties, or a combination of both. Methods that can derive and retain information on system-specific aspects of drug pharmacokinetics and use this in the translation of findings to drugs with different drug-specific properties should also be explored for both adult and pediatric obese patients.
4.1.5 Physiologically Based Pharmacokinetic Models
Another way to advance our knowledge would be a more extensive use of physiologically based pharmacokinetic (PBPK) models, where system-specific parameters truly reflect physiological or anatomical measures, in order to predict how drugs with specific physicochemical properties interact with the system (Johnson and Rostami-Hodjegan 2011). Although these models require a vast amount of data on changes in system-specific properties for specific patient subpopulations, the information that can be derived from this will be generalizable. An improved understanding of how, on top of maturational changes, physiological and anatomical parameters change with obesity will truly allow us to predict for all types of drugs how pharmacokinetic parameters will change in obese children of all ages. Although, contrary to population pharmacokinetic models, PBPK models cannot serve as a direct basis for drug dosing algorithms, information needed for the development of these algorithms can be derived from them. With appropriate PBPK models, we do not need to study every drug in every subpopulation separately anymore.
Application of PBPK approaches requires large amounts of data and specialized software or personnel. Therefore, simplified scaling methods to derive pharmacokinetic parameters for special patient populations are always sought after. Calvier et al. have illustrated how PBPK models can form the basis for the systematic evaluation of simplified scaling methods, to establish criteria required for accurate scaling with simplified methods (Calvier et al. 2017, 2018a). This concept was illustrated for pediatric populations with age-appropriate weights but could easily be extended to other populations for which PBPK parameters are known.
Another interesting application of PBPK modelling approaches is that these models can be used to assess the impact of changes in physiological or anatomical parameters in isolation. As such, it can be used for hypothesis testing, to establish which changes or combination of changes in the underlying physiology are likely to explain differences in observed primary pharmacokinetic parameters. This concept has been introduced for pediatric populations with age-appropriate weights (Krekels et al. 2012b) and could also be extended to other populations even when not all parameters are known in this population.
Finally, combinations of population pharmacokinetic modelling and PBPK modelling concepts can be used to quantify physiological or anatomical parameter from pharmacokinetic data, when direct measures of these parameters are not possible. Using such an approach has been successfully used to derive intrinsic clearance values for midazolam in the liver and gut wall in obese patients and patients after weight loss surgery (Brill et al. 2016), which allowed for the disentanglement of presystemic and systemic clearance of this drug. It has even be illustrated how optimal design principles in population modelling can be used to optimize the design of the clinical study to ascertain a priory that data obtained with a specific study design will indeed allow for precise and accurate estimation of the parameters of interest (Calvier et al. 2018b).
5 Conclusion
Although pharmacological studies in obese adults are increasingly being performed, they have not yet yielded a thorough understanding of the exact physiological changes with this disease and how these impact processes underlying drug pharmacokinetics and pharmacodynamics. This prevents the generalizability of findings. In obese pediatric patients, pharmacological studies are much more limited, but a number of these have shown that observations in adult patients cannot always be directly extrapolated to this younger population. Given the increased incidence of pediatric obesity worldwide, it is absolutely imperative that we perform more and smarter studies in this subpopulation, to increase our knowledge on the impact of this disease in combination with maturational changes on drug pharmacology, which is needed to provide safe and effective treatment for these children as well.
