
Abstract
Acalabrutinib, a selective Bruton’s tyrosine kinase inhibitor, is a biopharmaceutics classification system class II drug. The aim of this study was to develop a physiologically based pharmacokinetic (PBPK) model to mechanistically describe absorption of immediate release capsule formulation of acalabrutinib in humans. Integration of in vitro biorelevant measurements, dissolution studies and in silico modelling provided clinically relevant inputs for the mechanistic absorption PBPK model. The batch specific dissolution data were integrated in two ways, by fitting a diffusion layer model scalar to the drug product dissolution with integration of drug substance laser diffraction particle size data, or by fitting a product particle size distribution to the dissolution data. The latter method proved more robust and biopredictive. In both cases, the drug surface solubility was well predicted by the Simcyp simulator. The model using the product particle size distribution (P-PSD) for each clinical batch adequately captured the PK profiles of acalabrutinib and its active metabolite. Average fold errors were 0.89 for both Cmax and AUC, suggesting good agreement between predicted and observed PK values. The model also accurately predicted pH-dependent drug-drug interactions between omeprazole and acalabrutinib, which was similar across all clinical formulations. The model predicted acalabrutinib geometric mean AUC ratios (with omeprazole vs acalabrutinib alone) were 0.51 and 0.68 for 2 batches of formulations, which are close to observed values of 0.43 and 0.51~0.63, respectively. The mechanistic absorption PBPK model could be potentially used for future applications such as optimizing formulations or predicting the PK for different batches of the drug product.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Absorption of an orally administered drug is a complex process, which can be influenced by many factors including properties of the drug/formulation such as disintegration, solubility, permeability, supersaturation and precipitation as well as physiologically relevant factors such as gastric pH and emptying rate, uptake and efflux transporters, and metabolic enzymes in the intestine. In vitro dissolution experiments play an important role in understanding the drug release characteristics from the dosage form and should be used as an input to predict in vivo dissolution with a mechanistic modeling approach. Bridging in vitro dissolution and in vivo absorption has always been of great interest to regulators, academics and the pharmaceutical industry (1).
Physiologically based pharmacokinetic (PBPK) models that integrate both physiological characteristics and drug specific properties have gained increasing interest to describe the drug absorption processes. The gastro-intestinal tract can be described as a series of compartments, where volumes, pH, transit times, and surfaces that mimic the physiological situation and the absorption process can be described mechanically for dissolution, precipitation, permeation and transit (2). More specifically, in vitro solubility and dissolution data can be modeled mathematically and mechanistically using a theoretical approach to provide clinically relevant inputs for PBPK model (3). This approach has been applied to reasonably integrate dissolution in PBPK models for ketoconazole (2), lesinurad (4), acalabrutinib (5, 6), posaconazole (7), Naproxen (8), flurbiprofen (9), ibuprofen (10), BMS weak base (11), etoricoxib (12), ribociclib succinate (13, 14), warfarin and warfarin sodium (15), and entrectinib (16). Acid-reducing agents such as proton pump inhibitors may affect absorption for a drug exhibiting pH-dependent solubility. PBPK models have been able to qualitatively predict the effect of omeprazole in reducing the absorption of erlotinib and lack of effect on other weak base drugs such as BMS weak base (11), tapentadol, darunavir, and saxagliptin (17).
Bruton tyrosine kinase (BTK) is a key component of B-cell receptor signaling critical for cell proliferation, migration and survival (18). Acalabrutinib is a next-generation BTK inhibitor (19) approved for the treatment of adult patients with chronic lymphocytic leukemia in the United States and European Union, and small lymphocytic lymphoma (SLL) and mantle cell lymphoma (MCL) who have received at least one prior therapy in the United States (20). Acalabrutinib is rapidly absorbed with short oral half-life in healthy subjects, with an absolute oral bioavailability of 25% (21). Acalabrutinib is a Biopharmaceutics Classification System (BCS) class II drug and is predominantly metabolized by CYP3A enzymes (21).
Two PBPK models have been developed for acalabrutinib capsule formulation: one model was developed in Simcyp with a first order absorption model, which was mainly used to predict the potential CYP related drug-drug interactions for acalabrutinib (22); the other was developed in GastroPlus with consideration of mechanistic absorption (5, 6). The objectives of the current study were to further develop the PBPK model in Simcyp by incorporating mechanistic absorption using different batches of capsule formulations and to evaluate potential interaction between multiple doses of omeprazole and acalabrutinib.
METHODS
Modelling Strategy
A previously established PBPK model for acalabrutinib and its active metabolite, was used as starting point. The mechanistic absorption was incorporated to replace first order absorption while the distribution and elimination components were kept the same as the previous model. The updated PBPK model was then validated with multiple clinical studies conducted with different batches of acalabrutinib immediate release capsules. The PBPK model was finally used to predict the impact of omeprazole on the absorption of acalabrutinib as a result of the stomach pH change. The overall modeling strategy and individual clinical studies used in validation can be found in Fig. 1.

Overall modeling strategy and individual clinical studies used in validation.
