
Abstract
Physiologically based biopharmaceutics modeling (PBBM) emphasizes the integration of physicochemical properties of drug substance and formulation characteristics with system physiological parameters to predict the absorption and pharmacokinetics (PK) of a drug product. PBBM has been successfully utilized in drug development from discovery to postapproval stages and covers a variety of applications. The use of PBBM facilitates drug development and can reduce the number of preclinical and clinical studies. In this review, we summarized the major applications of PBBM, which are classified into six categories: formulation selection and development, biopredictive dissolution method development, biopharmaceutics risk assessment, clinically relevant specification settings, food effect evaluation and pH-dependent drug-drug-interaction risk assessment. The current state of PBBM applications is illustrated with examples from published studies for each category of application. Despite the variety of PBBM applications, there are still many hurdles limiting the use of PBBM in drug development, that are associated with the complexity of gastrointestinal and human physiology, the knowledge gap between the in vitro and the in vivo behavior of drug products, the limitations of model interfaces, and the lack of agreed model validation criteria, among other issues. The challenges and essential considerations related to the use of PBBM are discussed in a question-based format along with the scientific thinking on future research directions. We hope this review can foster open discussions between the pharmaceutical industry and regulatory agencies and encourage collaborative research to fill the gaps, with the ultimate goal to maximize the applications of PBBM in oral drug product development.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In recent decades, physiologically based pharmacokinetic (PBPK) modeling and simulation have been recognized as a valuable tool in drug discovery, formulation development, clinical trials, and drug approvals. In addition to the wide use of PBPK modeling and simulation in support of clinical pharmacology programs (e.g., drug-drug interactions (DDIs), special populations, pediatric study, etc.), biopharmaceutics applications of PBPK to support drug product development and quality are also rapidly evolving, especially for oral drug products. In October 2020, the United States (U.S.) FDA published draft guidance titled The Use of Physiologically Based Pharmacokinetic Analyses — Biopharmaceutics Applications for Oral Drug Product Development, Manufacturing Changes, and Controls (hereafter referred to as the FDA 2020 PBPK guidance) to provide general recommendations regarding PBPK model development and evaluation for regulatory biopharmaceutics applications [1]. The publication of the FDA 2020 PBPK guidance encourages the use of PBPK in the pharmaceutical industry for drug product development and approvals.
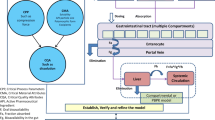
The new terminology, physiologically based biopharmaceutics modeling (PBBM), is increasingly being used in the biopharmaceutics field to replace PBPK. PBBM is defined as “physiologically based pharmacokinetic(s) absorption models including ACAT (Advanced Compartmental Absorption Transit) and ADAM (Advanced Dissolution, Absorption, and Metabolism) as well as other mechanistic models, which mimic physiological conditions and incorporate dissolution information while accounting for relevant physicochemical and physiological factors leading to a prediction of systemic exposure versus time” [2]. The term PBBM emphasizes the integration of physicochemical properties of drug substance, formulation characteristics of drug product, with system physiological parameters to predict the absorption and bioavailability (BA) of the drug product to understand the impact of formulation and manufacturing on drug bioavailability [1, 2]. PBBM is often used interchangeably with other terms, such as PBPK absorption modeling, physiologically based absorption modeling, and PBPK modeling for biopharmaceutics applications [1]. In this paper, we use PBBM in place of all other terms to avoid confusion.
PBBM provides the link between in vitro (related to product quality) and in vivo performance (BA and PK). It can be considered as a mechanistic framework to establish the in vitro-in vivo relationship (IVIVR). PBBM could also enable the risk assessment for formulation and manufacturing changes during drug development and throughout the drug life cycle. When combined with virtual bioequivalence (BE) studies, PBBM could be used to determine the BE safe space for certain quality attributes, thereby establishing clinically relevant product specifications (e.g., particle size distribution (PSD), in vitro dissolution specifications) while reducing the number of clinical trials needed for regulatory approvals and [3,4,5]. In addition, the use of PBBM for the evaluation of food effect and pH-dependent DDIs has also been actively explored [6,7,8].
The FDA 2020 PBPK guidance has outlined the general strategy for PBBM development and evaluation for biopharmaceutics applications [1]. The model structure of PBBM generally includes the mechanistic absorption model and the disposition PK model. The mechanistic absorption model is the key component for PBBM because the modeling is aimed to evaluate the impact on bioavailability due to the changes or variations in product quality attributes. Nevertheless, the model development for the disposition PK model (either full PBPK or reduced PBPK) is equally important. A good understanding of drug disposition after absorption is the prerequisite for PBBM development. General considerations on the use of PBBM were well summarized in a commentary paper, that discussed approaches to input drug product quality attributes, coupling with biorelevant dissolution data, parameter sensitivity analysis, and model validation [9]. The practice for PBBM model development has been extensively discussed [10, 11] and is not the focus of this review.
This review aims to depict the current state of PBBM by summarizing the major applications of PBBM and discussing the essential considerations for its successful use in oral drug product development. The challenges associated with the PBBM approach are also elucidated along with the scientific thinking on the future research directions to maximize the applications of PBBM in oral drug development and approval.
PBBM Applications in Oral Drug Development
PBBM applications received by U.S. FDA in New Drug Applications (NDAs) between January 2008 and December 2018 were surveyed by Wu et al. [9]. A total of 24 submissions were included in that report in which PBBM was used as an impactful tool in establishing the relationship between critical quality attributes, including formulation variables (e.g., in vitro dissolution) and the in vivo performance. It is worth noting that the regulatory submissions of PBBM generally aim to support product quality and biopharmaceutics with respect to drug approvals, which is considered as the late stage of drug development. In fact, PBBM can be used at all stages of drug development, and broad applications have been explored by generic and innovator drug companies [12]. Typical PBBM examples from published studies have been selected and presented in Table I to demonstrate a variety of PBBM applications in oral drug product development. The examples are classified into six major categories, including formulation selection and development, biopharmaceutics risk assessment, biopredictive dissolution method development, clinically relevant specifications, food effect assessment, and pH-dependent DDI evaluation. Summaries of the applications are included in the following subsections, accompanied by discussions about each one.
Formulation Selection and Development
Formulation selection and optimization are important activities in drug development. Optimizing a drug product’s bioperformance to provide best safety and efficacy profiles is the primary objective for formulation development. Particularly for immediate release (IR) oral drug products of Biopharmaceutics Classification System (BCS) class 2 and 4 drugs with low aqueous solubility, oral bioavailability improvement is often the aim of formulation optimization. Commonly used formulation strategies to enhance oral bioavailability include prodrug, particle engineering, emulsion, saltation, co-crystalline, and amorphous solid dispersion [24,25,26].
In the early drug development stage before moving to clinical studies, in vitro dissolution and preclinical PK studies are two major tools to evaluate formulation performance. However, commonly used in vitro dissolution tests are not necessarily biopredictive. PBBM is useful to streamline early formulation development by integrating both physiological conditions and physiochemical properties of drugs to project the PK performance and thereby inform the designs of preclinical and clinical studies. For example, in the study of Kaur et al., the selection of a co-crystal formulation and an optimal dose was supported by PBBM based on the prediction of drug exposure using the in vitro dissolution profile as an input [13]. Of note, a dissolution scaler constant (Kdiss) was calculated from in vitro dissolution data to account for the difference between in vitro and in vivo dissolution rate [13]. The use of PBBM for salt screening was demonstrated by Chiang and Wong, where PBBM coupled with sensitivity analysis was used to provide guidance to salt selection and to define a salt solubility requirement that was further confirmed by the animal PK studies [14].
PBPK modeling has been extensively applied to first-in-human trial design by incorporating the physiological and pharmacological differences among species [27, 28]. Combined with mechanistic absorption modeling, PBBM can help to translate preclinical data to predict clinical PK more accurately. One case study was presented in the paper of Kesisoglou et al., in which the human PK profile of Compound UCB-1 in an IR suspension was reasonably predicted by PBBM after adjusting metabolizing parameters by taking into account species differences between dogs and humans [15]. Additionally, in the Kesisoglou study, the PBBM incorporated the precipitation parameters derived from in vitro biorelevant dissolution testing, and the input particle size data was obtained from optimization using dog PK data [15]. The overall prediction accuracy of translational PBBM was evaluated by Zhang et al. by comparing the prospective prediction results with observed clinical PK data for 16 compounds across BCS classes in IR formulations [29]. In this study, PBBM in combination with various disposition parameter scaling methods were evaluated for prospective human PK projection [29]. For the disposition parameter scaling (e.g., clearance), the authors suggested that comprehensive analyses of all in vivo data from preclinical animals were useful to improve the prediction accuracy in humans [28]. The common challenges in translational PBBM were identified as nonlinearity (overdose proportionality or underdose proportionality), species disconnects, lack of in vitro-in vivo extrapolation (IVIVE), and formulation-dependent PK, among others [29].