References
Abarca-Gómez L, Abdeen ZA, Hamid ZA, Abu-Rmeileh NM, Acosta-Cazares B, Acuin C et al (2017) Worldwide trends in body-mass index, underweight, overweight, and obesity from 1975 to 2016: a pooled analysis of 2416 population-based measurement studies in 128·9 million children, adolescents, and adults. Lancet 390(10113):2627–2642
Abernethy DR, Greenblatt DJ, Smith TW (1981) Digoxin disposition in obesity: clinical pharmacokinetic investigation. Am Heart J 102(4):740–744
Abernethy DR, Divoll M, Greenblatt DJ, Ameer B (1982) Obesity, sex, and acetaminophen disposition. Clin Pharmacol Ther 31(6):783–790
Abernethy DR, Greenblatt DJ, Divoll M, Shader RI (1983) Enhanced glucuronide conjugation of drugs in obesity: studies of lorazepam, oxazepam, and acetaminophen. J Lab Clin Med 101(6):873–880
Abernethy DR, Greenblatt DJ, Divoll M, Smith RB, Shader RI (1984) The influence of obesity on the pharmacokinetics of oral alprazolam and triazolam. Clin Pharmacokinet 9(2):177–183
Adane ED, Herald M, Koura F (2015) Pharmacokinetics of vancomycin in extremely obese patients with suspected or confirmed Staphylococcus aureus infections. Pharmacotherapy 35(2):127–139
Alexander JK, Dennis EW, Smith WG, Amad KH, Duncan WC, Austin RC (1962–1963) Blood volume, cardiac output, and distribution of systemic blood flow in extreme obesity. Cardiovasc Res Cent Bull 1:39–44
Allard S, Kinzig M, Boivin G, Sörgel F, LeBel M (1993) Intravenous ciprofloxacin disposition in obesity. Clin Pharmacol Ther 54(4):368–373
Bardin C, Nobecourt E, Larger E, Chast F, Treluyer J-M, Urien S (2012) Population pharmacokinetics of metformin in obese and non-obese patients with type 2 diabetes mellitus. Eur J Clin Pharmacol 68(6):961–968
Bartelink IH, van Kesteren C, Boelens JJ, Egberts TCG, Bierings MB, Cuvelier GDE et al (2012) Predictive performance of a busulfan pharmacokinetic model in children and young adults. Ther Drug Monit 34(5):574–583
Bauer LA, Edwards WA, Dellinger EP, Simonowitz DA (1983) Influence of weight on aminoglycoside pharmacokinetics in normal weight and morbidly obese patients. Eur J Clin Pharmacol 24(5):643–647
Bauer LA, Black DJ, Lill JS (1998) Vancomycin dosing in morbidly obese patients. Eur J Clin Pharmacol 54(8):621–625
Benedek IH, Fiske WD, Griffen WO, Bell RM, Blouin RA, McNamara PJ (1983) Serum alpha 1-acid glycoprotein and the binding of drugs in obesity. Br J Clin Pharmacol 16(6):751–754
Blouin RA, Bauer LA, Miller DD, Record KE, Griffen WO (1982) Vancomycin pharmacokinetics in normal and morbidly obese subjects. Antimicrob Agents Chemother 21(4):575–580
Blouin RA, Kolpek JH, Mann HJ (1987) Influence of obesity on drug disposition. Clin Pharm 6(9):706–714
Bowman SL, Hudson SA, Simpson G, Munro JF, Clements JA (1986) A comparison of the pharmacokinetics of propranolol in obese and normal volunteers. Br J Clin Pharmacol 21(5):529–532
Brill MJE, Diepstraten J, van Rongen A, van Kralingen S, van den Anker JN, Knibbe CAJ (2012) Impact of obesity on drug metabolism and elimination in adults and children. Clin Pharmacokinet 51(5):277–304
Brill MJE, van Rongen A, Houwink API, Burggraaf J, van Ramshorst B, Wiezer RJ et al (2014a) Midazolam pharmacokinetics in morbidly obese patients following semi-simultaneous oral and intravenous administration: a comparison with healthy volunteers. Clin Pharmacokinet 53(10):931–941