Modelling of in vitro data was performed in the Simcyp In vitro data Analysis (SIVA) toolkit version 4, using the in-built feature to calculate the effect of surface pH on dissolution. The PBPK absorption model for acalabrutinib was developed in Simcyp, version 19. Both software are from Certara, Sheffield, UK.
Solubility and Dissolution
Acalabrutinib is treated as a diprotic base compound in the physiological pH range with pKas of 3.54 and 5.77, molecular weight of 465.5 and log P of 2.03 (22). Acalabrutinib is a BCS class II drug with intrinsic solubility of 48 μg/mL at pH 8. The solubilities in Fasted State Simulated Intestinal Fluid (FaSSIF v2), and Fed State Simulated Intestinal Fluid (FeSSIF) are 0.12 mg/mL (pH 6.5) and 0.67 mg/mL (pH 5) respectively (6). Acalabrutinib has a true density of 1.34 g/mL (6). Other parameters and all input data for the PBPK model can be found in supplementary Table S1. All dissolution experiments were performed in a Sotax AT7 or Distek 2100 USP apparatus 2 (paddle). Medium volume was fixed to 900 mL. Detailed experimental conditions have been published previously (6). Two batches of phase 1 capsules (NCZS and NVTF) used in early clinical studies and 2 batches of commercial capsules (L0505009 and W026394) used in later clinical studies were evaluated. Dissolution profiles of all 4 batches in 0.1 N HCl (pH 1) solution were obtained at paddle speed of 50 rpm. Dissolution profiles of batches L0505009 and W026394 were also evaluated in 0.001 N HCl (pH 3) solution and pH 6.8 phosphate buffer at paddle speed of 75 rpm. Dissolution profile of batch L0505009 was also available in pH 4.5 acetate buffer at paddle speed of 75 rpm (6).
Modelling In vitro Data
The solubility model in SIVA is a combination of aqueous phase solubility, governed by the Henderson-Hasselbalch equations for electrolytes, and a micelle-mediated solubility to account for partition of drug to surfactant micelles (2). The biorelevant media FaSSIF (pH 6.5) and FeSSIF (pH 5) solubility measurements along with intrinsic solubility were used to estimate acalabrutinib micelle:water partition coefficients for neutral and ionized molecular species. The sodium taurocholate concentrations were 3 and 15 mM, respectively, in FaSSIF and FeSSIF media, while lecithin concentration was 1 mM in both media.
Diffusion layer model (DLM) in SIVA uses the following equation to describe the dissolution of acalabrutinib particles (11).
where DR(t) is the overall rate of dissolution at time t, SS refers to different solid states, Ni,SS is the number of particles (in a given particle size bin), SDR,SS is the diffusion layer model scaling factor (default value 1), Deff,SS(t) is the effective diffusion coefficient, a(t) is particle radius at time t, heff,SS(t) is the effective diffusion layer thickness at time t, Ssurf,SS(t) is the concentration of drug at the particle surface at time t, and Cbulk,SS(t) is the concentration of the drug in the bulk solution at time t. The value for heff was estimated using the fluid dynamics sub-model.
A single product particle size distribution (P-PSD) was obtained for each batch of acalabrutinib capsule to explain all the in vitro dissolution profiles tested, in simple media throughout the physiological pH range. P-PSD was generated using the SIVA tool by fitting all dissolution profiles simultaneously for each batch of drug product (Figure S6) using the dissolution profiles specified in Table S3. The surface pH and solubility of acalabrutinib at different bulk pH values were also incorporated in the model using the built-in features of SIVA.
In addition, to test the DLM scalar approach on the drug substance particle size distribution, a DLM scalar was also fitted simultaneously to all dissolution profiles for batch L0505009 (Figure S4) using an 11-bin reduced drug substance particle size distribution as input (Figure S2). This unique DLM scalar for batch L0505009 was also used to predict the exposure to 100 mg acalabrutinib capsules (Figure S7). In addition, a DLM scalar is also fitted to each of the dissolution profiles for batch L0505009 (Figure S3), and the resulting 4 DLM scalars are presented in Table S3. The performance of the DLM scalar or the P-PSD fitted simultaneously to all dissolution data are assessed by calculating average fold error (AFE) and absolute average fold error (AAFE) for each timepoints of the dissolution (Table S5). These models are also compared to previous data generated by Pepin et al. using an Excel based tool for the P-PSD (Figure S6).
Precipitation rate constant (PRC) of acalabrutinib was assigned to be 0.036 h−1 based on the previous estimation of precipitation time of 100,000 s (5) and critical supersaturation ratio (CSR) was set to 10 (default value). The drug precipitation is unlikely in the model based on in vitro and in vivo evidence (5). Sensitivity analysis was conducted with respect to CSR from 1 to 100 and PRC from 0.001 to 1 h−1. The sensitivity analysis did not indicate a significant effect of PRC or CSR on acalabrutinib absorption.
