
Abstract
As products of living cells, biologics are far more complicated than small molecular-weight drugs not only with respect to size and structural complexity but also their sensitivity to manufacturing processes and post-translational changes. Most of the information on the manufacturing process of biotherapeutics is proprietary and hence not fully accessible to the public. This information gap represents a key challenge for biosimilar developers and plays a key role in explaining the differences in regulatory pathways required to demonstrate biosimilarity versus those required to ensure that a change in manufacturing process did not have implications on safety and efficacy. Manufacturing process changes are frequently needed for a variety of reasons including response to regulatory requirements, up scaling production, change in facility, change in raw materials, improving control of quality (consistency) or optimising production efficiency. The scope of the change is usually a key indicator of the scale of analysis required to evaluate the quality. In most cases, where the scope of the process change is limited, only quality and analytical studies should be sufficient while comparative clinical studies can be required in case of major changes (e.g., cell line changes). Biosimilarity exercises have been addressed differently by regulators on the understanding that biosimilar developers start with fundamental differences being a new cell line and also a knowledge gap of the innovator’s processes, including culture media, purification processes, and potentially different formulations, and are thus required to ensure that differences from innovators do not result in differences in efficacy and safety.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The development of biologic medicines has transformed the treatment of many serious diseases over the past decade [1]. However, the patents of a growing number of biologics have already expired or are due to expire [2–4] leading to an increased interest in the development of biosimilars (sometimes referred to as ‘follow-on biologics’) [3–5]. Figure 1 shows the period of market exclusivity for the top ten selling biologics, with the 2012–2019 patent cliff highlighted in the grey box.

Period of market exclusivity for the top ten selling biologics (Adapted from Calo-Fernandez and Martinez-Hurtado 2012 [5])
In contrast to generic small-molecule drugs, biosimilars are similar, but not identical, to their reference medicinal product. [1, 6, 7]. Biologics are diverse medicines derived from living systems (e.g., bacteria, yeast, mammalian cells), and modifications of the primary, secondary, tertiary or quaternary structure of an approved biologic may influence the quality, safety and efficacy [1]. Reproducing the complex structure of biologics remains a significant challenge to manufacturers of follow-on biologics [5].
In addition, biologics are subject to post-translational modifications (PTMs). Among the numerous types of PTMs of proteins, common ones are glycosylation (which includes galactosylation, fucosylation, high mannose derivatives and sialylation), oxidation, phosphorylation, sulphation, lipidation, disulphide bond formation and deamidation [8–10]. While most of these changes occur during cellular protein synthesis and secretion (i.e., at the level of the upstream process) some occur in the downstream process (e.g., purification, formulation and storage). Changes to proteins as a result of PTMs can affect protein activity [8–10], therefore there is a need to characterise and understand them when manufacturing biological products [8]. Furthermore, PTMs may have an impact on the immunogenicity of biologics [8, 11, 12].
Accordingly, the standard ‘generic’ approach is insufficient for the development, regulatory evaluation and licensing of biosimilars [8, 13, 14].
On another level it is known that developers of biotherapeutics need to implement some changes to their manufacturing processes that could vary in scope from minor changes in raw materials or manufacturing equipment and rarely a more significant change such as a formulation change or cell line. Such changes can be pursued in response to control of material supplies, regulatory requirements, move to a different facility, upscale of production, to improve quality or to optimise efficiency of production. They are well guided by interactions with regulatory authorities as well as by the vast in-house knowledge available to the innovator on the product history and process.
Lack of understanding of the difference between evaluation of a manufacturing change on the one hand and evaluation of a newly developed biosimilar on the other, often leads to confusion and to some misinterpreted assumptions that, by implementing a change to the manufacturing process, an innovator is creating a biosimilar of its own.
The main objective of this manuscript is to provide a better understanding of both pathways (i.e., manufacturing change versus biosimilarity) and the consequent differences in regulatory requirements. The healthcare community needs to understand the difference between ‘comparability’ and ‘biosimilarity’, as there is a trend to blur the distinction between the two terms.
What is a Biosimilar?