PBBM has also been used to aid the development of extended release (ER) formulations. One of the important objectives of ER formulations was to reduce the peak-to-trough ratio relative to the IR dosage forms and thus provide improved clinical tolerability. Jones et al. presented an example in which PBBM helped to design an ER formulation of a weakly basic lipophilic compound [16]. PBBM was used to estimate the release rate range that would produce a similar area under the plasma concentration-time curve (AUC) as that from an IR formulation, and the simulations were confirmed by the nonhuman primate study and single dose studies in humans [16]. Of note, the modeling indicated that drug absorption in the distal intestinal tract was considerable, and permeability parameter and transition time setting in the caecum and colon might be critical for drug exposure prediction for ER formulations [16].
Biopharmaceutics Risk Assessment
Biopharmaceutics risk assessment is the evaluation of the bioavailability/bioequivalence (BA/BE) impact attributed to physicochemical and biopharmaceutics properties of drug substances and drug products [30]. According to the presentation by FDA in April 2021 at CDER Small Business & Industry Assistance (SBIA) 2021 Generic Drugs Forum [29] the biopharmaceutics risk assessment framework was established mainly to guide in vitro dissolution specification setting for an oral drug product from a regulatory perspective [30]. Specifically, the amount of effort needed for in vitro dissolution development should be made depending on the BA/BE risk which assessed by a question-based framework consisting of critical questions relevant to a drug substance’s solubility, permeability, in vitro dissolution performance, the availability of Critical Bioavailability Attribute(s), and the feasibility of IVIVR establishment based on the submitted in vitro/in vivo/in silico data.
Generally, biopharmaceutics risk assessment is embedded in drug product development to determine how much BA/BE risk is associated with a drug product so that the necessary in vitro and in vivo studies can be performed for product development and an appropriate control strategy can be implemented to mitigate the risk [30]. PBBM can quantitatively assess BA/BE risk by considering the properties of the drug substance (solubility, permeability, particle size, etc.) and drug product in vitro performance (e.g., dissolution) as well as drug’s disposition and human physiology to determine rate-limiting factors (e.g., dissolution or permeability) for drug absorption and PK performance. This risk assessment is often conducted by parameter sensitivity analysis or PK simulations. In the study of Chow et al., the use of PBBM to evaluate the potential impact of excipients on oral drug absorption of several compounds across BCS classes was demonstrated, with parameter sensitivity analyses being performed on solubility, permeability, enzyme, and transporter activities to examine the risk of excipients’ impact on absorption in clinical settings [31]. Another example was demonstrated by Vaidhyanathan et al. using PBBM to identify the key factors that contributed to the failure to show bioequivalence between the oral suspension and tablet formulations in the bioequivalence study for dasatinib [19].
The recent progress on biopharmaceutics risk assessment using PBBM was also discussed in the FDA public workshop Regulatory Utility of Mechanistic Modeling to Support Alternative Bioequivalence Approaches held in the fall of 2021. Multiple case studies were presented by FDA speakers, including one on the use of the model to determine the following: 1) the impact of active pharmaceutical ingredient (API) PSD on the bioequivalence of a generic oral IR product to the reference listed drug; 2) the impact of faster drug release from lower strengths (compared with the highest strength) on the bioavailability to justify the biowaiver for lower strengths; 3) the feasibility to expand BCS class 3 biowaivers without qualitative and quantitative sameness for generic products; and 4) the risk of widening dissolution specifications on a drug product’s PK performance [32, 33]. PBBM has been widely accepted as a powerful tool to examine biopharmaceutics risks and is a promising approach that may be applied to support biowaivers.
Biopredictive Dissolution Method Development
The most commonly seen PBBM application in published studies is for biopredictive dissolution method development [34]. A biopredictive dissolution test is defined as “a set of testing conditions for which in vitro dissolution profiles are capable of predicting PK profiles” [1]. By definition, a biopredictive dissolution test can be used to predict PK profiles. Classical or mechanistic in vivo-in vitro correlation (IVIVC) or IVIVR establishment is generally accepted by regulatory agencies to demonstrate that an in vitro dissolution test is biopredictive. This could be a Level A IVIVC which allows the use of the in vitro dissolution profiles to predict the whole PK profiles, or a Level C IVIVC which allows in vitro dissolution data to be used to predict the key PK parameters, such as Cmax and AUC [35]. However, the establishment of conventional IVIVC is generally recognized as very challenging [2, 36]. PBBM provides an alternative interface to establish in vitro and in vivo link and aids in biopredictive dissolution method development [2].
In general, a biopredictive dissolution test supported by PBBM is expected to accurately predict systemic exposure based on direct or indirect input of in vitro dissolution data. The accuracy of the prediction might be determined by evaluating if the in vitro test can successfully predict the similarity and dissimilarity of bioavailability between formulations. There are generally two strategies for using PBBM for biopredictive dissolution method development. One strategy is to estimate the in vivo dissolution profiles by deconvolution based on PBBM and thereafter explore appropriate in vitro test methodology to match the in vivo profiles. In the study of Zhang et al., a PBBM of carbamazepine (CBZ) was developed and used to obtain the in vivo dissolution profile based on the observed PK data by deconvolution. Using the deconvolved in vivo percent dissolved versus time profile as a reference, a biopredictive dissolution approach was developed with a method using 900 mL 0.1% sodium lauryl sulfate as the medium for CBZ ER capsules [20]. Using a similar approach, a biopredictive dissolution method was developed for an IR product of AMG-Y, a weak base compound, as demonstrated by Kesisoglou et al. [27]. The in vivo dissolution profile generated via a PBBM model from the first-in-human PK data was used as a reference to develop the clinically relevant dissolution method [15].
The other strategy for biopredictive dissolution method development is using PBBM to verify if the in vitro dissolution profile(s) can effectively predict PK performance. This approach needs to directly or indirectly input in vitro dissolution profiles to the model. Direct input involves the use of in vitro dissolution profiles or after further data process as the in vivo dissolution profiles in the model, which is a standard practice for PBBM of ER formulations. Indirect input methods are more commonly used for IR formulations, primarily relying on semi-mechanistic dissolution models, such as the Takano model (z-factor) [37], diffusion layer model [21, 38,39,40], and product-PSD [3], to predict in vivo dissolution kinetics. The key semi-mechanistic dissolution model parameters are often obtained by fitting the in vitro dissolution data, and this approach can take into account the impact of excipients on drug release while allowing the simulation to dynamically adapt the dissolution kinetics to the local conditions of the gastrointestinal (GI) tract (such as transition, lumen fluid volume, and pH). Mechanistic dissolution model comparisons were well-summarized in a published review [31].
Biorelevant dissolution tests are believed to increase the chance to be biopredictive because the test conditions are intended to mimic the environment for the in vivo dissolution. As shown in the PBBM examples presented in Table I, 7 out of 15 studies incorporated the in vitro dissolution data obtained from biorelevant media using one- or two-stage dissolution methods. These methods include the use of biorelevant media and/or the apparatus to simulate the dynamic luminal conditions of the GI tract related to drug absorption, such as the mini-Gastrointestinal Simulator (GIS) system [41], artificial stomach-duodenum (ASD) model [42], BioGIT model [43], transfer model [44,45,46], and TNO gastro-intestinal model (TIM) [47]. PBBM provides an efficient tool to evaluate the biopredictive performance of biorelevant dissolution methods as demonstrated by Klumpp and Dressman for a weak base compound, dipyridamole, and a weak acid compound, glibenclamide [21]. Overall, PBBM offers more opportunities for IVIVC and IVIVR establishment with these biorelevant test methodologies, when added to mechanistic understanding of drug in vivo absorption to guide biopredictive dissolution method development.
Clinically Relevant Specifications
Clinically relevant specifications take into consideration the clinical impact of variations in the critical quality attributes and process parameters assuring a consistent safety and efficacy profile [48]. Recently, the term safe space has been used to define the boundaries of in vitro specifications, within which drug product variants are anticipated to be BE to one another [1]. The establishment of safe space offers a mechanistic understanding of the impact of in vitro testing on in vivo performance, and it may help to reduce unnecessary in vivo studies and accelerate drug approvals [3, 49].
PBBM establishes the relationship between in vitro characterizations and in vivo performance, and this approach for clinically relevant specifications was endorsed by FDA in the recently released guidance [1]. Heimbach et al. has compiled five case studies using PBBM to establish a BE safe space for BCS class 2 and 4 compounds with an IR formulation [5]. The BE safe space was developed by changing the dissolution rate parameters, such as z-factor or Weibull function parameters, to explore the failure edge of the dissolution test (or the range of dissolution rate) when the PK metrics (AUC and Cmax) would go beyond the BE acceptance margin.