Impact of Gastric pH Change
Acalabrutinib is a weak base with maximum basic pKa of 5.77, and the influence of surface pH on surface solubility can be important below the maximum basic pKa. Only the stomach compartment in the PBPK model is likely impacted by differences between surface pH and bulk pH of acalabrutinib. The approach presented previously (5, 23), i.e. use of micro-environmental pH to calculate in vivo dissolution in the gut compartmental model, was also used in the current simulations. Different doses of omeprazole could have different level of impact on gastric pH, and multiple doses of 40 mg omeprazole could increase gastric pH to the range of 4.4 to 6 (17). Therefore, a sensitivity analysis was conducted with respect to gastric pH from 5 to 7 to evaluate its impact on model prediction performance.
Clinical Studies
ACE-HV-001 was a phase 1 dose-escalation study in healthy subjects including a single dose of 25 mg, 50 mg, 75 mg, and 100 mg of acalabrutinib from cohort 3, 4, 5, and 6, respectively, as well as a single dose of 50 mg arm from cohort 7 (N = 17) (22, 24). ACE-HV-005 was a phase 1 study to evaluate effects of single therapeutic (100 mg) and supra-therapeutic (400 mg) oral doses of acalabrutinib on QTc interval in healthy subjects (N = 40) (22, 24). ACE-HV-113 was a phase 1 study to measure intrasubject variability in PK and determine gastric pH effects on acalabrutinib pharmacokinetics in healthy subjects (N = 13) after a single 100 mg dose was administered on 2 different occasions (22, 24). The batches of NCZS, L0505009 and W026394 were used in studies ACE-HV-001, ACE-HV-005 and ACE-HV-113, respectively. All 3 studies were used to validate the mechanistic absorption PBPK model of acalabrutinib.
ACE-HV-004 was a phase 1 drug interaction study in healthy subjects, where part 2 evaluated a 100 mg dose of acalabrutinib administered in the absence of omeprazole and again on the last day of 5 consecutive days of dosing omeprazole (40 mg QD) (N = 24) (24). ACE-HV-112 was a phase 1 study in healthy subjects to evaluate the effect of acidic conditions on the pharmacokinetic profile of acalabrutinib alone and when co-administered with omeprazole. In each of 3 parts of the study, 12 subjects were administered 100 mg acalabrutinib alone or in the presence of 5 consecutive days of dosing omeprazole (40 mg QD). The batches of NVTF and L0505009 were used in study ACE-HV-004 and ACE-HV-112, respectively. Both studies were used to evaluate PBPK prediction performance of omeprazole impact on acalabrutinib. Details on these 5 clinical studies including dose, formulation, number of subjects, age, sex can be found in supplementary Table S2. These studies were conducted in accordance with the Declaration of Helsinki and the International Conference on Harmonization/Good Clinical Practice. The final protocol and informed consent form were approved by the institutional review boards at the respective study sites. Informed consent was obtained from all volunteers before any study procedures were conducted.
Model Simulation Setup
Default Simcyp parameter values for creating a virtual Healthy Volunteer population (population, physiological parameters including liver volume and blood flows, enzyme abundances) were applied in the current analysis (25). All simulations were run as10 virtual replicate trials with number of subjects, age range and percentage of female matched with those in the corresponding actual clinical studies, and according to the same study design (dose, formulation) as summarized in Table S2. The model performance was evaluated using AFE and AAFE (5), which were calculated for the predicted geometric mean maximum plasma concentration (Cmax) or area under the concentration–time curve (AUC) over the observed values. AUC0-24 h were calculated in all simulations as close comparison to the reported AUC0-last in all clinical studies. The average fold error indicates whether the predicted values underestimate (less than one) or overestimate (more than one) the observed values. The AFE or AAFE of Cmax and AUC in the range of 0.8 ~ 1.25 were set as acceptance criteria for the model. The current model predictions were also compared to previous simulations for the same drug obtained by Pepin et al. (5) (Table S6 and Table S7).
RESULTS
The acalabrutinib micelle:water partition coefficients were estimated to be 4.385 and 2.228 for neutral and ionized molecular species, respectively, using solubility measurements in FaSSIF (pH 6.5) and FeSSIF (pH 5) media. The parameters were applied in the PBPK model to estimate the regional luminal pH and bile salt concentration variability in vivo.
The SIVA model predicted surface pH and measured surface pH (6) at different bulk pH are presented in Fig. 2. The predicted values are in reasonable agreement with those obtained experimentally. The surface pH deviates from bulk pH below the highest basic pKa of acalabrutinib, and there is a 2.5 pH unit difference between bulk and surface pH at pH of 1, indicating the surface solubility can be impacted. The prediction with SIVA for surface pH (and therefore surface solubility) are slightly lower compared to those obtained by Pepin et al. (6), which leads to higher estimated surface solubilities at pH 1, pH3 and pH 4.5 with SIVA.

Surface pH as a function of bulk pH for acalabrutinib. Open circle: measurements. Solid blue line: prediction with SIVA toolkit, solid green line: prediction from Pepin et al. (6).