Brill MJE, Houwink API, Schmidt S, Van Dongen EPA, Hazebroek EJ, van Ramshorst B et al (2014b) Reduced subcutaneous tissue distribution of cefazolin in morbidly obese versus non-obese patients determined using clinical microdialysis. J Antimicrob Chemother 69(3):715–723
Brill MJ, van Rongen A, van Dongen EP, van Ramshorst B, Hazebroek EJ, Darwich AS et al (2015) The pharmacokinetics of the CYP3A substrate midazolam in morbidly obese patients before and one year after bariatric surgery. Pharm Res 32(12):3927–3936
Brill MJE, Välitalo PAJ, Darwich AS, van Ramshorst B, van Dongen HPA, Rostami-Hodjegan A et al (2016) Semiphysiologically based pharmacokinetic model for midazolam and CYP3A mediated metabolite 1-OH-midazolam in morbidly obese and weight loss surgery patients. CPT Pharmacometrics Syst Pharmacol 5(1):20–30
Browning B, Thormann K, Donaldson A, Halverson T, Shinkle M, Kletzel M (2011) Busulfan dosing in children with BMIs ≥ 85% undergoing HSCT: a new optimal strategy. Biol Blood Marrow Transplant 17(9):1383–1388
Brussee JM, Vet NJ, Krekels EHJ, Valkenburg AJ, Jacqz-Aigrain E, van Gerven JMA et al (2018) Predicting CYP3A-mediated midazolam metabolism in critically ill neonates, infants, children and adults with inflammation and organ failure. Br J Clin Pharmacol 84(2):358–368
Calvier EAM, Krekels EHJ, Välitalo PAJ, Rostami-Hodjegan A, Tibboel D, Danhof M et al (2017) Allometric scaling of clearance in paediatric patients: when does the magic of 0.75 fade? Clin Pharmacokinet 56(3):273–285
Calvier EAM, Krekels EHJ, Yu H, Välitalo PAJ, Johnson TN, Rostami-Hodjegan A et al (2018a) Drugs being eliminated via the same pathway will not always require similar pediatric dose adjustments. CPT Pharmacometrics Syst Pharmacol 7(3):175–185
Calvier EAM, Nguyen TT, Johnson TN, Rostami-Hodjegan A, Tibboel D, Krekels EHJ et al (2018b) Can population modelling principles be used to identify key PBPK parameters for paediatric clearance predictions? An innovative application of optimal design theory. Pharm Res 35(11):209
Caraco Y, Zylber-Katz E, Berry EM, Levy M (1995) Caffeine pharmacokinetics in obesity and following significant weight reduction. Int J Obes Relat Metab Disord 19(4):234–239
Cardoso-Júnior A, Coelho LGV, Savassi-Rocha PR, Vignolo MC, Abrantes MM, de Almeida AM et al (2007) Gastric emptying of solids and semi-solids in morbidly obese and non-obese subjects: an assessment using the 13C-octanoic acid and 13C-acetic acid breath tests. Obes Surg 17(2):236–241
Chagnac A, Herman M, Zingerman B, Erman A, Rozen-Zvi B, Hirsh J et al (2008) Obesity-induced glomerular hyperfiltration: its involvement in the pathogenesis of tubular sodium reabsorption. Nephrol Dial Transplant 23(12):3946–3952
Chairat K, Jittamala P, Hanpithakpong W, Day NPJ, White NJ, Pukrittayakamee S et al (2016) Population pharmacokinetics of oseltamivir and oseltamivir carboxylate in obese and non-obese volunteers. Br J Clin Pharmacol 81(6):1103–1112
Cheymol G, Poirier JM, Barre J, Pradalier A, Dry J (1987) Comparative pharmacokinetics of intravenous propranolol in obese and normal volunteers. J Clin Pharmacol 27(11):874–879
Cheymol G, Weissenburger J, Poirier JM, Gellee C (1995) The pharmacokinetics of dexfenfluramine in obese and non-obese subjects. Br J Clin Pharmacol 39(6):684–687
Cheymol G, Poirier JM, Carrupt PA, Testa B, Weissenburger J, Levron JC et al (1997) Pharmacokinetics of beta-adrenoceptor blockers in obese and normal volunteers. Br J Clin Pharmacol 43(6):563–570