Exploration of PSD specification is a hot area for BE safe space establishment. A good example was presented by Pepin et al., with a PBBM being developed for a weak acid compound, lesinurad, to establish a BE safe space with respect to API particle size [3]. Of note, the theoretical PSD parameters derived from the in vitro dissolution data were input to the PBBM to replace the in vitro particle size measurement in order to describe the overall formulation effect on in vivo dissolution. In addition, the developed PBBM integrated the lag time, gastric transit time, and effective permeability fitted and optimized for individual subjects, as well as the water content in the small intestine and colon optimized from 40% and 10% to 7.5% and 2%, respectively.
The BE safe space is also useful to justify the clinical relevance of other product specifications, e.g., drug polymorphic purity. Such an example was demonstrated in the case study of canagliflozin which was included as a case study in the meeting report for ‘Developing Clinically Relevant Dissolution Specifications for Oral Drug Products—Industrial and Regulatory Perspectives’ convened by the APS Biopharmaceutics and Regulatory Sciences focus groups [50]. Specifically, the impact on bioavailability of a new form of drug substance in an IR formulation was evaluated by the PBBM using in vitro dissolution as the surrogate (via z-factor). The prediction showed that the new form of crystalline did not impact the bioavailability of the drug product compared to the reference form, and this result was confirm later by the clinical studies conducted using formulations containing 10%, 50%, and 100% of the new crystal form.
Food Effect Assessment
The food effect on a drug product’s bioavailability incorporates many mechanisms, including, but not limited to, direct interaction between meal components and drug compounds (e.g., chelation), and indirect interactions, such as change in GI tract physiology (e.g., prolonged gastric emptying time, changes in the pH and bile salt concentrations of lumen fluids, increased secretion of gastric and intestinal fluids, and increased splanchnic blood flow), inhibition on intestinal drug transporters or metabolizing enzymes, and increase on biliary excretion [7, 51]. FDA recommends assessing the food effect of oral drug products from the early stages of drug development to aid in further clinical study designs [52]. In vitro characterizations and animal studies are often used to estimate the food effect prior to conducting a clinical trial, such as evaluation of physicochemical properties (solubility, dose number, LogD, etc.) [53], prediction based on BCS classification [54, 55], in vitro dissolution in biorelevant media (e.g., FeSSGF and FaSSIF), dissolution-permeation systems for micelle entrapment evaluation, Caco-2 cell model for permeability evaluation at fasted/fed status, and assessment in preclinical models (mouse, rat, dog or monkey) [56, 57]. However, due to the complexity of the food-drug interactions in humans and the lack of translation from in vitro and/or preclinical to humans, the overall prediction accuracy requires improvement. PBBM has advantages over in vitro approaches, as it can provide a quantitative prediction of the food effect by integrating the changes in physiology, drug product-related properties, and elimination parameters due to food intake.
The majority of published food effect assessments by PBBM are for IR products. We selected two examples of PBBM applications (presented in Table I) for prospective food effect predictions. In one example, PBBM with biorelevant solubility input was able to correctly predict no considerable food effect for the nanosized form of aprepitant, while describing the positive food effect of its micronized form where food increases the drug exposure [22]. In the other example, PBBM has successfully predicted the lack of food effect for etoricoxib, in which the model was adjusted for the gastric emptying time and absorption scale factor in the duodenum and jejunum to reflect the food effect [23].
Li et al. summarized the published case studies as well as FDA NDAs up to April 30, 2016, that used PBPK models to evaluate food effect, with the objective to assess the overall predictive performance of the modeling approach [6]. Kesisoglou compiled the published studies of PBBM to assess food effect with clinical data and proposed a workflow of PBBM to streamline food effect assessment during clinical development for different BCS classes [7]. Both reports noted that the published PBBM examples for food effect assessment focused on the impact of BA associated with the changes in the GI environment (primarily changes in bile salt concentrations for luminal solubilization) and gastric emptying time with standard high-calorie/high-fat food intake. When the food effect involves complexity such as transporter or intestinal metabolizing enzyme interactions, drug precipitation, and interactions with excipient, the use of PBBM for food effect is very limited and confidence in the prediction is low [7]. The use of PBBM for food effect assessment of ER formulations is generally more challenging due to the lack of understanding of in vivo dissolution of ER formulations and the food interaction with rate-controlling excipients [7].
Recently, scientists from the International Consortium for Innovation and Quality in Pharmaceutical Development (IQ Consortium) food effect PBPK modeling working group used a holistic approach to investigate the performance of PBBM for food effect prediction of IR products by generating de novo mechanistic absorption models for 30 compounds (across BCS classes) using physicochemical and in vitro data obtained in accordance with a predefined methodology and an aligned decision tree for workflow [23]. This analysis focused on the absorption-related mechanisms of food effect; therefore, compounds lacking clinical intravenous PK data or population PK-based data, as well as compounds with high hepatic extraction, were excluded [23]. With standard practice for PBBM model development, 24 of the models could correctly capture the likelihood of food effect without any optimization, and 23 of them could predict the food impact of PK within 2 fold differences compared with the observed data [23]. High confidence in PBBM prediction of food effects was typically observed for compounds in which the mechanism of food effect was related to physiology, including composition and volume changes in the GI luminal fluids, GI motility, pH, ion pairing, and bile salts. Furthermore, for the food effect predictions with low confidence (i.e., prediction errors PK parameters more than 2 fold), the root causes of misprediction were analyzed and the failure to capture food effect was attributed to inaccurate extrapolation from in vitro to in vivo solubility (including simulation of salt solubility and common ion effects), hydrodynamics under fed conditions (including the viscosity of GI fluids), and micelle–drug interactions [58]. In a follow-up paper from the same working group of IQ Consortium, it recommended that model improvement should focus on the PBBM model functionality, especially better reflecting fasted- and fed-state gastric solubility, gastric re-acidification, and complex mechanisms related to gastric emptying of drugs, as well as the capability to accurately capture common ion effect and drug precipitation [59].
The regulatory use of PBBM in lieu of human food effect studies is limited. FDA guidance recommends that the in vivo food effect evaluation should be conducted with the to-be-marketed productto inform final product labeling [52]. Nevertheless, we can see PBBM for food effect assessment is evolving, and experiences with it can guide clinical development with respect to food effect assessment in support of formulation and dose selection as well as dosing instruction. In the future, PBBM development for food effect assessment might focus on more dedicated in vitro and in vivo testing to provide understanding on the drive of food effect to be incorporated by PBBM. With more mechanistic understanding in food-drug interactions and more experiences in the PBBM for food effect prediction, confidence in using PBBM for this application is expected to increase.
pH-Dependent DDI Evaluation
Acid-reducing agents (ARAs), including proton-pump inhibitors (PPIs), H2-receptor antagonists, antacids, and histamine, are commonly used for gastric protection by inhibiting gastric acid excretion [60, 61]. As a consequence, ARAs can result in elevated gastric pH [62, 63] and may impact coadministered drugs’ bioavailability (especially for weak acid and weak base compounds) [64,65,66,67]. The FDA draft guidance Evaluation of Gastric pH-Dependent Drug Interactions With Acid-Reducing Agents: Study Design, Data Analysis, and Clinical Implications acknowledges the use of PBPK simulation as an alternative approach for evaluating pH-dependent DDI [67]. Although there are only limited regulatory experiences with the PBBM approach to assess pH-dependent DDI risk or support labeling recommendations, the potential of PBBM for pH-dependent DDI has been explored actively in the pharmaceutical society at different stages of drug development [68,69,70].
The case studies of pH-dependent DDI using PBBM are well summarized and discussed in Mitra et al. and Dong et al., representing the perspectives of industry and regulators, respectively [8, 71]. In Mitra et al., seven case studies from five pharmaceutical companies were presented demonstrating cross-industry experiences in PBBM prediction of pH-dependent DDIs for weakly basic drugs, and a pragmatic PBBM workflow was proposed to inform clinical development and regulatory decisions in pH-dependent DDI risk assessment [8]. Similarly, in Dong et al., the performance of PBBM to predict the lack of pH-dependent DDI was demonstrated by case studies of weakly basic drug products, while it was suggested that the prediction performance on the presence of pH-dependent DDI and risk potential be confirmed by more studies [71].
In general, the PBBM approach is advantageous over solubility and dissolution framework in predicting pH-dependent DDI, considering the quantitative prediction of PK impact by PBBM. Such assessments are generally performed by elevating gastric pH and/or incorporating dissolution/solubility data under an ARA-induced condition for PBBM simulation to evaluate the impact on absorption of investigated drugs with respect to PK parameters (Cmax and AUC). The PBBM approach is also promising to take into account the effect of precipitation for pH-dependent DDI assessment, while the precipitation parameter setting needs to be further verified. In addition to the adjustment of gastric pH, PBBM can account for additional mechanisms of DDI, such as the impact on the metabolizing enzymes and transporters by ARAs. It was demonstrated in Merdy et al. that the exposure change of nifedipine (a CYP3A substrate) and its major metabolites with coadministration of omeprazole (a PPI drug and CYP3A inhibitor) was successfully described using PBBM by incorporating the impact of omeprazole on both the gastric pH and the CYP3A4 activity [68].