The dissolution profiles of batch L0505009 in 0.1 N HCl (pH 1), 0.001 N HCl (pH 3), acetate buffer (pH 4.5) as well as phosphate buffer (pH 6.8) were used simultaneously to estimate the P-PSD. A polydispersed P-PSD with a mean radius of 122.4 µm with range of 9.9 to 332.7 µm and coefficient of variation of 78.8% can reasonably describe L0505009 dissolution profiles in the pH range of 1 to 6.8 (Figure S5). The mean radius of 152.3, 148.7 and 193.6 µm were estimated for batch W026394, NCZS and NVTF, respectively. Other P-PSD parameters can be found in supplementary Table S1. To illustrate the differences in dissolution rate between batches L0505009, W026394, NCZS and NVTF, the dissolution rate measured in USP2, 900 mL, 0.1 N HCl, 50 rpm is shown in Fig. 3A. The fitted cumulative particle size distribution for all 4 batches are presented in Fig. 3B. P-PSD values of batch L0505009 were implemented in the PBPK model for the simulation of studies ACE-HV-005 and ACE-HV-112. P-PSD values of batches of NCZS, NVTF and W026394 were used in the simulation of studies ACE-HV-001, ACE-HV-004 and ACE-HV-113, respectively. The performance of the acalabrutinib PBPK model with mechanistic absorption was assessed by simulating plasma concentration–time profiles following a single 25, 50, 75, 100 mg dose in healthy subjects using the same study designs in ACE-HV-001, ACE-HV-005 and ACE-HV-113. Figure 4 shows the predicted pharmacokinetic profiles of acalabrutinib and its metabolite in comparison with the observed data from study ACE-HV-005 and ACE-HV-113. Predicted PK profiles of acalabrutinib overlaid with observed values for ACE-HV-001 are presented in supplementary Figure S1. The PBPK model reasonably recovered the PK profiles for acalabrutinib, and the predicted geometric mean Cmax and AUC compared to observed values for 13 arms of clinical studies in the dose range of 25 to 100 mg are presented in Fig. 5. The AFE and AAFE values were 0.89 and 1.22, respectively for both Cmax and AUC, suggesting good agreement between predicted and observed PK values. The model was compared to previously published data obtained with GastroPlus using a fixed 8-subject representative population and where only the drug product P-PSD and stomach pH were changed across the different scenarios (5). Both model performance indicators are shown in Table S6 for AUC and Table S7 for Cmax. The GastroPlus underestimation of AUC is more pronounced for the lower clinical doses compared to Simcyp. However, the Cmax is better predicted throughout the scenarios tested in GastroPlus compared to Simcyp. For both AUC and Cmax the AAFE are comparable across the two software platforms.

(a) Dissolution rate of batches NVTF, NCZS, L0505009 and W026394 in USP2, 900 mL, 0.1 N HCL, 50 rpm, 37 °C. (b) Cumulative product particle size distribution of different batches of acalabrutinib.

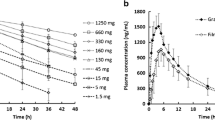
Plasma concentrations of acalabrutinib and its metabolite ACP-5862 after single oral dosing of 100 mg acalabrutinib in healthy volunteers for study ACE-HV-005 (Batch L0505009) (A) and ACE-HV-113 (Batch W026394) (B). Simulated values are displayed as lines, including individual trials (color lines) and 5% and 95% percentiles (gray lines). Observed data from clinical studies are displayed as solid dots, both arms of study ACE-HV-113 are presented. h, hour.

Predicted vs observed Cmax (top) and AUC (bottom) of acalabrutinib in ACE-HV-001 (Batch NCZS), ACE-HV-004 (Batch NVTF), ACE-HV-005 (Batch L0505009), ACE-HV-112 (Batch L0505009) and ACE-HV-113 (Batch W026394) for various dosing conditions. Open and closed symbols represent for 100 mg dose and <100 mg dose of acalabrutinib respectively.
After the validation of the acalabrutinib PBPK model using capsules with various dissolution rates and different P-PSD values, it was applied to predict the impact of gastric pH change on acalabrutinib absorption. The default pH of stomach compartment for an average healthy volunteer subject in fasted condition is 1.5. A sensitivity analysis was first conducted by adjusting the stomach pH from 1 to 7. As the pH increases, both the Cmax and AUC of acalabrutinib decreased dramatically. It has been reported stomach pH could increase to the range of 4.4 to 6 after multiple doses of 40 mg omeprazole. Therefore a stomach pH of 5.2 was targeted in the current simulation to mimic omeprazole effect on gastric pH. Figure 6 shows pH values in each segment of gastro-intestinal tract in default stomach setting and the stomach with multiple doses treatment of omeprazole. The mean simulated stomach pH values from 120 subjects were 1.23 with 90% confidence interval (CI) of 0.78 to 2.19 and 5.23 with 90% CI of 3.60 to 6.83 in the default setting and in the presence of omeprazole, respectively. By applying such adjustment of pH in stomach compartment, the PBPK model could reasonably predict the acalabrutinib PK profile change in the presence of multiple doses of omeprazole (Fig. 7). Batch NVTF capsule was used in ACE-HV-004 study, where 24 healthy subjects by crossover design was conducted to evaluate omeprazole impact and a relatively large variability was observed with 383% CV for Cmax in the omeprazole co-administered arm. The observed geometric mean Cmax and AUC0-last were approximately 0.21-fold lower and 0.43-fold lower when acalabrutinib was co-administered with omeprazole as compared to acalabrutinib alone. The model-predicted geometric mean AUC and Cmax ratio were 0.51 and 0.21, which are close to observed values. Batch L0505009 capsule was used in ACE-HV-112 study to evaluate omeprazole impact. The observed geometric ratios (omeprazole vs alone) of Cmax and AUC0-last were 0.27, 0.24, 0.34 and 0.58, 0.51, 0.63, respectively for each arm. The model predicted geometric mean Cmax and AUC ratio were 0.34 and 0.68, which are close to observed values. The comparison of predicted and observed values is presented in Table I.