In the EU, a ‘biosimilar’ is defined by the European Medicines Agency (EMA) as ‘A biological medicinal product that contains a version of the active substance of an already authorised original biological medicinal product (reference medicinal product). A biosimilar demonstrates similarity to the reference medicinal product in terms of quality characteristics, biological activity, safety and efficacy based on a comprehensive comparability’ [15].
The EMA also reinforces the point that the foundation of biosimilar development is wide-ranging structural and functional characterisation and comparison of the biosimilar and its reference product, with high similarity to the reference product in terms of physicochemical and functional characteristics, and clinical performance being paramount. They also state that although the amino acid sequence (primary structure) should be the same for the biosimilar and its reference product, small differences in the micro-heterogeneity pattern of the molecule might be permitted if suitably justified with regard to its potential effect on safety and efficacy [16].
Regulators and biosimilar developers have a good understanding of the challenges associated with the development of biosimilars without full access to the innovator’s information on the cell line, cultures, fermentation temperature, pH, growth media, filtration, purification etc. This knowledge gap, together with the understanding of the structural complexity and process sensitivity of biologics, explains the prudent approach and the width of regulatory requirements needed to demonstrate biosimilarity. Some biosimilar sponsors have struggled to design products with adequate structural similarity, and others have been challenged to confirm safety and efficacy in the required clinical studies. The EU biosimilarity pathway has therefore proven to be efficient in filtering out some potential, but inadequate, biosimilar development programs.
Next generation biosimilar marketing applications will include more complicated monoclonal antibodies and fusion proteins which are more structurally complex with inherent activity such as effector functions (antibody-dependent cell-mediated cytotoxicity [ADCC], complement-dependent cytotoxicity [CDC]) and may have higher likelihood for heterogeneity. Accordingly these applications are likely to be evaluated based on the totality of evidence and would mostly require analytical, non-clinical and clinical studies to demonstrate biosimilarity.
Biosimilar Applications in Europe
Omnitrope (somatropin) was the first product approved in the EU as a biosimilar in 2006. As of September 2014 the EMA has recommended the authorisation of more than 23 biosimilars within the product classes of human growth hormone, granulocyte colony-stimulating factor, erythropoietin, follitropin, TNF-inhibitor and insulin for use in the EU (Table I). Two biosimilar approvals were subsequently withdrawn by the manufacturers; one for Filgrastim ratiopharm (filgrastim) in April 2011 and one for Valtropin (somatropin) in May 2012 [17].
Demonstrating Biosimilarity
Evaluation of the molecular weight and size differences between biosimilars and reference medicinal product may be achieved with the use of size-exclusion chromatography and liquid chromatography electrospray ionisation mass spectrometry. Differences in secondary, tertiary and quaternary structures may be determined with highly-advanced analytical methods such as Fourier transformed infrared spectroscopy (FTIR), circular dichroism differential scanning calorimetry, isothermal calorimetry, nuclear magnetic resonance, hydrogen-deuterium exchange, mass spectroscopy and x-ray diffraction methods. Discrepancy in glycosylation patterns, aggregation and purity may be assessed by utilising high-resolution HILIC UPLC, capillary and gel electrophoresis, reversed-phase HPLC, mass spectrometry and size-exclusion chromatography [5, 8, 18, 19]. Unfortunately, there remain gaps in the sensitivities of these methods for certain structural variants, including denatured forms and higher order aggregates (i.e., particulates), such that clinically relevant differences in impurities may go undetected. Furthermore, there will be differences identified in the molecules (particularly PTMs and also an inability to predict which ones matter, or how much they matter. Hence, even with these high-technology capabilities there will still be questions about the differences identified in addition to still knowing there can be differences not detected. Therefore, these methods are not able to predict all biological activity in patients and even advanced in vivo models are unable to adequately predict human immunogenicity, as many immune responses are species-specific [5, 20]. Hence the need for functional assays, clinical trials and effective post-marketing pharmacovigilance for biosimilars – only these data will guarantee the same safety and efficacy as the originator product [5, 21].