Christoff PB, Conti DR, Naylor C, Jusko WJ (1983) Procainamide disposition in obesity. Drug Intell Clin Pharm 17(7–8):516–522
Cortínez LI, Anderson BJ, Penna A, Olivares L, Muñoz HR, Holford NHG et al (2010) Influence of obesity on propofol pharmacokinetics: derivation of a pharmacokinetic model. Br J Anaesth 105(4):448–456
Davis RL, Quenzer RW, Bozigian HP, Warner CW (1990) Pharmacokinetics of ranitidine in morbidly obese women. DICP 24(11):1040–1043
De Cock RFW, Piana C, Krekels EHJ, Danhof M, Allegaert K, Knibbe CAJ (2011) The role of population PK-PD modelling in paediatric clinical research. Eur J Clin Pharmacol 67(Suppl. 1):5–16
De Cock RFW, Allegaert K, Sherwin CMT, Nielsen EI, de Hoog M, van den Anker JN et al (2014) A neonatal amikacin covariate model can be used to predict ontogeny of other drugs eliminated through glomerular filtration in neonates. Pharm Res 31(3):754–767
de Hoogd S, Välitalo PAJ, Dahan A, van Kralingen S, Coughtrie MMW, van Dongen EPA et al (2017) Influence of morbid obesity on the pharmacokinetics of morphine, morphine-3-glucuronide, and morphine-6-glucuronide. Clin Pharmacokinet 56(12):1577–1587
Diepstraten J, Chidambaran V, Sadhasivam S, Blussé van Oud-Alblas HJ, Inge T, van Ramshorst B et al (2013) An integrated population pharmacokinetic meta-analysis of propofol in morbidly obese and nonobese adults, adolescents, and children. CPT Pharmacometrics Syst Pharmacol 2(9):e73
Dong D, Peng X, Liu J, Qian H, Li J, Wu B (2016) Morbid obesity alters both pharmacokinetics and pharmacodynamics of propofol: dosing recommendation for anesthesia induction. Drug Metab Dispos 44(10):1579–1583
Dzierlenga AL, Clarke JD, Hargraves TL, Ainslie GR, Vanderah TW, Paine MF et al (2015) Mechanistic basis of altered morphine disposition in nonalcoholic steatohepatitis. J Pharmacol Exp Ther 352(3):462–470
Emery M, Fisher JM, Chien JY, Kharasch ED, Dellinger EP, Kowdley KV et al (2003) CYP2E1 activity before and after weight loss in morbidly obese subjects with nonalcoholic fatty liver disease. Hepatology 38(2):428–435
Farrell GC, Teoh NC, Mccuskey RS (2008) Hepatic microcirculation in fatty liver disease. Anat Rec Adv Integr Anat Evol Biol 291(6):684–692
Fisher CD, Lickteig AJ, Augustine LM, Oude Elferink RPJ, Besselsen DG, Erickson RP et al (2009) Experimental non-alcoholic fatty liver disease results in decreased hepatic uptake transporter expression and function in rats. Eur J Pharmacol 613(1–3):119–127
Flechner SM, Kolbeinsson ME, Tam J, Lum B (1989) The impact of body weight on cyclosporine pharmacokinetics in renal transplant recipients. Transplantation 47(5):806–810
Gade C, Christensen HR, Dalhoff KP, Holm JC, Holst H (2018) Inconsistencies in dosage practice in children with overweight or obesity: a retrospective cohort study. Pharmacol Res Perspect 6(3):e00398
Gibbs JP, Gooley T, Corneau B, Murray G, Stewart P, Appelbaum FR et al (1999) The impact of obesity and disease on busulfan oral clearance in adults. Blood 93(12):4436–4440
Greenblatt DJ, Abernethy DR, Locniskar A, Harmatz JS, Limjuco RA, Shader RI (1984) Effect of age, gender, and obesity on midazolam kinetics. Anesthesiology 61(1):27–35
Greenblatt DJ, Friedman H, Burstein ES, Scavone JM, Blyden GT, Ochs HR et al (1987) Trazodone kinetics: effect of age, gender, and obesity. Clin Pharmacol Ther 42(2):193–200
Greenblatt DJ, Harmatz JS, Chow CR (2018a) Vortioxetine disposition in obesity. J Clin Psychopharmacol 38(3):172–179
