
Abstract
The fastest-growing category of biopharmaceuticals is known as a “biosimilar,” which refers to a biological medication that is replicated and sold at a lower price than the original biological product. Treatment with such biologics has additional benefits over conventional medication due to the involvement of a specific target, high efficacy, and fewer adverse effects. In addition to preventive use of biologics being used to avoid the return of the illness condition, diseases like cancer, autoimmune diseases, and inflammatory ailments can be cured. However, their exorbitant price places a heavy load on health care. Biosimilars are created as a result of the biologics’ patents expiring, with the intention of giving more patients access to cutting-edge treatment at a reasonable price. Biosimilars are not only identical to the reference standard used by the original creator, but also very identical in terms of efficacy and safety. The WHO sets internationally recognized norms and criteria that are widely accepted for the assessment of biotherapeutics as part of its obligation to confirm the global safety, efficacy and quality of products. The regulatory agencies have put a high priority on safety, and the development process follows a step-by-step methodology that is thoroughly explained in this chapter. Global regulations are contrasted, and suggestions are made for developing at the lowest possible expense. To accelerate the development process, the key components to establishing biosimilarity are outlined, including analytical and bioanalytical characterisation, nonclinical testing, clinical pharmacology testing, and clinical efficacy testing. There is also a summary of FDA-approved products. The goal of the current chapter is to deliver a brief compilation of biosimilars, their process of manufacturing, regulatory requirements, and to discuss both their current and prospective future roles in the field of medical sciences/biotherapeutics.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Biological therapeutic agents, also known as biologics, are a diverse group of substances that can be produced by cells or other living organisms through a number of different biological processes. Some examples of these processes include controlled transcription and translation process of protein synthesis, immunoglobulin technologies, and genetic engineering technologies (Humphreys 2022). Biologics have had a significant impact in the treatment of a variety of acute and chronic illnesses, including hormonal imbalance, a variety of inflammatory diseases, diabetes (Zhang et al. 2020) and autoimmune conditions, as well as cancer and haematological malignancies (Schiestl et al. 2020). In addition, the biologics sector of the pharmaceutical industry accounts for fifty percent of all products currently on the market for the administration and treatment of cancers. However, developing biological drugs requires a significant financial investment and a significant amount of time; as a result, the entire pharmaceutical industries is changing emphasis to the development of “Biosimilars.” Biosimilars are a type of biopharmaceutical product comparable with its purity, efficacy, and safety, is very similar to other reference products that are already on the market. In spite of the fact that their amino acid sequences are comparable to those of their reference products, may still biosimilars possesses distinguishing characteristics (De Mora 2015). Some of these characteristics include their three-dimensional structures, protein aggregation isoform profiles and glycosylation sites. However, various parameters, such as their therapeutic indication, route of administration, mechanism of action, dosage form, and strength, required to be comparable with their reference product (Ruppach 2020). In the year 1980 marked the beginning of the treatment and management of cancer utilizing biological substances as the primary method. Interferon alfa-2b (INTRONATM, Schering Corporation, Kenilworth, USA) was the pioneer industry of biopharmaceutical received approval from the Food and Drug Administration (FDA) in June 1986. It is presently marketed under seven different brand names. After that, in October of 2005, EMA- European Medicines Agency initiate the guidelines for biosimilars was the very first regulatory authority. In 2015, the US-FDA (United States Food and Drug Administration) granted approval to Filgrastim under the brand name Zarxio®. In addition, FDA released the guidelines affiliated with biosimilars in the year 2015. These guidelines address the quality and scientific aspects of demonstrating biosimilarity to the original product. Both acquiring a license to sell on the market and providing direction regarding how the FDA will determine whether or not two products are biosimilar were primary goals of this project. In addition, the FDA issued guideline documents in the years 2016 and 2018 with the aim of addressing challenges such as evidences of clinical pharmacological to substantiate biosimilarity and labelling guidelines within the confines of Sect. 351(k) of the Public Health Service Act (42 U.S.C. 262(k)) (Lemery et al. 2017).