pH values in each segment of gastro-intestinal tract in default stomach setting (left) as well as the stomach with multiple doses treatment of omeprazole (right) Blue lines with circle are the mean pH values in each segment, and dashed lines are 5% and 95% percentiles of pH values in simulated subjects.

Plasma concentrations of acalabrutinib alone or in the presence of multiple doses of omeprazole after single oral dosing of 100 mg acalabrutinib in healthy volunteers for study ACE-HV-004 (Batch NVTF) (A) and ACE-HV-112 (Batch L0505009) (B). Simulated values are displayed as lines, including individual trials (color lines) and 5% and 95% percentiles (gray lines). Mean observed data from clinical studies are displayed as solid dots with standard deviation as error bars, 3 arms of study ACE-HV-112 are presented. h, hour.
DISCUSSION
PBPK modeling has been widely utilized in drug development to support development decisions, pediatric dose selection, as well as for regulatory submissions and labelling (26,27,28). A PBPK model of acalabrutinib was developed previously for the purpose of predicting CYP related drug-drug interactions (22). A simple first order absorption model was used in the previous model, and the model was updated with mechanistic absorption using the in vitro solubility and dissolution results. Regulators currently require that models also integrate a mechanistic part for absorption in order to be relevant to the batches used in the clinical trials and allow a translation of clinical findings to support the evaluation of the intended commercial formulations. Recently the US FDA has published a draft guidance on the use of PBPK models to support biopharmaceutics analyses (29). The updated PBPK model was able to capture mechanistically the absorption and disposition of acalabrutinib. Validation with clinical data from healthy volunteer subjects confirmed that the model was able to predict acalabrutinib PK profiles in various studies using different batches of capsule formulations. The PBPK model was further validated for its ability to accurately predict the impact of an acid-reducing agent (omeprazole) on acalabrutinib PK profiles. It is interesting to observe that batch L0505009 (commercial capsule representative comprising a dry granulated powder) and batch NVTF (phase 1 capsule comprising a binary blend of drug and excipient) have approximately the same drug-drug interaction with omeprazole, despite the fact that they are not bioequivalent to one another. The slow dissolution observed for batch NVTF (and NCZS) was ascribed to the choice of formulation and poor drug wettability in the dissolution medium. The P-PSD was able to capture this phenomenon by reducing the surface area available for dissolution by increasing the particle size for batch NVTF, demonstrating that the clinical prediction is accurate in both the normal stomach conditions or following omeprazole administration. In vivo, the NVTF capsule batch could also have issues of wetting in the stomach and this could lead to a reduced exposure. Indeed after stomach emptying, the particles which have not dissolved would be transferred into the intestine where the drug solubility could be limiting for absorption. The PBPK model is therefore able to mechanistically explain the lack of bioequivalence for batches NVTF and L0505009 in the fasted state or when omeprazole is co-administered. In addition, the batches L0505009 and W026394 showed in vitro dissolution differences but such difference did not translate into differences in vivo which was also predicted by the model.