Challenges in PBBM Development, Validation, and Applications
Despite the variety of PBBM applications, there are many challenges faced by the pharmaceutical industry and regulatory agencies that limit the use of PBBM. The major challenges are associated with the incomplete understanding on the complexity of GI and human physiology, the knowledge gap between the in vitro and the in vivo behavior of drug products, the limitations of model interfaces, and the lack of agreed-upon model validation criteria. In this section, these challenges are further discussed in a question-and-answer format, with the intention of promoting additional effort to improve PBBM platforms and increase the utilization of PBBM for oral drug product development.
Question 1: Which Approaches Should Be Taken to Effectively Incorporate Formulation and Manufacturing Process Properties to the PBBM Interfaces?
The formulation of an oral drug product and manufacturing process parameters, in addition to physicochemical properties of the drug substance, could significantly influence a drug’s bioavailability. The commonly used PBBM platforms provide limited options for oral drug product quality parameter incorporation and hardly take into consideration either the effects of excipients or the manufacturing process on drug in vivo dissolution and absorption. The incorporation of quality parameters can be categorized as directly (e.g., PSD, solubility, in vitro dissolution), or indirectly (e.g., a surrogate parameter to represent the quality attribute). In the commonly seen mechanistic absorption models, solubility, particle size of drug substance, and dissolution are usually the parameters to incorporate directly, while other product quality attributes (formulation or process) need to be linked to the abovementioned parameters to represent the to-be-addressed quality issue(s).
In vitro dissolution is often used as a surrogate for assessing formulation and process effects on an oral drug’s bioavailability. However, the biopredictive capability of an in vitro dissolution is often difficult to demonstrate, even though the in vitro dissolution test might be able to discriminate the formulation and process variants. The use of in vitro dissolution profiles as direct inputs (or after further processing, e.g., modeled by Weibull function or Hill function) assumes that the in vitro dissolution represents the in vivo dissolution both in terms of extent and time-course dissolution process. This assumption is challenging to verify as there is a very limited understanding of in vivo drug dissolution, due to the technical difficulties in the direct GI sampling in humans and the lack of understanding of the GI tract hydrodynamics [72]. It is worth noting that recent advances in intubation techniques have enabled direct luminal sampling in the GI tract in humans to determine local concentrations of dissolved and undissolved drug for a couple of drug products [73,74,75]. Due to the high cost and advanced technique requirements for these type of studies, limited research has been conducted. In addition, it is challenging to translate the luminal concentration data to in vivo drug dissolution kinetics due to high variability of the concentration data and the interplay of GI motility and hydrodynamics. Moreover, the use of in vitro dissolution profiles as direct input would mask the influence from the GI tract (e.g., transit time, the impact of luminal fluid composition, etc.). The prediction of in vivo dissolution by in vitro biorelevant dissolution methodologies is also challenging due to the limitations of the available techniques to simulate time-course in vivo dissolution in the GI tract, which involves the complexity of GI tract physiology. Especially for ER dosage forms, the confidence with bioavailability predictions from a PBBM is considered low in general, and extensive model validation with PK data from different release rates would need to demonstrate the model’s robustness.
For IR dosage forms, the interaction of GI tract physiology with the drug’s dissolution is generally simulated on top of many key assumptions associated with GI tract transition and hydrodynamics, in vivo drug disintegration and dissolution, precipitation, as well as drug’s permeation [76, 77]. To simulate the in vivo dissolution, one approach is to fit the in vitro dissolution data to generate a z-factor [37] to derive in vivo dissolution rate constants at different pH and predict in vivo dissolution along with the GI tract. Another approach is to simulate in vivo dissolution based on predefined dissolution theories (mechanistic or empirical). The commonly used mechanistic dissolution models are generally based on the first principle dissolution theories, such as the Noyes-Whitney model [78], Nernst-Brunner model [79, 80], and Wang-Flanagan model [40, 81]. Of note, these dissolution theories commonly need the input of drug physicochemical properties, such as diffusion coefficient, diffusion layer thickness, solubility, drug particle radius and density of the drug particles, to calculate dissolution rate.
Although API PSD is used to predict drug in vivo dissolution in the mechanistic dissolution models, it has been questioned whether the API particle size measurement could reasonably represent the size of disintegrated drug particles in the GI tract [82]. It is scientifically reasonable to question the reliability of using API particle size in vitro measurement to directly predict in vivo dissolution, because this approach likely ignores the effect of formulation on drug dissolution. Hence, the predicted in vivo dissolution rate based on API particle size measurement might not be realistic. However, it is also often evident for many drug products that the API particle size is correlated with drug bioavailability, especially for low solubility drug substances [83]. Considering that the in vivo dissolution could not be easily measured, the use of API particle size combined with mechanistic dissolution theories could be a good starting point to predict in vivo dissolution. Ideally, the validity of a selected mechanistic dissolution model needs further evaluation by clinical PK data from the same formulations with different API particle sizes. When such data is not available, in vitro dissolution data with drug products manufactured with API lots with different PSD could be supportive to validate the mechanistic dissolution model.
In the research by Pepin et al., in vitro dissolution data was fit to a product-PSD that would match observed in vitro dissolution factoring in the volume, solubility, and doses used in the in vitro experiments. Subsequently, this “theoretical” PSD data was input to the PBBM to replace the in vitro particle size measurement [3]. This innovative approach has been effectively used to predict the PK of lesinurad tablets. The essence of this approach is to use in vitro dissolution to verify the dissolution theory and select appropriate surrogate parameters (such as particle size input) to represent the overall formulation effect for in vivo dissolution prediction. This approach provides an opportunity to relate input parameters to characterize the overall drug product performance rather than only focusing on drug substance properties. Such innovative approaches should be greatly encouraged, while justification for a selected surrogate and the verification for the assumptions (e.g., the selected dissolution theory) would be needed for model development in aregulatory-related PBBM application along with the validation of the PBBM against clinical data with different bioavailability.
For drug products containing weak base drug substances and any supersaturating system in the formulation, the incorporation of precipitation kinetics in the absorption model is another challenge. The risk of drug precipitation and how it can impact a drug’s bioavailability are difficult to evaluate by in vitro studies. Although there are some biorelevant dissolution tests targeting for such evaluation, the translation of in vitro data to in vivo precipitation parameter setting (such as precipitation time, redissolving) is challenging. In addition, formulation and process could greatly affect the potential of drug precipitation, and it is challenging to determine how to incorporate these factors in the model to accurately reflect in vivo drug precipitation risk and kinetics. In recent reach from OriBiTo, the mechanistic modeling of precipitation was explored and used to predict human systemic and intraluminal concentrations of the lipophilic weak base, posaconazole, in two different suspension formulations [43]. This in silico modeling of suppuration and precipitation used an empirical approach based upon in vitro experiments to extend the mechanistic understanding. To resolve uncertainty related to supersaturation and precipitation, more research is highly recommended for PBBM to elucidate the gap between in vitro and in vivo in this regard.
Question 2: How Much Clinical and In Vitro Data are Needed to Develop and Validate a PBBM?
The goal of PBBM development is to establish a mechanistically sound model structure to reasonably describe the in vivo process of a drug product. The model structure and parameters are generally set to reflect the understanding of drug in vivo dissolution, absorption, disposition, and elimination. Therefore, certain in vitro and in vivo data for model setting are generally necessary, such as solubility, permeability, dissolution, absorption, distribution, metabolism, and elimination. PBBM can be developed by bottom-up, top-down, or middle-out approaches [84]. The amount of data needed for model development primarily depends on the intended purpose. For instance, in the early drug development stage, a bottom-up approach might be taken with very limited in vitro and in vivo data. Such a model might aid in drug discovery, formulation selection, or nonclinical or clinical study design. As the model at that stage is generally for information purposes, the accuracy of the model prediction is not as risky. During the late stage of the clinical pharmacology program, the objectives of a PBBM might be to support a formulation bridge, dose selection and dosing instructions for the clinical studies, and drug product quality specifications. These applications would likely involve communications with regulatory agencies. The quantity and type of data needed for model development and validation may vary depending on the clinical risk of to-be-addressed issues.
It is possible that there is insufficient information to support parameter settings or there are uncertainties with the parameters that are extrapolated from in vitro, in silico, or nonclinical data. In these cases, model validation might help in verifying the assumptions. Planning the PBBM development in the early stages and trying to optimize and validate the model throughout the clinical pharmacology program would increase the chances of a successful application of PBBM in the regulatory context. To fulfill PBBM development and validation, establishing a solid mechanistic understanding on the drug release, absorption, and disposition is important. A thoughtful plan combining the in vitro and in vivo studies could possibly reduce the need of additional dedicated BA/BE studies for PBBM validation. For example, if drug precipitation has been incorporated in the model, a thorough in vitro investigation of the drug precipitation behavior at different doses and under different conditions taking into consideration the GI environment for drug dissolution may be needed. In addition, PK data from various doses (e.g., single dose ascending studies) might help justify the parameter setting of drug precipitation. .