Till the date, due to the many bottlenecks which not allowing biosimilars fully accepted in the clinical practice. For instance, the most significant risk associated with biosimilars is known as immunogenicity. Immune reactions have a propensity to cause adverse effects, the majority of which have an influence on how effectively the product works (Joshi et al. 2022). As a result, it is necessary to conduct ongoing assessments of the product's efficacy and safety during both the clinical trials and the post-marketing stages. Lack of knowledge is another challenge that must be overcome in the case of biosimilars. Research has shown that implementation of biosimilars into clinical practice, there is a need to raise awareness of the concept of biosimilars among medical professionals (De Mora 2015) (Fig. 1).

Correlation between biologics, originator biologics and biosimilars
This is necessary so that medical professionals should understand the biosimilar concepts based on trustworthy scientific data, generated from clinical trials. According to the findings of one research carried out by Cook and colleagues, around 26% of oncologists and approximate 21% of doctors only are aware with the concept of biosimilars. Although a number of regulatory authorities have established and published standardized guidelines along with approval procedures for biosimilars, the primary concern is still the transition from expensive biologics to less expensive biosimilars, particularly in terms of the safety of the treatment (Wiland et al. 2018).
2 Biosimilar Primer
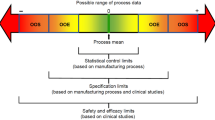
An identical biological drug developed by the originator is developed as a low-cost competitor to the first biological product, and this is what is known as a biosimilar. The biosimilars category is the one within the biopharmaceuticals industry that is expanding at the fastest rate (Humphreys 2022). The regulatory agencies have placed an extreme emphasis on safety, and the process of development takes a stepwise strategy, stated in the chapter. The regulations that are in place all over the world are analysed, and recommendations are made. In the interest of accelerating the development process, the essential components necessary to establish biosimilarity, such as analytical and bioanalytical assessment, nonclinical testing, clinical pharmacology testing, and clinical efficacy testing, have been broken down and explained (Ishii-Watabe and Kuwabara 2019). In addition, a summary of FDA-licensed products along with additional details on the studies that were sent in and an update on the status of biosimilars provided here (Fig. 2).

Biosimilar official definitions
Large and complicated pharmaceuticals known as biologic drugs have structures, physicochemical and biochemical properties, and manufacturing processes that directly affect their organic action. The development of biologics during the 1980s completely changed. How doctors handled their patients, particularly those who had diseases for which there was no effective treatment at the time (Iskit 2021). Ankylosing spondylitis, psoriasis, rheumatoid arthritis (Chadwick et al. 2018), and psoriatic arthritis are just a few of the chronic inflammatory illnesses that biologic medications have helped treat better, they also include some cancers (Joshi et al. 2022). Biologics have a high cost due to their complex manufacturing process, which places extra strain on the healthcare system. However, after their market-exclusivity patents have expired, Biosimilars have emerged as an alternative, cost-effective therapeutic option to reference product (the existing innovator biological therapies) and their active components. This is done to reduce healthcare spending and access to biological medicines promote greater. Notably, other words have also been used to refer to biosimilars, including follow-on biologics, similar biotherapeutic products, and biocomparables. The word “biosimilars” is now widely used in place of the latter. In comparison to generic versions of synthetic molecules, regulatory agencies around the globe demand a more and different involved procedure for the approval of biosimilars (Liu et al. 2022).
This is founded on a sophisticated set of tests for similarity called a biosimilarity exercise. Worldwide, a biosimilar must be comparably potent, pure, safe, and effective to the reference/ standard molecules based on a thorough process of comparability, with no clinically significant differences (Mysler et al. 2021). The regulatory pathway for proving biosimilarity is more stringent but shorter than that for an originator biologic in the Europe, United States, and globally based on WHO's standards. The regulation procedures are designed to determine if the new molecule is sufficiently comparable to the reference product in relation of purity, molecular structure, pharmacological characteristics, and clinical efficacy. It is common observation that slight variations can appear over time, even between batches of the same standard product (Lyu et al. 2022). Due to the potential for even minute differences to affect pharmacokinetics (PK), pharmacodynamics (PD), efficacy, and safety, a lot-to-lot assessment of biosimilars must be performed in comparison to the reference product as part of the similarity exercise. In most cases, a product can be considered a biosimilar only if all the criteria in the resemblance exercise are satisfied. Intended copies may exist when a molecule claims to be highly similar to an existing innovator molecule but fails to provide proof that it does so in accordance with the biosimilars regulatory pathway in its entirety. In addition to “biomimic” and “nonregulated biologic,” other words have been used to describe these items (Niazi 2022).