When evaluating the efficacy of a complex biosimilar, such as a monoclonal antibody, primary clinical endpoints that focus on detecting potential differences in efficacy, rather than demonstrating efficacy per se, is more appropriate [22]. For example, objective response rate or change in tumour mass may be more suitable than overall survival (the gold standard for establishing patient benefit) when evaluating anticancer monoclonal antibodies [22–24]. With regard to safety, the risk of detecting new serious adverse effects after licensing is considered much lower for a biosimilar product than for a biologic containing a new or modified active substance, but immunogenicity remains an ongoing concern because this can be altered, even by apparently minor changes – this is why human immunogenicity data should always be required for the licensing of biological medicines, including biosimilars [22].
Changing Manufacturing Process
Manufacturers commonly implement changes in manufacturing processes over the life cycle of an approved biological product for a multiplicity of reasons that may include responding to regulatory commitments, tightening limits of specifications, up-scaling yield, changing raw materials or manufacturing equipment, reducing impurities and optimising production efficiency. Furthermore, the scope of changes may also vary from raw materials or equipment changes, to the rare but more extreme change in cell line or formulation.
However, while implementing these changes the innovator usually operates from a strong position of knowledge of the product development history, cell lines and different processes that are owned by the developer and are not accessible to the public or biosimilar manufacturers.
With that said it is also important to mention that biologics manufacturing process changes are well controlled by manufacturers and regulators, and guided by The International Conference on Harmonisation Q5E (ICH Q5E) [25].
Accordingly, regulators worldwide acknowledge that ‘comparability’ can lead to the conclusion that products have highly similar attributes before and after manufacturing process changes and that no adverse impact on safety or efficacy (including immunogenicity) of the drug product occurred. In other words, if evaluation of all analytical data before and after a change of a manufacturing step leads to the conclusion that quality attributes are virtually unchanged, no additional clinical studies will be required. In this context it is important to note that no absolute criteria (e.g., how much difference, if any, is allowed for a particular parameter) exist for such a conclusion but that this has to be considered on a case-by-case basis.
Hence, demonstrating comparability means assessing the potential impact of a specific change to the performance of the process and the quality of the product, and then conducting a risk-based comparison of product quality before and after the change. This exercise includes defining the structural parameters that should be assessed, and understanding how any changes in these parameters might impact safety and efficacy. The foundation of a comparability exercise includes physicochemical characterisations, biological assays, and stability degradation profiles of the biologic product; the need for additional animal and clinical studies (e.g., pharmacokinetic, efficacy, immunogenicity) depends on the ability to establish highly similar analytical profiles [25, 26] and the type or extent of manufacturing changes being implemented.
The importance of clinical investigations in determining the potential effects of manufacturing changes in cases where the risks cannot be fully evaluated, based solely on analytical studies, are highlighted by the unexpected clinical findings following a major manufacturing change seen with several biologics (see Table II) [27–31].
Are Comparability and Biosimilarity the Same?
Although ‘comparability’ (evaluation of an original biologic after a manufacturing change) and ‘biosimilarity’ (evaluation of a biosimilar made by a different manufacturer compared to its reference product) have related scientific and regulatory concepts [4, 22], there are important distinctions to be made.
Differences Between Biosimilarity and Comparability
Demonstrating that a new product is biosimilar to its reference product is much more complex than assessing the comparability of an originator before and after manufacturing changes made by the same manufacturer [32]. The major differences between the two pathways are represented in Table III.
Biosimilars all start with a new cell line since the manufacturers do not have access to the innovator’s cell line. Different cell lines have inherent differences affecting the glycosylation patterns [5, 21], and differing levels of product oxidation and aggregation changing a product’s three-dimensional structure [5, 33]. Such structural alterations may have consequences for patient outcomes, with undesired immunogenicity being of particular concern [5, 22, 34]. For example, differences in glycosylation, particularly the presence or absence of core fucose in the Fc domain of therapeutic antibodies, have been shown to affect the effector functions of ADCC and CDC [35–37]. In addition, aggregation has been recently implicated in the immunogenicity of epoetins [22, 31] and currently there is significant interest in how the biopharmaceutical industry monitors and evaluates aggregation in terms of detection, quantification and characterisation [8, 38, 39].