Greenblatt DJ, Harmatz JS, Ryan MJ, Chow CR (2018b) Sustained impairment of lurasidone clearance after discontinuation of posaconazole. J Clin Psychopharmacol 38(4):289–295
Harnois F, Msika S, Sabaté J-M, Mechler C, Jouet P, Barge J et al (2006) Prevalence and predictive factors of non-alcoholic steatohepatitis (NASH) in morbidly obese patients undergoing bariatric surgery. Obes Surg 16(2):183–188
Hohmann N, Kocheise F, Carls A, Burhenne J, Haefeli WE, Mikus G (2015) Midazolam microdose to determine systemic and pre-systemic metabolic CYP3A activity in humans. Br J Clin Pharmacol 79(2):278–285
Huttunen R, Syrjänen J (2013) Obesity and the risk and outcome of infection. Int J Obes 37(3):333–340
Jain R, Chung SM, Jain L, Khurana M, Lau SWJ, Lee JE et al (2011) Implications of obesity for drug therapy: limitations and challenges. Clin Pharmacol Ther 90(1):77–89
Johnson TN, Rostami-Hodjegan A (2011) Resurgence in the use of physiologically based pharmacokinetic models in pediatric clinical pharmacology: parallel shift in incorporating the knowledge of biological elements and increased applicability to drug development and clinical practice. Paediatr Anaesth 21(3):291–301
Jusko WJ, Gardner MJ, Mangione A, Schentag JJ, Koup JR, Vance JW (1979) Factors affecting theophylline clearances: age, tobacco, marijuana, cirrhosis, congestive heart failure, obesity, oral contraceptives, benzodiazepines, barbiturates, and ethanol. J Pharm Sci 68(11):1358–1366
Kees MG, Weber S, Kees F, Horbach T (2011) Pharmacokinetics of moxifloxacin in plasma and tissue of morbidly obese patients. J Antimicrob Chemother 66(10):2330–2335
Knibbe CAJ, Krekels EHJ, Danhof M (2011) Advances in paediatric pharmacokinetics. Expert Opin Drug Metab Toxicol 7(1):1–8
Knibbe CAJ, Brill MJE, van Rongen A, Diepstraten J, van der Graaf PH, Danhof M (2015) Drug disposition in obesity: toward evidence-based dosing. Annu Rev Pharmacol Toxicol 55(1):149–167
Kolwankar D, Vuppalanchi R, Ethell B, Jones DR, Wrighton SA, Hall SD et al (2007) Association between nonalcoholic hepatic steatosis and hepatic cytochrome P-450 3A activity. Clin Gastroenterol Hepatol 5(3):388–393
Krekels EHJ, Neely M, Panoilia E, Tibboel D, Capparelli E, Danhof M et al (2012a) From pediatric covariate model to semiphysiological function for maturation: part I – extrapolation of a covariate model from morphine to zidovudine. CPT Pharmacometrics Syst Pharmacol 1(10):e9
Krekels EHJ, Johnson TN, den Hoedt SM, Rostami-Hodjegan A, Danhof M, Tibboel D et al (2012b) From pediatric covariate model to semiphysiological function for maturation: part II – sensitivity to physiological and physicochemical properties. CPT Pharmacometrics Syst Pharmacol 1:e10
Lee WH, Kramer WG, Granville GE (1981) The effect of obesity on acetaminophen pharmacokinetics in man. J Clin Pharmacol 21(7):284–287
Lemmens H, Bernstein D, Brodsky J (2006) Estimating blood volume in obese and morbidly obese patients. Obes Surg 16(6):773–776
Lloret-Linares C, Hirt D, Bardin C, Bouillot J-L, Oppert J-M, Poitou C et al (2014) Effect of a Roux-en-Y gastric bypass on the pharmacokinetics of oral morphine using a population approach. Clin Pharmacokinet 53(10):919–930
Machado M, Marques-Vidal P, Cortez-Pinto H (2006) Hepatic histology in obese patients undergoing bariatric surgery. J Hepatol 45(4):600–606
Michalaki MA, Gkotsina MI, Mamali I, Markantes GK, Faltaka A, Kalfarentzos F et al (2011) Impaired pharmacokinetics of levothyroxine in severely obese volunteers. Thyroid 21(5):477–481