This analysis illustrates the importance of in vitro clinically relevant dissolution measurements, and a mechanistic absorption PBPK model. A few approaches could be applied to integrate drug product dissolution data with PBPK (3). The simplest is to directly use an in vitro dissolution profile as in vivo dissolution in the simulations. This approach could work for BCS class I drugs, where the in vitro and in vivo dissolution are expected to be independent of volume or pH in the gastrointestinal tract. Another approach is to fit observed a dissolution profile with mathematical models, such as Weibull functions, to provide clinically relevant inputs. However, this approach would not allow prediction of the impact of stomach pH by changing the physiological parameters of the PBPK model. Mechanistic models of in vitro dissolution data to predict in vivo dissolution are the approach of choice for immediate release formulations. There are two methods to obtain relevant inputs for PBPK using the DLM model in Simcyp. One method is to use experimentally measured drug substance particle size distribution (Figure S2, obtained by laser diffraction data) together with drug product dissolution profiles to estimate a DLM scaling factor (Figure S4), while the other method is to keep DLM scaling factor as 1 and estimate the P-PSD using dissolution profiles (Figure S5). Both methods were evaluated in current analysis. In the DLM scaling factor approach, one DLM scaling factor could be obtained to fit each drug product in vitro dissolution profile reasonably well (Figure S3). However, the estimated DLM scaling factors varied over 80 fold for the 4 dissolution profiles of batch L0505009 (Table S3). A single DLM scaling factor (0.052) was also attempted using all 4 dissolution profiles of batch L0505009 in combination with drug substance measured particle size (Figure S4), but this single DLM scaling factor significantly underestimated Cmax and overall PK profile of study ACE-HV-005 (Figure S7). Therefore, the P-PSD method was selected in current analysis as this approach has more adjustable parameters (10 bins) compared to the DLM scalar approach (1 scalar applied to measured 11 bins drug substance PSD). The AFE and AAFE for prediction of in vitro dissolution data for the same drug product batch were compared in Table S5. These performance indicators show that the P-PSD in SIVA is superior to the DLM scaling factor since it has reduced the model error by 1.8 fold (the model error is defined here by AAFE-1). In comparison the P-PSD obtained in Excel by Pepin et al. (6) reduced the model error by twofold compared to the P-PSD in SIVA and by 3.8 fold compared to the DLM scalar in SIVA. It is believed that the differences in surface solubility predictions are responsible for these differences in the two P-PSD models. There are several theoretical limitations which prevent the use of a single DLM scaling factor to be applied to all the bins of a drug substance particle size distribution as explained by Pepin et al. (30). For example, fine drug substance particles with a large surface to volume ratio, may not necessarily wet in a dry blend or when the pure drug is placed in a capsule. With a more elaborate formulation as is the case for acalabrutinib commercial capsules, the presence of excipients and the application of dry granulation manufacturing process ensures that the drug substance is well mixed and in contact with excipients helping with capsule disintegration and drug substance wetting through wicking effect. There were no major differences of drug substance particle size between batches that comprised drug products NVTF or L0505009. Yet, these products dissolve differently (Fig. 2) and are not bioequivalent in the clinic (Table I). This work illustrates the importance of introduction of batch level information in PBPK modelling when assessing the clinical data and to propose physiologically based biopharmaceutics models (PBBMs) which will translate from the various clinical phases to the evaluation of commercial formulations pre or post approval. This work also demonstrates the role of dissolution testing as the primary source of information to assess drug product quality in a PBPK model. It also shows the limitation of utilizing drug substance particle size to predict the in vivo performance of a dry solid oral dosage form.
Acid-reducing agents are among the most frequently used medicines. For a drug exhibiting pH-dependent solubility, gastric pH increase by an acid-reducing agent may affect its absorption, leading to reduced exposures in many patients which can lead to decreased therapeutic benefit (31). Evaluation of the pH-dependent drug-drug interactions potential becomes an important step in drug development, which may impact the drug dosing regimen (32). PBPK model approaches have been applied to evaluate such pH-dependent drug-drug interactions (11, 16, 17, 23), indicating that PBPK could be used to quantitatively predict the effect of acid-reducing agents in reducing the absorption of entrectinib, erlotinib, ketoconazole and posaconazole or lack of the effect on tapentadol, darunavir, and saxagliptin. The current analysis provided an additional case for the interaction between omeprazole and acalabrutinib. The gastric pH was increased to 5.23 (90% CI 3.60—6.83) in this analysis, which are in the similar range used in entrectinib (pH 4.5) (16) and erlotinib (pH 6.0) (17) analysis. The clinical studies presented in this work using 2 different batches of capsules exhibited a small different impact by omeprazole although the absolute exposure for these formulations was very different. The current PBPK model using P-PSD approach could incorporate different dissolution rates to fit the batch specific P-PSD and to predict the pharmacokinetics of specific batches of drug products.
CONCLUSIONS
The integration of in vitro biopharmaceutical properties, batch specific P-PSD from in vitro drug product dissolution and in silico modelling provided clinical relevant inputs for a mechanistic absorption PBPK model for acalabrutinib in Simcyp®. The lessons learnt from this exercise were that for acalabrutinib immediate release capsules:
-
1-
The P-PSD approach is superior to the DLM scalar applied to the drug substance particle size distribution to predict in vitro dissolution and human PK using Simcyp and provides similar prediction performance as in GastroPlus.
-
2-
The inclusion of surface pH is key and slight model difference can impact the prediction of in vitro dissolution (comparison between SIVA and in-house Excel tool)
-
3-
The use of a single batch P-PSD and the mechanistic model for surface solubility in Simcyp allowed to predict formulation differences and the impact of pH dependent drug-drug interactions between omeprazole and acalabrutinib.
In conclusion, the PBPK model could be potentially used for future applications such as formulation optimization or bridging between formulations.
References
Abend A, Heimbach T, Cohen M, Kesisoglou F, Pepin X, Suarez-Sharp S. Dissolution and translational modeling strategies enabling patient-centric drug product development: the M-CERSI Workshop Summary Report. The AAPS journal. 2018;20(3).
Pathak SM, Ruff A, Kostewicz ES, Patel N, Turner DB, Jamei M. Model-based analysis of biopharmaceutic experiments to improve mechanistic oral absorption modeling: an integrated in vitro in vivo extrapolation perspective using ketoconazole as a model drug. Mol Pharm. 2017;14(12):4305–20.