The diagnosis, prevention, treatment, and management of many serious and chronic illnesses have been revolutionized by the development of biological products. They vary from more conventional, small-molecule medications like acetaminophen or acetylsalicylic acid [aspirin] in that biologic agents are substances that are naturally present in your body, such as sugars, proteins, nucleic acids, or particular cells or tissues. This is what sets them apart from more traditional, small-molecule drugs. In the process of treating illnesses such as cancer, various concentrations or formulations of these naturally occurring substances can be used, which ultimately results in the creation of biologic medicines. Since biosimilars are a relatively new entity, we are having this conversation at a very opportune moment because the very first biosimilar to be approved for use did so in Europe in 2006 and in the United States only very recently, in 2015 (Kang et al. 2023). The use of biologics is not something novel; in fact, decades have passed since the discovery of human growth hormone, insulin, and agents that stimulate red blood cell production. Patients with diabetes were required to use insulin that had been extracted and purified from the pancreas of cows or swine prior to the year 1982. After that, researchers found a way to modify cells in the laboratory so that they would express insulin. With this new technique, insulin could be manufactured and distributed to patients. The disciplines of science known as genomics and proteomics, in addition to microarray, cell culture, and monoclonal antibody technology, are utilized in the process of developing biologics. As more genetic information and a deeper comprehension of disease processes have become available, the number of diseases that can be targeted by biologic therapies has expanded exponentially (Declerck et al. 2017). We are able to investigate the illness or condition more thoroughly and discover what is going on on the inside of each of our cells, in addition to the components that are responsible for making up each cell. Increasing our understanding of genetics and cellular processes has led to the discovery of potential new biologic (and drug) targets at each stage in the process of protein synthesis. This paves the way for brand new therapies that are extremely specific, which in turn leads to a better comprehension of diseases (Bachu et al. 2022).
The sequence of manufacturing biosimilars begins at the end, using a process that is referred to as “reverse engineering.” This is done rather than starting from what could be considered the beginning of a traditional drug development practice (which would be the beginning of the process) (Wolff-Holz et al. 2019). Before beginning the manufacturing process, this method ensures that the biosimilar molecule will be very similar to the reference molecule in relation of safety, quality, and effectiveness. This method is called step-by-step manufacturing. Once the period of exclusivity for the innovator product has passed, the biosimilar is then reverse-engineered from the innovator product. This is done because the manufacturing specifics of the innovator product are proprietary information and a closely guarded secret. This indicates that the developer of the biosimilar must first acquire the product that was developed by the innovator, then work backward from the completed product using sophisticated analytical tools and previous clinical knowledge in order to design their own procedure/ process that will result in a molecule that is highly similar to the molecule that was originally developed. In the context of biologics and biosimilars, the product is extremely dependent on the process. The structure, function, and quality of these medications are all directly attributable to the manufacturing process that was used to produce them. Because of these reasons, regulatory agencies like the FDA acknowledge that a biosimilar cannot be structurally identical to the product that it is compared to (the reference product) because of differences in the manufacturing process that change the final product (Lemery et al. 2017). The FDA requires that a biosimilar not be “clinically different” from an originator biologic rather than requiring that a biosimilar be fundamentally identical to an originator biologic.