Mooij MG, van Duijn E, Knibbe CAJ, Windhorst AD, Hendrikse NH, Vaes WHJ et al (2014) Pediatric microdose study of [14C]paracetamol to study drug metabolism using accelerated mass spectrometry: proof of concept. Clin Pharmacokinet 53(11):1045–1051
Morgan DJ, Bray KM (1994) Lean body mass as a predictor of drug dosage. Implications for drug therapy. Clin Pharmacokinet 26(4):292–307
Ng M, Fleming T, Robinson M, Thomson B, Graetz N, Margono C et al (2014) Global, regional, and national prevalence of overweight and obesity in children and adults during 1980-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet 384(9945):766–781
Nguyen L, Leger F, Lennon S, Puozzo C (2006) Intravenous busulfan in adults prior to haematopoietic stem cell transplantation: a population pharmacokinetic study. Cancer Chemother Pharmacol 57(2):191–198
Pai MP, Norenberg JP, Anderson T, Goade DW, Rodvold KA, Telepak RA et al (2007) Influence of morbid obesity on the single-dose pharmacokinetics of daptomycin. Antimicrob Agents Chemother 51(8):2741–2747
Park EJ, Pai MP, Dong T, Zhang J, Ko C-W, Lawrence J et al (2012) The influence of body size descriptors on the estimation of kidney function in normal weight, overweight, obese, and morbidly obese adults. Ann Pharmacother 46(3):317–328
Rainone V, Schneider L, Saulle I, Ricci C, Biasin M, Al-Daghri NM et al (2016) Upregulation of inflammasome activity and increased gut permeability are associated with obesity in children and adolescents. Int J Obes 40(6):1026–1033
Reilly JJ, El-Hamdouchi A, Diouf A, Monyeki A, Somda SA (2018) Determining the worldwide prevalence of obesity. Lancet 391(10132):1773–1774
Reiss RA, Haas CE, Karki SD, Gumbiner B, Welle SL, Carson SW (1994) Lithium pharmacokinetics in the obese. Clin Pharmacol Ther 55(4):392–398
Ribstein J, du Cailar G, Mimran A (1995) Combined renal effects of overweight and hypertension. Hypertension 26(4):610–615
Schwartz AE, Matteo RS, Ornstein E, Young WL, Myers KJ (1991) Pharmacokinetics of sufentanil in obese patients. Anesth Analg 73(6):790–793
Shibutani K, Inchiosa MA, Sawada K, Bairamian M (2004) Accuracy of pharmacokinetic models for predicting plasma fentanyl concentrations in lean and obese surgical patients: derivation of dosing weight (“pharmacokinetic mass”). Anesthesiology 101(3):603–613
Smit C, Wasmann RE, Goulooze SC, Hazebroek EJ, Van Dongen EPA, Burgers DMT et al (2019) A prospective clinical study characterizing the influence of morbid obesity on the pharmacokinetics of gentamicin: towards individualized dosing in obese patients. Clin Pharmacokinet. https://doi.org/10.1007/s40262-019-00762-4
Smit C, Wasmann RE, Wiezer MJ, van Dongen HPA, Mouton JW, Brüggemann RJM, Knibbe CAJ (2019) Tobramycin clearance is best described by renal function estimates in obese and non-obese individuals: results of a prospective rich sampling pharmacokinetic study. Pharm Res 36(8):112. https://doi.org/10.1007/s11095-019-2651-2
Sparreboom A, Wolff AC, Mathijssen RHJ, Chatelut E, Rowinsky EK, Verweij J et al (2007) Evaluation of alternate size descriptors for dose calculation of anticancer drugs in the obese. J Clin Oncol 25(30):4707–4713
Tamborlane WV, Haymond MW, Dunger D, Shankar R, Gubitosi-Klug R, Bethin K et al (2016) Expanding treatment options for youth with type 2 diabetes: current problems and proposed solutions. Diabetes Care 39(3):323–329