Jamei M, Abrahamsson B, Brown J, Bevernage J, Bolger MB, Heimbach T, Karlsson E, Kotzagiorgis E, Lindahl A, McAllister M, Mullin JM, Pepin X, Tistaert C, Turner DB, Kesisoglou F. Current status and future opportunities for incorporation of dissolution data in PBPK modeling for pharmaceutical development and regulatory applications: OrBiTo consortium commentary. Eur J Pharm Biopharm. 2020;155:55–68.
Pepin XJ, Flanagan TR, Holt DJ, Eidelman A, Treacy D, Rowlings CE. Justification of drug product dissolution rate and drug substance particle size specifications based on absorption PBPK modeling for lesinurad immediate release tablets. Mol Pharm. 2016;13(9):3256–69.
Pepin XJH, Moir AJ, Mann JC, Sanderson NJ, Barker R, Meehan E, Plumb AP, Bailey GR, Murphy DS, Krejsa CM, Andrew MA, Ingallinera TG, Slatter JG. Bridging in vitro dissolution and in vivo exposure for acalabrutinib. Part II. A mechanistic PBPK model for IR formulation comparison, proton pump inhibitor drug interactions, and administration with acidic juices. European journal of pharmaceutics and biopharmaceutics. 2019;142:435–448.
Pepin XJH, Sanderson NJ, Blanazs A, Grover S, Ingallinera TG, Mann JC. Bridging in vitro dissolution and in vivo exposure for acalabrutinib. Part I. Mechanistic modelling of drug product dissolution to derive a P-PSD for PBPK model input. European Journal of Pharmaceutics and Biopharmaceutics. 2019;142:421–434.
Hens B, Pathak SM, Mitra A, Patel N, Liu B, Patel S, Jamei M, Brouwers J, Augustijns P, Turner DB. In silico modeling approach for the evaluation of gastrointestinal dissolution, supersaturation, and precipitation of posaconazole. Mol Pharm. 2017;14(12):4321–33.
Loisios-Konstantinidis I, Cristofoletti R, Fotaki N, Turner DB, Dressman J. Establishing virtual bioequivalence and clinically relevant specifications using in vitro biorelevant dissolution testing and physiologically-based population pharmacokinetic modeling. case example: Naproxen. European Journal of Pharmaceutical Sciences. 2020;143:105170.
Loisios-Konstantinidis I, Cristofoletti R, Jamei M, Turner D, Dressman J. Physiologically based pharmacokinetic/pharmacodynamic modeling to predict the impact of CYP2C9 genetic polymorphisms, co-medication and formulation on the pharmacokinetics and pharmacodynamics of flurbiprofen. Pharmaceutics. 2020;12(11).
Cristofoletti R, Hens B, Patel N, Esteban VV, Schmidt S, Dressman J. Integrating drug- and formulation-related properties with gastrointestinal tract variability using a product-specific particle size approach: case example ibuprofen. J Pharm Sci. 2019;108(12):3842–7.
Chirumamilla SK, Banala VT, Jamei M, Turner DB. Mechanistic PBPK modelling to predict the advantage of the salt form of a drug when dosed with acid reducing agents. Pharmaceutics. 2021;13(8).
Heimbach T, Kesisoglou F, Novakovic J, Tistaert C, Mueller-Zsigmondy M, Kollipara S, Ahmed T, Mitra A, Suarez-Sharp S. Establishing the bioequivalence safe space for immediate-release oral dosage forms using physiologically based biopharmaceutics modeling (PBBM): Case studies. Journal of Pharmaceutical Sciences. 2021.
Heimbach T, Suarez-Sharp S, Kakhi M, Holmstock N, Olivares-Morales A, Pepin X, Sjögren E, Tsakalozou E, Seo P, Li M, Zhang X, Lin H-P, Montague T, Mitra A, Morris D, Patel N, Kesisoglou F. Dissolution and translational modeling strategies toward establishing an in vitro-in vivo link—a workshop summary report. The AAPS Journal. 2019;21(2).
Laisney M, Heimbach T, Mueller-Zsigmondy M, Blumenstein L, Costa R, Ji Y. Physiologically based biopharmaceutics modeling to demonstrate virtual bioequivalence and bioequivalence safe-space for ribociclib which has permeation rate-controlled absorption. Journal of Pharmaceutical Sciences. 2021.
Zhang X, Wen H, Fan J, Vince B, Li T, Gao W, Kinjo M, Brown J, Sun W, Jiang W, Lionberger R. Integrating in vitro, modeling, and in vivo approaches to investigate warfarin bioequivalence. CPT: pharmacometrics & systems pharmacology. 2017.
Parrott N, Stillhart C, Lindenberg M, Wagner B, Kowalski K, Guerini E, Djebli N, Meneses-Lorente G. Physiologically based absorption modelling to explore the impact of food and gastric pH changes on the pharmacokinetics of entrectinib. The AAPS Journal. 2020;22(4).