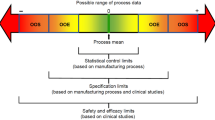
Figure 2 summarises what data are required for comparability exercises for manufacturing changes by the same manufacturer versus a biosimilar manufacturing process, and the 2012 FDA draft guidance entitled ‘Scientific Considerations in Demonstrating Biosimilarity to a Reference Product’ clearly states that ‘even though some of the principles described in ICH Q5E may also apply in the demonstration of biosimilarity, in general, more data and information will be needed to establish biosimilarity than would be needed to establish that a manufacturer’s post-manufacturing change product is comparable to the pre-manufacturing change product’ [32]. As can be seen, high-risk manufacturing changes require clinical trials, and biosimilar development typically goes beyond the highest risk of manufacturing change in terms of the comprehensive nature of the change and the limited access to historical data for the reference product. Biosimilar manufacturers must demonstrate analytical and preclinical biosimilarity to the reference medicinal product and are also required to conduct clinical studies for approval in the EU [16].

Data requirements for comparability versus biosimilarity.
Thus, declaring a reference biologic ‘comparable’ with its original state following a manufacturing change will be different from declaring a similar biological medicinal product ‘biosimilar’ to its reference product. Although the applicability of the comparability exercise described in ICH Q5E is explicitly limited to the same manufacturer process changes, the EU guidelines use the terms ‘similarity’ and ‘comparability’ interchangeably within the same regulatory documents, as they acknowledge that the two terms refer to the same scientific principle [8]. However, this is likely to create confusion in the clinical community and it is important for healthcare professionals to understand that originator molecules that undergo changes over time are not ‘biosimilars of themselves’.
From the intrinsic difference between the two approaches it is clear that upon introduction of a biosimilar in clinical practice healthcare professionals should realise that interchangeability, substitution or switching between biosimilar and reference product (or vice versa), needs to be considered as new, currently unexplored, situations. Consequently, in the absence of sufficient clinical evidence the appropriateness of such ‘actions’ should first be studied by adequately powered clinical studies (cf. FDA statements on the fact that biosimilarity does not imply interchangeability [32]). In addition, upon introduction of new biosimilars one should realise that adequate pharmacovigilance is of the utmost importance and the risk management plan should not (fully) rely on the longstanding experience of the reference medicinal product, nor on the possible longstanding experience gained with an earlier introduced biosimilar of the same reference medicinal product.
Conclusions
Although ‘comparability’ (evaluation of an original biologic after a manufacturing change) and ‘biosimilarity’ (evaluation of a new biosimilar against its reference product) are related scientific and regulatory concepts, there are important distinctions to be made, which should be understood by the healthcare community. Because a biosimilar can never exactly replicate the innovator manufacturer‘s development process, and because of the need to develop a new cell line (and the associated processes involved in that) a ‘knowledge gap’ in manufacturing data will always exist and should be acknowledged to avoid oversimplification of the stepwise biosimilar development pathway.
Abbreviations
- ADCC:
-
Antibody-dependent cell-mediated cytotoxicity (ADCC)
- CDC:
-
Complement-dependent cytotoxicity
- CHMP:
-
Committee for medicinal products for human use
- EMA:
-
European medicines agency
- FTIR:
-
Fourier transform infrared spectroscopy
- ICH Q5E:
-
The international conference on harmonisation Q5E
- PTM:
-
Post-translational modifications
References
Lee JF, Litten JB, Grampp G. Comparability and biosimilarity: considerations for the healthcare provider. Curr Med Res Opin. 2012;28:1053–8.
Schellekens H. Biosimilar therapeutics – what do we need to consider? NDT Plus. 2009;2 Suppl 1:i27–36.
Simoens S. Biosimilar medicines and cost-effectiveness. Clinicoeconomic Outcome Res. 2011;3:29–36.