3 Biosimilars Approval and Regulatory Requirements
In general, the regulatory requirements for the approval of biosimilars are the same across all three of these organizations: the WHO, the EMA, and Health Canada, as well as the guidelines released by the FDA (Administration 2018). All of these regulatory bodies demand a methodical, step-by-step process in order to determine whether or not two products are biologically comparable, despite the fact that these guidelines might have some inconsequential, sometimes even terminological, differences. Comparative evaluations incorporating analytical, nonclinical, and clinical studies are a standard part of these well-established regulatory processes (Barbier et al. 2022). The European Medicines Agency (EMA) has assumed a leadership role in the global regulatory community by initiating the first legislation. This move has allowed the EMA to assume a position of authority within the global regulatory community. When it comes to requiring identical head-to-head comparison research, it was the EMA that paved the way for other agencies to follow suit (Jimenez and Brake 2011). When all of the evidence from each evaluation has been compiled, biosimilarity can be taken into consideration. However, each stage of this procedure needs to be supported by the stage that came before it. 1. The first step is ensuring the biosimilar's quality is grounded in its structural and functional similarities to the reference product using analytical analyses that employ numerous orthogonal approaches. The biosimilar must be compared to the standard product to establish its resemblance. Next, the biosimilar needs approval from the appropriate regulatory agency. 2. The second step, the biosimilar agent must be shown to have the same target or physiologic process as the reference product and to be equally as hazardous through nonclinical trials. In order for the biosimilar agent to be approved by authorities, certain studies are required. 3. The evaluation of a biosimilar product reaches its climax with the third step, which is also the most crucial stage of the procedure. It is a specialized clinical study program that evaluates biosimilar regard to pharmacokinetics, clinical effectiveness and safety, as well as immunogenicity (Anon 2022).
4 Biosimilar Manufacturing Process
The production of biosimilars involves a process that involves sequentially demonstrating that they are comparable to the Reference Biologic through comprehensive characterization studies that reveal the molecular and quality characteristics of the Reference molecule of biologics (Schiestl et al. 2020). In order to protect the general public's health and to adhere to the standards set forth by international organizations, biosimilar medications need to be able to demonstrate that they are secure, effective, and of high enough quality. The nonclinical and clinical assessment of the biosimilar is probably little less than that which is required for the reference product; however, it is absolutely necessary to sufficiently test biosimilars. These requirements cannot be fulfilled for quality components because they require a demonstration of comparability, which is not possible. If the studies reveal that reference product and biosimilars not identical, they are significantly different in the aspect of efficacy, safety and purity, then the product will not be regarded as biosimilar. If the Reference product is used to treat more than one clinical condition, the biosimilar will only be eligible for all indications if it can be explained and if it satisfies the conditions that are outlined in the section that is titled “Extrapolation of Efficacy and Safety Data to other Indications.” In the event that the Reference Biologic is used to treat more than one clinical condition, the biosimilar will only be eligible for all indications if it can be explained (McKinnon et al. 2018).
4.1 Choosing a Reference Biological
Reference Biologic is a product created by an innovator that has been authorized following review of the entire dossier, which is essential for the creation of biosimilar. Every comparability experiment involving quality, nonclinical, and clinical factors must use the Reference Biologic (Kang et al. 2023). The following considerations should be considered while choosing reference biological product:
-
The Reference Biologic must be the creator's creation and must be licensed or authorized in India or one of the ICH nations. A complete set of safety, efficacy, and quality statistics should be used to license the Reference Biologic.
-
The same Reference Biologic should be used throughout the research and development process of biosimilars to support the product's efficacy, safety, and quality.
-
The Reference Biologic's dosage form, strength, and method should be used to administer the biosimilar. It must be demonstrated that the comparable biologic and the standard biologic's active medication component are equivalent.
4.2 Process of Manufacturing
The biosimilar producer must create an exact manufacturing process to create a product that is identical to the reference product in terms of identity, purity, and potency. The production of biosimilars must be verified in order to show that it is dependable and incredibly consistent. It is advised to use the same host primary cell line for the production of the biosimilar if the host primary cell line used to produce the Reference Biologic is made available. This prevents certain kinds of process-related impurities from being incorporated that might have a negative effect on clinical outcomes and immunogenicity, as well as the possibility of major changes in the product's quality attributes (QAs) (European Medicines Agency 1995; Galbraith 2017).
4.2.1 Upstream Process Development
-
The upstream process needs to be fully described, till down to the elements of the media used for cell development.
-
Data on reproducible fermentation from at least three batches during the pilot period. (Sufficient amount of purified product should be generated from the bache size, to generate nonclinical data).
-
Carefully controlling and monitoring the main process is necessary. Information on pH, temperature, dissolved oxygen, cell growth, product formation, the pattern of primary nutrient consumption, and agitation rate are just a few examples of the specific details about upstream process kinetics that can be noticed from consistency batches.