Dong Z, Li J, Wu F, Zhao P, Lee SC, Zhang L, Seo P, Zhang L. Application of physiologically-based pharmacokinetic modeling to predict gastric pH-dependent drug-drug interactions for weak base drugs. CPT: pharmacometrics & systems pharmacology. 2020;9(8):456–465.
Küppers R. Mechanisms of B-cell lymphoma pathogenesis. Nat Rev Cancer. 2005;5(4):251–62.
Covey T, Barf T, Gulrajani M, Krantz F, van Lith B, Bibikova E, van de Kar B, de Zwart E, Hamdy A, Izumi R, Kaptein A. Abstract 2596: ACP-196: a novel covalent Bruton’s tyrosine kinase (Btk) inhibitor with improved selectivity and in vivo target coverage in chronic lymphocytic leukemia (CLL) patients. Can Res. 2015;75(15 Supplement):2596.
CDER F. Highlights of Prescribing Information. CALQUENCE® (acalabrutinib) capsules, for oral use Initial U.S. Approval: 2017. 2017 12/12/2017. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/210259s000lbl.pdf.
Podoll T, Pearson PG, Evarts J, Ingallinera T, Bibikova E, Sun H, Gohdes M, Cardinal K, Sanghvi M, Slatter JG. Bioavailability, biotransformation, and excretion of the covalent bruton tyrosine kinase inhibitor acalabrutinib in rats, dogs, and humans. Drug Metab Dispos. 2019;47(2):145–54.
Zhou D, Podoll T, Xu Y, Moorthy G, Vishwanathan K, Ware J, Slatter JG, Al-Huniti N. Evaluation of the drug-drug interaction potential of acalabrutinib and its active metabolite, ACP-5862, using a physiologically-based pharmacokinetic modeling approach. CPT: pharmacometrics & systems pharmacology. 2019;8(7):489–499.
Cristofoletti R, Patel N, Dressman JB. Assessment of bioequivalence of weak base formulations under various dosing conditions using physiologically based pharmacokinetic simulations in virtual populations. Case Examples: Ketoconazole and Posaconazole. J Pharm Sci. 2017;106(2):560–569.
Izumi R, Pearson PG, Hamdy A, Rodriquez S, Yang YYL, Ulrich RG, Podoll T, Andrew M, Krejsa CM, Ingallinera T. CYP3A-mediated drug interaction profile of Bruton Tyrosine Kinase inhibitor. Acalabrutinib. In.: American Society of Hematology Washington, DC; 2017.
Jamei M, Marciniak S, Feng K, Barnett A, Tucker G, Rostami-Hodjegan A. The Simcyp population-based ADME simulator. Expert Opin Drug Metab Toxicol. 2009;5(2):211–23.
Johnson TN, Zhou D, Bui KH. Development of physiologically based pharmacokinetic model to evaluate the relative systemic exposure to quetiapine after administration of IR and XR formulations to adults, children and adolescents. Biopharmaceutics & Drug Disposition;35(6):341–352.
Chen Y, Zhou D, Tang W, Zhou W, Al-Huniti N, Masson E. Physiologically based pharmacokinetic modeling to evaluate the systemic exposure of gefitinib in CYP2D6 ultrarapid metabolizers and extensive metabolizers. J Clin Pharmacol. 2018;58(4):485–93.
Zhou D, Bui K, Sostek M, Al‐Huniti N. Simulation and prediction of the drug‐drug interaction potential of naloxegol by physiologically based pharmacokinetic modeling. CPT: Pharmacometrics & Systems Pharmacology. 2016;5(5):250–257.
CDER F. The use of physiologically based pharmacokinetic analyses — biopharmaceutics applications for oral drug product development, manufacturing changes, and controls. In: CDER, editor.; 2020.
Pepin X, Goetschy M, Abrahmsén-Alami S. Mechanistic models for USP2 dissolution apparatus, including fluid hydrodynamics and sedimentation. Journal of Pharmaceutical Sciences. 2021.
Budha NR, Frymoyer A, Smelick GS, Jin JY, Yago MR, Dresser MJ, Holden SN, Benet LZ, Ware JA. Drug absorption interactions between oral targeted anticancer agents and PPIs: Is pH-dependent solubility the achilles heel of targeted therapy? Clin Pharmacol Ther. 2012;92(2):203–13.
Zhang L, Wu F, Lee SC, Zhao H. pH-dependent drug-drug interactions for weak base drugs: potential implications for new drug development. Clin Pharmacol Ther. 2014;96(2):266–77.
ACKNOWLEDGMENTS AND DISCLOSURES
The authors would like to acknowledge David B. Turner and Sumit Arora of Certara UK Limited for their technical support and discussion.
All authors are employees of AstraZeneca at the study was conducted. All authors have stock ownership and/or stock interests or options in AstraZeneca.
Funding
This study was supported by AstraZeneca.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zhou, D., Chen, B., Sharma, S. et al. Physiologically Based Absorption Modelling to Explore the Formulation and Gastric pH Changes on the Pharmacokinetics of Acalabrutinib. Pharm Res 40, 375–386 (2023). https://doi.org/10.1007/s11095-022-03268-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-022-03268-0