McCamish M, Woollett G. The state of the art in the development of biosimilars. Clin Pharmacol Ther. 2012;91:405–17.
Calo-Fernández B, Martínez-Hurtado JL. Biosimilars: company strategies to capture value from the biologics market. Pharmaceuticals (Basel). 2012;5:1393–408.
Boone N, van der Kuy H, Scott M, et al. How to select a biosimilar. Eur J Hosp Pharm. 2013;20:275–86.
Blackstone EA, Fuhr Jr JP. Innovation and competition: will biosimilar succeed? Biotechnol Healthc. 2012;9:24–7.
Berkowitz SA, Engen JR, Mazzeo JR, Jones GB. Analytical tools for characterizing biopharmaceuticals and the implications for biosimilars. Nat Rev Drug Discov. 2012;11:527–40.
Farley AR, Link AJ. Identification and quantification of protein posttranslational modifications. Methods Enzymol. 2009;463:725–63.
Walsh G, Jefferis R. Post-translational modifications in the context of therapeutic proteins. Nat Biotechnol. 2006;24:1241–52.
Büttel IC, Chamberlain P, Chowers Y, et al. Taking immunogenicity assessment of therapeutic proteins to the next level. Biologicals. 2011;39:100–9.
Baker MP, Reynolds HM, Lumicisi B, Bryson CJ. Immunogenicity of protein therapeutics: the key causes, consequences and challenges. Self Nonself. 2010;1:314–22.
Beck A, Reichert JM. Approval of the first biosimilar antibodies in Europe: a major landmark for the biopharmaceutical industry. MAbs. 2013;5:621–3.
Chow S-H. Assessing biosimilarity and interchangeability of biosimilar products. Stat Med. 2012;32:361–3.
Guideline on similar biological medicinal products. European Medicines Agency 2013. Available at: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2013/05/WC500142978.pdf.
Guideline on similar biological medicinal products containing monoclonal antibodies: non-clinical and clinical issues. European Medicines Agency 2012. Available at: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/06/WC500128686.pdf.
European Public Assessment Reports. European Medicines Agency. Available at: http://www.ema.europa.eu/ema/index.jsp?curl = pages%2Fmedicines%2Flanding%2Fepar_search.jsp&mid = WC0b01ac058001d124&searchTab = searchByAuthType&alreadyLoaded = true&isNewQuery = true&status = Authorised&status = Withdrawn&keyword = Enter + keywords&searchType = name&taxonomyPath = &treeNumber = &searchGenericType = biosimilars.
Beck A, Wagner-Rousset E, Ayoub D, Van Dorsselaer A, Sanglier-Cianférani S. Characterization of therapeutic antibodies and related products. Anal Chem. 2013;85:715–36.
Stöckmann H, Adamczyk B, Hayes J, Rudd PM. Automated, high-throughput IgG-antibody glycoprofiling platform. Anal Chem. 2013;85:8841–9.
Roger SD, Mikhail A. Biosimilars: opportunity or cause for concern? J Pharm Pharm Sci. 2007;10:405–10.
Roger SD. Biosimilars: how similar or dissimilar are they? Nephrology (Carlton). 2006;11:341–6.
Weise M, Bielsky MC, De Smet K, et al. Biosimilars: what clinicians should know. Blood. 2012;120:5111–7.
Schneider CK, Kalinke U. Toward biosimilar monoclonal antibodies. Nat Biotechnol. 2008;26:985–90.
Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: quality issues (revision 1). European Medicines Agency 2012. Available at: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/05/WC500127960.pdf.
Q5E Comparability of biotechnological/biological products subject to changes in their manufacturing process. Rockville, MD: FDA/Center for Drug Evaluation and Research (CDER)/Center for Biologics Evaluation and Research (CBER), 2005. Available at: http://www.fda.gov/downloads/ RegulatoryInformation/Guidances/ucm128076.pdf.
Alsenaidy MA, Jain NK, Kim JH, et al. Protein comparability assessments and potential applicability of high throughput biophysical methods and data visualization tools to compare physical stability profiles. Front Pharmacol. 2014;5:39.