Volumetric productivity and product per litre yield are the metrics that will be used to determine concentration. Information to demonstrate the consistency of the specific protein yield, or the quantity of protein produced for each unit of cell mass, across all upstream batches. Showcase ways to scale up replication and economic growth in general (Kesik-Brodacka 2018).
4.2.2 Construction of Downstream Processes
-
Complete in-depth breakdown of the steps taken to gather the cells and remove the unwanted protein.
-
The amount of protein per lot that needs to be purified.
-
A precise breakdown of each stage that makes up a unit operation in the purification and recovery of proteins, as well as a quantitative evaluation of the quantity of protein recovered at each level (ICH 2010).
-
The consistency of the recovery over three separate batches of cell culture or fermentation that were purified from three separate batches of those processes of manufacturing.
-
Describe any variants that emerged following the translation.
-
Information on how to remove impurities, such as host cells, impurities associated with the manufacturing process, and variations and impurities related to the product that are believed to pose an immunogenicity risk. (EMEA 1997), research to show that the pathogen has been eradicated.
4.3 Quality Control Consideration
4.3.1 Analytical Methods
The appropriate methods of analysis should be selected in accordance with the crucial product quality attributes in order to show product comparability. Various orthogonal methods, like product aggregation, are frequently used to describe specific characteristics. Analytical method should be very sensitive even minor differences should be able to detect during the quality characterisation studies. If available Indian Pharmacopoeia should be referred for the quality attributes during the process of characterisations, properly qualified assays that are capable of reproducibility and dependability are needed. For batch release stability studies, in-process controls, and method validation in accordance with ICH standards (ICH Q27, Q5C8, and Q6B9) are required for quality attributes characteristics. The characterization studies should involve examples of the derived DNA product, control Reference Biologic, a known positive and negative control standard. Each quantitative experiment must be carried out a minimum of three times, and the results must be stated with mean and standard deviation, in order to give confidence in the statistical analysis’ accuracy. The proper representation of the statistical significance in the appropriate formats must be included with all characterization data (Galbraith 2017; ICH 2010).
4.3.2 Characterization of Product
Functional assays, physicochemical properties, biological activity, immunological properties, purity (including impurities related to the manufacturing process and the product itself), contamination, strength, and substance are among the studies used to describe biosimilars. It is crucial to follow the guidelines outlined in the ICH Q6B rule.
Physicochemical and Structural Properties: The estimation of the structures of the biosimilar components and the finished product, as well as the measurement of any other important physicochemical properties, should be considered when analysing physicochemical characteristics (Kirchhoff et al. 2017). Biosimilar amino acid sequence must be confirmed because it is expected that sequence will be same as that of the Reference biologics. The analytical methods must be precise and accurate to a level that is appropriate. Identification and measurement of the post-translational modifications that have occurred are required to capture in cases if they are occurring. If any significant differences are found, it is essential that they are thoroughly examined in nonclinical studies as well as clinical experiments and supported by scientific data.
Biological Activities: Biological products may contain a variety of biological activities. In such situations, suitable biological assays will be used to illustrate the activities, ascertain the mechanism of action of the product, and identify the clinical activity (Kirchhoff et al. 2017). A national or international Reference standard should be used to validate biological assays when it is suitable and available. An internal Reference standard must be developed in accordance with ICH suggestions if there are no such standards. The methods of the bioassay(s) may be used for tests if they are mentioned in the specification.
Immunological Properties: It is well known that the production process has an impact on the number of process-related impurities and post-translational modifications in biosimilars. Such characteristics changes leads to effect of product sensitivity or immunogenicity. Sufficient nonclinical studies need to perform to generate the data for affinity, specificity, binding, antibody products and different immunogenic parameters etc., and should be used in the evaluation process (Bielsky et al. 2020).
Purity and Impurities: The following must be assessed using a range of diagnostic methods when describing a biosimilar:
-
Specific product variants (e.g., isomers etc.)
-
Impurities related to products (e.g., oxidized, or aggregated)
-
Contaminants connected to host cells (e.g., host cell protein and DNA etc.)
-
Contaminants linked to processes (e.g., resin leachates or residual media components etc.).
Different nonclinical and clinical studies are chosen to conduct for assessment based on the existence of impurities in the biosimilar products (Galbraith 2017; ICH 2010; Jimenez and Brake 2011).