Tchernof A, Lévesque E, Beaulieu M, Couture P, Després JP, Hum DW et al (1999) Expression of the androgen metabolizing enzyme UGT2B15 in adipose tissue and relative expression measurement using a competitive RT-PCR method. Clin Endocrinol 50(5):637–642
Teixeira TFS, Souza NCS, Chiarello PG, Franceschini SCC, Bressan J, Ferreira CLLF et al (2012) Intestinal permeability parameters in obese patients are correlated with metabolic syndrome risk factors. Clin Nutr 31(5):735–740
Ulvestad M, Skottheim IB, Jakobsen GS, Bremer S, Molden E, Åsberg A et al (2013) Impact of OATP1B1, MDR1, and CYP3A4 expression in liver and intestine on interpatient pharmacokinetic variability of atorvastatin in obese subjects. Clin Pharmacol Ther 93(3):275–282
van Kralingen S, Diepstraten J, Peeters MYM, Deneer VHM, van Ramshorst B, Wiezer RJ et al (2011a) Population pharmacokinetics and pharmacodynamics of propofol in morbidly obese patients. Clin Pharmacokinet 50(11):739–750
van Kralingen S, van de Garde EMW, Knibbe CAJ, Diepstraten J, Wiezer MJ, van Ramshorst B et al (2011b) Comparative evaluation of atracurium dosed on ideal body weight vs. total body weight in morbidly obese patients. Br J Clin Pharmacol 71(1):34–40
van Rongen A, Välitalo PAJ, Peeters MYM, Boerma D, Huisman FW, van Ramshorst B et al (2016) Morbidly obese patients exhibit increased CYP2E1-mediated oxidation of acetaminophen. Clin Pharmacokinet 55(7):833–847
van Rongen A, van der Aa MP, Matic M, van Schaik RHN, Deneer VHM, van der Vorst MM et al (2018a) Increased metformin clearance in overweight and obese adolescents: a pharmacokinetic substudy of a randomized controlled trial. Pediatr Drugs 20(4):365–374
van Rongen A, Brill MJE, Vaughns JD, Välitalo PAJ, van Dongen EPA, van Ramshorst B et al (2018b) Higher midazolam clearance in obese adolescents compared with morbidly obese adults. Clin Pharmacokinet 57(5):601–611
Van Wart S, Phillips L, Ludwig EA, Russo R, Gajjar DA, Bello A et al (2004) Population pharmacokinetics and pharmacodynamics of garenoxacin in patients with community-acquired respiratory tract infections. Antimicrob Agents Chemother 48(12):4766–4777
Varin F, Ducharme J, Théorêt Y, Besner JG, Bevan DR, Donati F (1990) Influence of extreme obesity on the body disposition and neuromuscular blocking effect of atracurium. Clin Pharmacol Ther 48(1):18–25
Wellen KE, Hotamisligil GS (2003) Obesity-induced inflammatory changes in adipose tissue. J Clin Invest 112(12):1785–1788
Woolsey SJ, Mansell SE, Kim RB, Tirona RG, Beaton MD (2015) CYP3A activity and expression in nonalcoholic fatty liver disease. Drug Metab Dispos 43(10):1484–1490
Xing J, Chen JDZ (2004) Alterations of gastrointestinal motility in obesity. Obes Res 12(11):1723–1732
Xiong Y, Fukuda T, Knibbe CAJ, Vinks AA (2017) Drug dosing in obese children. Pediatr Clin N Am 64(6):1417–1438
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Krekels, E.H.J., Knibbe, C.A.J. (2019). Pharmacokinetics and Pharmacodynamics of Drugs in Obese Pediatric Patients: How to Map Uncharted Clinical Territories. In: Kiess, W., Schwab, M., van den Anker, J. (eds) Pediatric Pharmacotherapy . Handbook of Experimental Pharmacology, vol 261. Springer, Cham. https://doi.org/10.1007/164_2019_250
Download citation
DOI: https://doi.org/10.1007/164_2019_250
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-50493-9
Online ISBN: 978-3-030-50494-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)