As discussed in Question 1, mechanistic absorption models are based on many assumptions that are technically difficult to verify by the in vivo studies. These assumptions include the following: the in vitro dissolution represents the in vivo dissolution; the in vivo dissolution kinetics agree with the dissolution theories derived from in vitro; the permeability predicted by in silico model based on in vitro measurement, among others. As there is still a knowledge gap between the in vitro dissolution and in vivo dissolution, the uncertainty of the model parameters or assumptions should be considered, especially for high impact uses of the model (related to clinical risk, such as safety and efficacy).
The PK data from similar formulations, different doses, or multiple doses are often used for model validation. While these data might help in verifying the distribution and elimination parameter setting, it is not possible to validate the absorption model if the model inputs for these dosing scenarios are same or similar. There has been ongoing debate over whether the PK data from formulations with different bioavailability are needed for model validation. From a pharmaceutical industry perspective, the feasibility to plan these BA/BE studies has raised ethical and cost-related questions. Occasionally, PK data from different formulations might be available; however, such studies aim to select the best performance formulation for subsequent efficacy and safety studies and are usually conducted on a small scale. Once the formulation has been selected, formulation optimization — if necessary — is not intended to change the bioavailability of the drug product in general. In addition, a small-scale PK study for relative bioavailability comparison of multiple formulations might not ideally provide data for model validation. In case PK exhibits high variability, it might only see a trend of bioavailability change, while a statistical bioavailability difference is often not possible to conclude due to insufficient statistical power.
To help solve this dilemma, collaboration among the pharmaceutical industry, regulatory agencies, and academia is needed to build up confidence in the translation of in vitro to in vivo (especially regarding dissolution). Collaborative efforts are desirable to evaluate the validity of the core assumptions based on available experiences. Encouraging additional dedicated studies would increase the understanding of GI tract physiology and drug in vivo dissolution kinetics for some typical formulations (e.g., IR formulations with poorly soluble API and ER formulations). In addition, a more risk-based framework for model validation can be considered for PBBM applications. The risk of model applications might take into consideration pharmacodynamics and clinical response rather than only considering PK parameters (e.g., to meet BE criteria) [85]. The link of PBBM with pharmacodynamics (PD) and exposure-response relationship might possibly provide more flexibility in PBBM validation.
Question 3: Which Criteria Should Be Used to Validate a PBBM?
A lack of agreed-upon validation criteria is one of the major challenges for promoting PBBM applications. It is generally agreed that model validation criteria should depend on the intended purpose of the modeling and the clinical impact of such an application. Unfortunately, there is no regulatory guidance that discusses or provides recommended acceptance criteria for PBBM or PBPK model validation for any type of application. There were extensive discussions on the model validation acceptance criteria for PBBM applications [6, 11, 86]. The BE 80–125% range was a widely adopted criterion for evaluating predictive performance of PBBMs in the regulatory context [11]. However, it should be noted that the BE limit of 80–125% is based on the statistical analysis of average BE for the ratio of the population geometric means of the measures for the test and reference product [87]. This BE limit as validation criteria might be applicable only when a virtual trial is simulated and compared to the observed PK data.
The IVIVC guidance-recommended model validation criteria are also often adopted for PBBM validation, and a maximum difference of 10–20% in the predicted Cmax and AUC from the PBBM compared with the average observation in the PK studies was considered as a validation criterion [1, 5, 35]. It is worth noting that meeting the prediction error within 10–20% is not easy in general as independent PK data (other than those used for model development) is recommended for validation. For prediction error calculation, PBBM usually simulates a representative subject’s PK profile, and this is compared to the average observed PK profile. For the PK profile averaged from individual subjects of selected PK data for validation, the Cmax from the average PK profile might deviate from the Cmax of the studied population, as the Tmax for individual subjects varies. As a result, the average PK profile might provide an underestimated Cmax compared with the average of all subjects’ Cmax. In addition, the Cmax could exhibit high variability and easily be affected by dosing conditions. How to take into consideration data variability in model validation is a critical question to be addressed in the future.
In a recently published FDA white paper, a risk-informed credibility assessment framework to PBPK modeling and simulation has been proposed and the implementation tor PBPK applications in clinical pharmacology was discussed in a regulatory setting.. Specifically, the model risk levels determined by model influence and decision consequence were suggested to be used to select verification and validation activities and define outcomes that will provide evidence to demonstrate model credibility for a context of use [10]. This approach should also be applicable for PBBM applications. The details of model risk assessment regarding each PBBM application and recommended corresponding model validation activity and criteria should be further discussed among all stakeholders. Guidance from regulatory agencies is highly encouraged.
Question 4: How Do Stakeholders Leverage PBPK Model Development for Clinical Pharmacology with PBBM Development for Biopharmaceutics Applications?
PBPK has been extensively utilized in regulatory submissions in support of clinical pharmacology programs, and the applications cover a wide array of topics. Most of these applications are to support DDI risk assessment, followed by applications in pediatrics dose selection, and hepatic impairment and renal impairment evaluation [70, 88]. The PBPK predictions are also combined with PK/PD and/or exposure- response (E-R) relationship for risk assessment in clinical pharmacology program.
The model development for clinical pharmacology (PBPK) and biopharmaceutics (PBBM) have commonalities, with both emphasizing model plausibility regarding physiology and drug absorption, distribution, metabolism, and excretion [58]. For PBPK models to support clinical pharmacology, the drug’s disposition parameters (metabolism and elimination) are often extensively verified by clinical PK data, such as single and multiple dose PK studies, PK data with different dosing regimens, available clinical DDI studies, PK data in special populations (e.g., with organ impairment, poor or extensive metabolizers), etc. On the other hand, PBBM focuses more on the mechanistic absorption model development to provide an estimation on a drug’s bioavailability based on physicochemical properties, in vitro performance, and quality attributes and is validated by BA/BE studies. It will benefit both clinical pharmacology and biopharmaceutics applications if the knowledge base can be efficiently shared for the activities in PBPK or PBBM model development.
Regulatory agencies might also leverage the knowledge and lessons learned from model development and validation in the clinical pharmacology and biopharmaceutics fields. Discussions between disciplines and collaborative efforts provide an efficient path to improve the general PBPK and PBBM interfaces for future advances. Although model development and validation are fit for purposes, the best practice for model development and validation should be similar, such as the principles in model risk assessment and predictive performance evaluation. Drafting guidance to provide best practices for PBPK model development and validation might be initiated by both parties to encourage continuous discussions between these two major application categories.
For the applications of PBBM, the consideration on PK/PD and E-R relationship (often analyzed in the clinical pharmacology program) would potentially provide more flexibility in drug product quality standards. A published study by Parasio et al. demonstrated the use of f PBPK/PD to establish clinically relevant dissolution specifications for an immediate release tablet product of zolpidem, a non-benzodiazepine hypnotic agent [89]. Such use of PBBM/PD was also shown in the study by Cristofoletti and Dressman where the standard bioequivalence criteria of generic ibuprofen IR products were evaluated by taking into account PD endpoints: antipyresis and dental pain relief [85]. PBBM coupled with PD may further establish a truly clinically driven “safe space” and patient-focused specifications.
Conclusions and Future Research Opportunities
PBBM is a promising tool to guide oral drug development throughout the product life cycle. Specifically, PBBM has been successfully utilized to aid in formulation selection and development, biopredictive dissolution method development, biopharmaceutics risk assessment, clinically relevant specification settings, food effect evaluation, and pH-dependent DDI risk assessment. While PBBM is strongly endorsed by health authorities [1, 90], the associated challenges are also evident, especially in model development to incorporate formulation and manufacturing process properties (as model input), as well as model validation. The agreed-upon practice for PBBM model development and validation is lacking. It appears as if considerable experience with PBBM has been obtained by the pharmaceutical industry and regulatory agencies. It might be a good time to develop best practices for PBBM model development and validation to promote PBBM applications.
It is also important to understand the hurdles in PBBM so that stakeholders can work together to fill the gaps, with the goal of increasing confidence in the use of PBBM. These gaps include, but are not limited to, the following: the knowledge gap in the translation of in vitro dissolution to in vivo dissolution, the understanding of the in vivo drug dissolution kinetics in the GI tract, and the determination of model risk for each typical PBBM application to guide model development and validation activities. We believe open discussions between the pharmaceutical industry and regulatory agencies and initiation of collaborative research to fill the gaps would pave the way for routine use of PBBM in drug product development.