Avonex. Summary Basis for Approval. Rockville, MD: FDA/Center for Biologics Evaluation and Research (CBER)/Center for Drug Evaluation and Research (CDER), 2004. Available at: http://www.fda.gov/downloads/Drugs/ DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ ApprovalApplications/TherapeuticBiologicApplications/ucm086056.pdf.
Raptiva_ European Public Assessment Report Scientific Discussion. London: European Medicines Agency, 2004. Available at: http://www.ema.europa.eu/ docs/en_GB/document_library/EPAR_-_Scientific_Discussion/human/000542/ WC500057849.pdf.
Meeting of the Endocrinologic and Metabolic Drugs Advisory Committee (EMDAC) to Evaluate Myozyme (alglucosidase alfa) for the treatment of late onset Pompe Disease. Rockville, MD: FDA/Center for Biologics Evaluation and Research (CBER)/Center for Drug Evaluation and Research (CDER), 2008. Available at: http://www.fda.gov/ohrms/dockets/ac/08/briefing/2008- 4389b1-01-FDA.pdf.
Omnitrope. European Public Assessment Report. European Medicines Agency 2006. Available at: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_Scientific_Discussion/human/000607/WC500043692.pdf. Last accessed October 2014.
Seidl A, Hainzl O, Richter M, et al. Tungsten-induced denaturation and aggregation of epoetin alfa during primary packaging as a cause of immunogenicity. Pharm Res. 2012;29:1454–67.
Scientific Considerations in Demonstrating Biosimilarity to a Reference Protein Product; Draft Guidance; 9 February 2012. Rockville, MD: FDA/Center for Biologics Evaluation and Research (CBER)/Center for Drug Evaluation and Research (CDER), 2012. Available at: http://www.fda.gov/downloads/ Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291128.pdf.
Schellekens H. Immunogenicity of therapeutic proteins: clinical implications and future prospects. Clin Ther. 2002;24:1720–40.
Fernandes D. Demonstrating comparabiclity of antibody glycosylation during biomanufacturing. Euro Biopharma Rev 2005; 106–110.
Yu M, Brown D, Reed C, et al. Production, characterization, and pharmacokinetic properties of antibodies with N-linked mannose-5 glycans. MAbs. 2012;4:475–87.
Kanda Y, Yamada T, Mori K, et al. Comparison of biological activity among nonfucosylated therapeutic IgG1 antibodies with three different N-linked Fc oligosaccharides: the high-mannose, hybrid, and complex types. Glycobiology. 2007;17:104–18.
Chung S, Quarmby V, Gao X, et al. Quantitative evaluation of fucose reducing effects in a humanized antibody on Fcγ receptor binding and antibody-dependent cell-mediated cytotoxicity activities. MAbs. 2012;4:326–40.
Carpenter JF, Randolph TW, Jiskoot W, et al. Overlooking subvisible particles in therapeutic protein products: gaps that may compromise product quality. J Pharm Sci. 2009;98:1201–5.
Philo JS. A critical review of methods for size characterization of non-particulate protein aggregates. Curr Pharm Biotechnol. 2009;10:359–72.
Lawrence S, Lahteenmaki R. Public biotech 2013 – the numbers. Nat Biotechnol. 2014;32:626–32.
Biogen Gets Patent Extension on Multiple Sclerosis Drug Avonex to 2026. Pharmaceutical Processing. Available at: http://www.pharmpro.com/news/2009/09/biogen-gets-patent-extension-multiple-sclerosis-drug-avonex-2026.
ACKNOWLEDGMENTS AND DISCLOSURES
The Authors would like to thank Diane Rees who provided medical writing support for the preparation of this article which was funded by Amgen. The Authors did not receive any personal financial support from Amgen and the funding for writing assistance had no influence on the content of the article or the decision to submit it for publication.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Declerck, P., Farouk-Rezk, M. & Rudd, P.M. Biosimilarity Versus Manufacturing Change: Two Distinct Concepts. Pharm Res 33, 261–268 (2016). https://doi.org/10.1007/s11095-015-1790-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-015-1790-3