References
Food and Drug Admistration. The Use of Physiologically Based Pharmacokinetic Analyses — Biopharmaceutics Applications for Oral Drug Product Development, Manufacturing Changes, and Controls https://www.fda.gov/media/142500/download. 2020.
Heimbach T, Suarez-Sharp S, Kakhi M, Holmstock N, Olivares-Morales A, Pepin X. Dissolution and translational modeling strategies toward establishing an in vitro-in vivo link—a workshop summary report. AAPS J. 2019;21(2):29. https://doi.org/10.1208/s12248-019-0298-x.
Pepin XJ, Flanagan TR, Holt DJ, Eidelman A, Treacy D, Rowlings CE. Justification of drug product dissolution rate and drug substance particle size specifications based on absorption PBPK modeling for Lesinurad immediate release tablets. Mol Pharm. 2016;13(9):3256–69. https://doi.org/10.1021/acs.molpharmaceut.6b00497.
Laisney M, Heimbach T, Mueller-Zsigmondy M, Blumenstein L, Costa R, Ji Y. Physiologically based biopharmaceutics modeling to demonstrate virtual bioequivalence and bioequivalence safe-space for Ribociclib which has permeation rate-controlled absorption. J Pharm Sci 2022;111(1):274–284. doi: https://doi.org/10.1016/j.xphs.2021.10.017.
Heimbach T, Kesisoglou F, Novakovic J, Tistaert C, Mueller-Zsigmondy M, Kollipara S, et al. Establishing the bioequivalence safe space for immediate-release oral dosage forms using physiologically based biopharmaceutics modeling (PBBM): case studies. J Pharm Sci. 2021;110(12):3896–906. https://doi.org/10.1016/j.xphs.2021.09.017.
Li M, Zhao P, Pan Y, Wagner C. Predictive performance of physiologically based pharmacokinetic models for the effect of food on oral drug absorption: current status. CPT Pharmacometrics Syst Pharmacol. 2018;7(2):82–9. https://doi.org/10.1002/psp4.12260.
Kesisoglou F. Can PBPK modeling streamline food effect assessments? J Clin Pharmacol. 2020;60(S1):S98–S104. https://doi.org/10.1002/jcph.1678.
Mitra A, Parrott N, Miller N, Lloyd R, Tistaert C, Heimbach T, et al. Prediction of pH-dependent drug-drug interactions for basic drugs using physiologically based biopharmaceutics modeling: industry case studies. J Pharm Sci. 2020;109(3):1380–94. https://doi.org/10.1016/j.xphs.2019.11.017.
Wu F, Shah H, Li M, Duan P, Zhao P, Suarez S, et al. Biopharmaceutics applications of physiologically based pharmacokinetic absorption modeling and simulation in regulatory submissions to the US food and drug administration for new drugs. AAPS J. 2021;23(2):1–14. https://doi.org/10.1208/s12248-021-00564-2.
Kuemmel C, Yang Y, Zhang X, Florian J, Zhu H, Tegenge M, et al. Consideration of a credibility assessment framework in model-informed drug development: potential application to physiologically-based pharmacokinetic modeling and simulation. CPT Pharmacometrics Syst Pharmacol. 2020;9(1):21–8. https://doi.org/10.1002/psp4.12479.
Parrott N, Suarez-Sharp S, Kesisoglou F, Pathak SM, Good D, Wagner C, et al. Best practices in the development and validation of physiologically based biopharmaceutics modeling. A workshop summary report. J Pharm Sci. 2021;110(2):584–93. https://doi.org/10.1016/j.xphs.2020.09.058.
Yuvaneshwari K, Kollipara S, Ahmed T, Chachad S. Applications of PBPK/PBBM modeling in generic product development: an industry perspective. J Drug Deliv Sci Technol. 2022;69:103152. https://doi.org/10.1016/j.jddst.2022.103152.
Kaur M, Yardley V, Wang K, Masania J, Arroo RRJ, Turner DB, et al. Artemisinin cocrystals for bioavailability enhancement. Part 2: in vivo bioavailability and physiologically based pharmacokinetic modeling. Mol. Pharmaceutics. 2021;18(12):4272–89. https://doi.org/10.1021/acs.molpharmaceut.1c00385.
Chiang P-C, Wong H. Incorporation of physiologically based pharmacokinetic modeling in the evaluation of solubility requirements for the salt selection process: a case study using phenytoin. AAPS J. 2013;15(4):1109–18. https://doi.org/10.1208/s12248-013-9519-x.
Kesisoglou F, Chung J, van Asperen J, Heimbach T. Physiologically based absorption modeling to impact biopharmaceutics and formulation strategies in drug development—industry case studies. J Pharm Sci. 2016;105(9):2723–34. https://doi.org/10.1016/j.xphs.2015.11.034.
Jones HM, Chen Y, Gibson C, Heimbach T, Parrott N, Peters SA, et al. Physiologically based pharmacokinetic modeling in drug discovery and development: a pharmaceutical industry perspective. Clin Pharmacol Ther. 2015;97(3):247–62. https://doi.org/10.1002/cpt.37.
Kesisoglou F, Balakrishnan A, Manser K. Utility of PBPK absorption modeling to guide modified release formulation development of gaboxadol, a highly soluble compound with region-dependent absorption. J Pharm Sci. 2016;105(2):722–8. https://doi.org/10.1002/jps.24674.
Kesisoglou F, Xia B, Agrawal NG. Comparison of deconvolution-based and absorption modeling IVIVC for extended release formulations of a BCS III drug development candidate. AAPS J. 2015;17(6):1492–500. https://doi.org/10.1208/s12248-015-9816-7.
Vaidhyanathan S, Wang X, Crison J, Varia S, Gao JZH, Saxena A, et al. Bioequivalence comparison of pediatric Dasatinib formulations and elucidation of absorption mechanisms through integrated PBPK modeling. J Pharm Sci. 2019;108(1):741–9. https://doi.org/10.1016/j.xphs.2018.11.005.
Zhang X, Lionberger RA, Davit BM, Yu LX. Utility of physiologically based absorption modeling in implementing quality by design in drug development. AAPS J. 2011;13(1):59–71. https://doi.org/10.1208/s12248-010-9250-9.
Klumpp L, Dressman J. Physiologically based pharmacokinetic model outputs depend on dissolution data and their input: case examples glibenclamide and dipyridamole. Eur J Pharm Biopharm. 2020;151:105380. https://doi.org/10.1016/j.ejps.2020.105380.
Pepin XJH, Huckle JE, Alluri RV, Basu S, Dodd S, Parrott N, et al. Understanding mechanisms of food effect and developing reliable pbpk models using a middle-out approach. AAPS J. 2021;23(1):1–14. https://doi.org/10.1208/s12248-020-00548-8.
Riedmaier AE, DeMent K, Huckle J, Bransford P, Stillhart C, Lloyd R, et al. Use of physiologically based pharmacokinetic (PBPK) modeling for predicting drug-food interactions: an industry perspective. AAPS J. 2020;22(6):123. https://doi.org/10.1208/s12248-020-00508-2.
Kawakami K. Modification of physicochemical characteristics of active pharmaceutical ingredients and application of supersaturatable dosage forms for improving bioavailability of poorly absorbed drugs. Adv Drug Deliv Rev. 2012;64(6):480–95. https://doi.org/10.1016/j.addr.2011.10.009.
Carrier RL, Miller LA, Ahmed I. The utility of cyclodextrins for enhancing oral bioavailability. J Control Release. 2007;123(2):78–99. https://doi.org/10.1016/j.jconrel.2007.07.018.
Fasinu P, Pillay V, Ndesendo VM, du Toit LC, Choonara YE. Diverse approaches for the enhancement of oral drug bioavailability. Biopharm Drug Dispos. 2011;32(4):185–209. https://doi.org/10.1002/bdd.750.
Jones HM, Dickins M, Youdim K, Gosset JR, Attkins NJ, Hay TL, Gurrell IK, Logan YR, Bungay PJ, Jones BC, Gardner IB. Application of PBPK modelling in drug discovery and development at Pfizer. Xenobiotica. 2012;42(1):94–106. https://doi.org/10.3109/00498254.2011.627477.
Miller NA, Reddy MB, Heikkinen AT, Lukacova V, Parrott N. Physiologically based pharmacokinetic modelling for first-in-human predictions: an updated model building strategy illustrated with challenging industry case studies. Clin Pharmacokinet. 2019;58(6):727–46. https://doi.org/10.1007/s40262-019-00741-9.
Zhang T, Heimbach T, Lin W, Zhang J, He H. Prospective predictions of human pharmacokinetics for eighteen compounds. J Pharm Sci. 2015;104(9):2795–806. https://doi.org/10.1002/jps.24373.
Li M. Biopharmaceutics risk assessment to guide dissolution method development for solid oral dosage forms. 2021. https://www.youtube.com/watch?v=tjfbT7ThWn8
Wu D, Sanghavi M, Kollipara S, Ahmed T, Saini AK, Heimbach T. Physiologically based pharmacokinetics modeling in biopharmaceutics: case studies for establishing the bioequivalence safe space for generic and innovator drugs (submitted). Pharm Res. 2022. https://doi.org/10.1007/s11095-022-03319-6.
Raines K. PBPK biopharmaceutics guidance and progress on risk assessment, in regulatory utility of mechanistic modeling to support alternative bioequivalence approaches workshop. 2021. https://www.complexgenerics.org/media/SOP/complexgenerics/pdf/Conference-Slides/D2-04%20Kimberly%20Raines_PBPKGuidanceRiskAssessment.pdf
Wu F. PBPK 2021: Regulatory Utility of Mechanistic Modeling to Support Alternative Bioequivalence Approaches; Oral PBPK as alternative BE approach, risk assessment/biowaiver. , in PBPK Absorption Modeling to Support Risk Assessment and Biowaiver for Generic Oral Products. 2021 https://complexgenerics.org/media/SOP/complexgenerics/pdf/Conference-Slides/D2-03%202021_PBPK_workshop_Fang%20Wu_Presentation_Final_Modified_for_Posting.pdf
Aburub A, Chen Y, Chung J, Gao P, Good D, Hansmann S, et al. An IQ consortium perspective on connecting dissolution methods to in vivo performance: analysis of an industrial database and case studies to propose a workflow. AAPS J. 2022;24(3):49. https://doi.org/10.1208/s12248-022-00699-w.
Food and Drug Admistration. Guidance for Industry Extended Release Oral Dosage Forms: Development, Evaluation, and Application of In Vitro/In Vivo Correlations. 1997. https://www.fda.gov/media/70939/download
Suarez-Sharp S, Li M, Duan J, Shah H, Seo P. Regulatory experience with in vivo in vitro correlations (IVIVC) in new drug applications. AAPS J. 2016;18(6):1379–90. https://doi.org/10.1208/s12248-016-9966-2.
Takano R, Furumoto K, Shiraki K, Takata N, Hayashi Y, Aso Y, et al. Rate-limiting steps of oral absorption for poorly water-soluble drugs in dogs; prediction from a miniscale dissolution test and a physiologically-based computer simulation. Pharm Res. 2008;25(10):2334–44. https://doi.org/10.1007/s11095-008-9637-9.
Pathak SM, Schaefer KJ, Jamei M, Turner DB. Biopharmaceutic IVIVE—mechanistic modeling of single-and two-phase in vitro experiments to obtain drug-specific parameters for incorporation into PBPK models. J Pharm Sci. 2019;108(4):1604–18. https://doi.org/10.1016/j.xphs.2018.11.034.
Jamei M, Turner D, Yang J, Neuhoff S, Polak S, Rostami-Hodjegan A, et al. Population-based mechanistic prediction of oral drug absorption. AAPS J. 2009;11(2):225–37. https://doi.org/10.1208/s12248-009-9099-y.
Wang J, Flanagan DR. General solution for diffusion-controlled dissolution of spherical particles. 1. Theory. J Pharm Sci. 1999;88(7):731–8. https://doi.org/10.1021/js980236p.
Tsume Y, Takeuchi S, Matsui K, Amidon GE, Amidon GL. In vitro dissolution methodology, mini-gastrointestinal simulator (mGIS), predicts better in vivo dissolution of a weak base drug, dasatinib. Eur J Pharm Sci. 2015;76:203–12. https://doi.org/10.1016/j.ejps.2015.05.013.
Ding X, Gueorguieva I, Wesley JA, Burns LJ, Coutant CA. Assessment of in vivo clinical product performance of a weak basic drug by integration of in vitro dissolution tests and physiologically based absorption modeling. AAPS J. 2015;17(6):1395–406. https://doi.org/10.1208/s12248-015-9797-6.
Hens B, Pathak SM, Mitra A, Patel N, Liu B, Patel S, et al. In silico modeling approach for the evaluation of gastrointestinal dissolution, supersaturation, and precipitation of Posaconazole. Mol Pharm. 2017;14(12):4321–33. https://doi.org/10.1021/acs.molpharmaceut.7b00396.
Wagner C, Jantratid E, Kesisoglou F, Vertzoni M, Reppas C, Dressman JB. Predicting the oral absorption of a poorly soluble, poorly permeable weak base using biorelevant dissolution and transfer model tests coupled with a physiologically based pharmacokinetic model. Eur J Pharm Biopharm. 2012;82(1):127–38. https://doi.org/10.1016/j.ejpb.2012.05.008.
Kambayashi A, Yasuji T, Dressman JB. Prediction of the precipitation profiles of weak base drugs in the small intestine using a simplified transfer (“dumping”) model coupled with in silico modeling and simulation approach. Eur J Pharm Biopharm. 2016;103:95–103. https://doi.org/10.1016/j.ejpb.2016.03.020.
Patel S, Zhu W, Xia B, Sharma N, Hermans A, Ehrick JD, et al. Integration of precipitation kinetics from an in vitro, multicompartment transfer system and mechanistic Oral absorption modeling for pharmacokinetic prediction of weakly basic drugs. J Pharm Sci. 2019;108(1):574–83. https://doi.org/10.1016/j.xphs.2018.10.051.
Dickinson PA, Abu Rmaileh R, Ashworth L, Barker RA, Burke WM, Patterson CM, et al. An investigation into the utility of a multi-compartmental, dynamic, system of the upper gastrointestinal tract to support formulation development and establish bioequivalence of poorly soluble drugs. AAPS J. 2012;14(2):196–205. https://doi.org/10.1208/s12248-012-9333-x.
Sharp SS, Presentation: establishing clinically relevant drug product specifications: FDA Perspective, AAPS Annual meeting and exposure
Kato T, Nakagawa H, Mikkaichi T, Miyano T, Matsumoto Y, Ando S. Establishment of a clinically relevant specification for dissolution testing using physiologically based pharmacokinetic (PBPK) modeling approaches. Eur J Pharm Biopharm. 2020;151:45–52. https://doi.org/10.1016/j.ejpb.2020.03.012.
McAllister M, Flanagan T, Boon K, Pepin X, Tistaert C, Jamei M, Abend A, Kotzagiorgis E, Mackie C. Developing clinically relevant dissolution specifications for oral drug products—industrial and regulatory perspectives. https://doi.org/10.3390/pharmaceutics12010019.
Xiao J, Tran D, Zhang X, Zhang T, Seo S, Zhu H, et al. Biliary excretion–mediated food effects and prediction. AAPS J. 2020;22(6):124. https://doi.org/10.1208/s12248-020-00509-1.
Food and Drug Administration. Assessing the Effects of Food on Drugs in INDs and NDAs. 2020. https://www.fda.gov/media/121313/download
Gu CH, Li H, Levons J, Lentz K, Gandhi RB, Raghavan K, et al. Predicting effect of food on extent of drug absorption based on physicochemical properties. Pharm Res. 2007;24(6):1118–30. https://doi.org/10.1007/s11095-007-9236-1.
Fleisher D, Li C, Zhou Y, Pao LH, Karim A. Drug, meal and formulation interactions influencing drug absorption after oral administration. Clin Pharmacokinet. 1999;36(3):233–54. https://doi.org/10.2165/00003088-199936030-00004.
Wu C-Y, Benet LZ. Predicting drug disposition via application of BCS: transport/absorption/elimination interplay and development of a biopharmaceutics drug disposition classification system. Pharm Res. 2005;22(1):11–23. https://doi.org/10.1007/s11095-004-9004-4.
Zhang T, Wells E. A review of current methods for food effect prediction during drug development. Curr Pharmacol Rep. 2020;6(5):267–79. https://doi.org/10.1007/s40495-020-00230-9.
Lentz KA. Current methods for predicting human food effect. AAPS J. 2008;10(2):282–8. https://doi.org/10.1208/s12248-008-9025-8.
Zhao P, Rowland M, Huang S-M. Best practice in the use of physiologically based pharmacokinetic modeling and simulation to address clinical pharmacology regulatory questions. Clin Pharmacol Ther. 2012;92(1):17–20. https://doi.org/10.1038/clpt.2012.68.
Wagner C, Kesisoglou F, Pepin XJH, Parrott N, Emami RA. Use of physiologically based pharmacokinetic modeling for predicting drug–food interactions: recommendations for improving predictive performance of low confidence food effect models. AAPS J. 2021;23(4):85. https://doi.org/10.1208/s12248-021-00601-0.
Lewis JM, Stott KE, Monnery D, Seden K, Beeching NJ, Chaponda M, et al. Managing potential drug-drug interactions between gastric acid-reducing agents and antiretroviral therapy: experience from a large HIV-positive cohort. Int J STD AIDS. 2016;27(2):105–9. https://doi.org/10.1177/0956462415574632.
Smelick GS, Heffron TP, Chu L, Dean B, West DA, Duvall SL, et al. Prevalence of acid-reducing agents (ARA) in cancer populations and ARA drug–drug interaction potential for molecular targeted agents in clinical development. Mol Pharm. 2013;10(11):4055–62. https://doi.org/10.1021/mp400403s.
Fallingborg J. Intraluminal pH of the human gastrointestinal tract. Dan Med Bull. 1999;46(3):183–96.
Segregur D, Flanagan T, Mann J, Moir A, Karlsson EM, Hoch M, et al. Impact of acid-reducing agents on gastrointestinal physiology and design of biorelevant dissolution tests to reflect these changes. J Pharm Sci. 2019;108(11):3461–77. https://doi.org/10.1016/j.xphs.2019.06.021.
Del Re M, Omarini C, Diodati L, Palleschi M, Meattini I, Crucitta S, et al. Drug-drug interactions between palbociclib and proton pump inhibitors may significantly affect clinical outcome of metastatic breast cancer patients. ESMO Open. 2021;6(5):100231. https://doi.org/10.1016/j.esmoop.2021.100231.
Budha NR, Frymoyer A, Smelick GS, Jin JY, Yago MR, Dresser MJ, et al. Drug absorption interactions between oral targeted anticancer agents and PPIs: is pH-dependent solubility the Achilles heel of targeted therapy? Clin Pharmacol Ther. 2012;92(2):203–13. https://doi.org/10.1038/clpt.2012.73.
Numico G, Fusco V, Franco P, Roila F. Proton pump inhibitors in cancer patients: how useful they are? A review of the most common indications for their use. Crit Rev Oncol Hematol. 2017;111:144–51. https://doi.org/10.1016/j.critrevonc.2017.01.014.
Food and Drug Administration. Evaluation of Gastric pH-Dependent Drug Interactions With Acid-Reducing Agents: Study Design, Data Analysis, and Clinical Implications. 2020 https://www.fda.gov/media/144026/download
Le Merdy M, Tan ML, Sun D, Ni Z, Lee SC, Babiskin A, et al. Physiologically based pharmacokinetic modeling approach to identify the drug–drug interaction mechanism of Nifedipine and a proton pump inhibitor, Omeprazole. Eur J Drug Metab Pharmacokinet. 2021;46(1):41–51. https://doi.org/10.1007/s13318-020-00649-x.
Chirumamilla SK, Banala VT, Jamei M, Turner DB. Mechanistic PBPK modelling to predict the advantage of the salt form of a drug when dosed with acid reducing agents. Pharmaceutics. 2021;13(8):1169. https://doi.org/10.3390/pharmaceutics13081169.
Lin W, Chen Y, Unadkat JD, Zhang X, Wu D, Heimbach T. Applications, challenges, and outlook for PBPK modeling and simulation: a regulatory, industrial and academic perspective. Pharm Res. 2022;39:1701–31. https://doi.org/10.1007/s11095-022-03274-2.
Dong Z, Li J, Wu F, Zhao P, Lee SC, Zhang L, et al. Application of physiologically-based pharmacokinetic modeling to predict gastric pH-dependent drug–drug interactions for weak base drugs. CPT Pharmacometrics Syst Pharmacol. 2020;9(8):456–65. https://doi.org/10.1002/psp4.12541.
Vinarov Z, Abdallah M, Agundez JA, Allegaert K, Basit AW, Braeckmans M, Ceulemans J, Corsetti M, Griffin BT, Grimm M, Keszthelyi D. Impact of gastrointestinal tract variability on oral drug absorption and pharmacokinetics: an UNGAP review. Eur J Pharm Sci. 2021;162:105812. https://doi.org/10.1016/j.ejps.2021.105812.
Koenigsknecht MJ, Baker JR, Wen B, Frances A, Zhang H, Yu A, et al. In vivo dissolution and systemic absorption of immediate release ibuprofen in human gastrointestinal tract under fed and fasted conditions. Mol Pharm. 2017;14(12):4295–304. https://doi.org/10.1021/acs.molpharmaceut.7b00425.
Yu A, Baker JR, Fioritto AF, Wang Y, Luo R, Li S, et al. Measurement of in vivo gastrointestinal release and dissolution of three locally acting Mesalamine formulations in regions of the human gastrointestinal tract. Mol Pharm. 2017;14(2):345–58. https://doi.org/10.1021/acs.molpharmaceut.6b00641.
Yu A, Koenigsknecht MJ, Hens B, Baker JR, Wen B, Jackson TL, et al. Mechanistic deconvolution of oral absorption model with dynamic gastrointestinal fluid to predict regional rate and extent of GI drug dissolution. AAPS J. 2019;22(1):3. https://doi.org/10.1208/s12248-019-0385-z.
Yu LX, Lipka E, Crison JR, Amidon GL. Transport approaches to the biopharmaceutical design of oral drug delivery systems: prediction of intestinal absorption. Adv Drug Deliv Rev. 1996;19(3):359–76. https://doi.org/10.1016/0169-409x(96)00009-9.
Huang W, Lee SL, Yu LX. Mechanistic approaches to predicting oral drug absorption. AAPS J. 2009;11(2):217–24. https://doi.org/10.1208/s12248-009-9098-z.
Noyes AA, Whitney WR. The rate of solution of solid substances in their own solutions. J Am Chem Soc. 1897;19(12):930–4. https://doi.org/10.1021/ja02086a003.
Nernst W. Theorie der Reaktionsgeschwindigkeit in heterogenen Systemen. Z Phys Chem. 1904;47(1):52–5. https://doi.org/10.1515/zpch-1904-4704.
Dokoumetzidis A, Macheras P. A century of dissolution research: from Noyes and Whitney to the biopharmaceutics classification system. Int J Pharm. 2006;321(1–2):1–11. https://doi.org/10.1016/j.ijpharm.2006.07.011.
Wang J, Flanagan DR. General solution for diffusion-controlled dissolution of spherical particles. 2. Evaluation of experimental data. J Pharm Sci. 2002;91(2):534–42. https://doi.org/10.1002/jps.10039.
Kesisoglou F, Wu Y. Understanding the effect of API properties on bioavailability through absorption modeling. AAPS J. 2008;10(4):516–25. https://doi.org/10.1208/s12248-008-9061-4.
Kumar R, Thakur AK, Chaudhari P, Banerjee N. Particle size reduction techniques of pharmaceutical compounds for the enhancement of their dissolution rate and bioavailability. J Pharm Innov. 2021:1–20. https://doi.org/10.1007/s12247-020-09530-5.
Peters SA, Dolgos H. Requirements to establishing confidence in physiologically based pharmacokinetic (PBPK) models and overcoming some of the challenges to meeting them. Clin Pharmacokinet. 2019;58(11):1355–71. https://doi.org/10.1007/s40262-019-00790-0.
Cristofoletti R, Dressman JB. Use of physiologically based pharmacokinetic models coupled with pharmacodynamic models to assess the clinical relevance of current bioequivalence criteria for generic drug products containing ibuprofen. J Pharm Sci. 2014;103(10):3263–75. https://doi.org/10.1002/jps.24076.
Tsakalozou E, Alam K, Babiskin A, Zhao L. Physiologically-based pharmacokinetic modeling to support determination of bioequivalence for dermatological drug products: scientific and regulatory considerations. Clin Pharmacol Ther. 2022;111(5):1036–49. https://doi.org/10.1002/cpt.2356.
Food and Drug Administration, Statistical Approaches to Establishing Bioequivalence. 2001. https://www.fda.gov/media/70958/download
Grimstein M, Yang Y, Zhang X, Grillo J, Huang S-M, Zineh I, et al. Physiologically based pharmacokinetic modeling in regulatory science: an update from the U.S. Food and Drug Administration’s Office of Clinical Pharmacology. J Pharm Sci. 2019;108(1):21–5. https://doi.org/10.1016/j.xphs.2018.10.033.
Paraiso RL, Rose RH, Fotaki N, McAllister M, Dressman JB. The use of PBPK/PD to establish clinically relevant dissolution specifications for zolpidem immediate release tablets. Eur J Pharm Sci. 2020;155:105534. https://doi.org/10.1016/j.ejps.2020.105534.
European Medicines Agency, Guideline on the reporting of physiologically based pharmacokinetic (PBPK) modelling and simulation. 2020. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-reporting-physiologically-based-pharmacokinetic-pbpk-modelling-simulation_en.pdf
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclaimer
This article reflects the views of the authors and should not be construed to represent FDA’s views or policies. DW is an employee of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA and may hold equity stock in Merck & Co., Inc., Kenilworth, NJ, USA.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of Interest
The authors have no conflicts to declare.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Wu, D., Li, M. Current State and Challenges of Physiologically Based Biopharmaceutics Modeling (PBBM) in Oral Drug Product Development. Pharm Res 40, 321–336 (2023). https://doi.org/10.1007/s11095-022-03373-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-022-03373-0