5 Specification
To guarantee a constant standard of product quality and that it is comparable to the Reference Biologic in accordance with the relevant guideline, the Specifications of Biosimilar (for drug substances and drug products) are organized around QAs (ICH Q6B). The analytical techniques used to characterize products and establish their comparability may or may not be the same as those employed to define product standards (Lemery et al. 2017).
6 Stability
The shelf life of drug substances and drug products, as well as the ideal storage conditions, should be determined using real-time stability tests, based on the relevant regulations (such as WHO TRS 822, ICH Q1 A(R2), and ICH Q5C). This is done to guarantee the validity of the stable study findings. Studies comparing the structure's accelerated and strained stability should be conducted side by side. The products similarity can be proven by showing degradation patterns that are similar to those of the reference biologic.
7 Comparison and Quality Analysis
It is absolutely necessary to compare the quality of Reference Biologic items to those of Biosimilar ones. Before beginning clinical trials, the applicant is required to first submit a complete quality dossier that is in accordance with the Central Drug Standard Control Organization (CDSCO) guideline for industry, 2008. This dossier must include the outcomes of a comparability exercise that compares the biosimilar to the Reference Biologic. Generating and reporting the comparability data of biosimilars with reference is required. It is advised to use the first three standardized quantities that have been used in a row to show consistency in the process of manufacturing. It is essential to confirm that the active drug substance in the biosimilar has a chemical makeup identical to that of the active drug substance in the Reference Biologic. If it is found that the similarities and variations between the Reference Biologic and the biosimilar may affect the efficacy and safety of the biosimilar, further studies might be necessary to characterization similarities and differences.
Critical quality attributes (CQA) and Key quality attributes (KQA) are subcategories of quality attributes of a biosimilar.
-
(1)
Critical quality attributes, or CQAs, are characteristics of a product that directly affect its therapeutic safety or efficacy. All the characteristics of the substance that directly affect the known mechanism(s) of action that it holds are included in this category. It is necessary to control CQAs within the parameters that must be established as appropriate based on the Reference Biologic.
-
(2)
“Key Quality Attributes,” or KQA for short, are Quality Attributes that are important from the standpoint of product and process consistency but are not known to have an impact on clinical safety and effectiveness. This category contains a molecule's characteristics that don't affect any of its known mechanisms of action. KQAs must be kept within allowable bounds, but these bounds must be carefully abided by the rules.
8 Nonclinical Studies Data Requirements
8.1 Prerequisite Before Conducting Nonclinical Studies
All of the RCGM's requirements, including proving that the process and product are consistent with one another, describing the product, and giving product specifications, must be met by the applicant. To obtain approval, the applicant must send the generated data to RCGM along with the following fundamental clinical data and nonclinical study protocols. After receiving approval from the RCGM, the toxicity research needs to start. The following details about the Reference Biologic and the Biosimilar may be regarded as some of the essential information:
Information on the fundamentals of Reference Biochemical.
-
Information pertaining to the medication, including but not limited to dosage, delivery mechanism, route of administration, rate of absorption and excretion, therapeutic index, dose response, and so forth.
-
The range of bioequivalence, if appropriate. if such knowledge can be obtained, tissue-specific localization.
-
The latest toxicology information for the Reference Biologic. Details about the Biosimilars’ core concepts
-
The developer must send the application to RCGM with the permission of the Institutional BioSafety Committee (IBSC) and, if applicable, the Institutional Animal Ethics Committee (IAEC). Toxicity study detail protocol and the location of execution of toxicity study along with the detail of personals involved like principal investigator, study director, histopathologist, quality assurance person and researchers should be provided by the applicant.
8.2 Early-Stage Research Pharmacodynamic and Toxicology Studies
Nonclinical studies are required before starting a clinical trial. Non-clinical research comparing the Reference Biologic and the Biosimilar should identify any discrepancies between the two. Therapeutic index, indication spectrum, and other clinical factors can all influence the methodology of preclinical studies. The nonclinical overview needs to be 100% behind the method being used. Unless there is a good reason not to, nonclinical research should be done with both the Reference Biologic and the final version of the Biosimilar that will be used in the clinic.
Research that is not done in a clinical setting needs to be done before starting any kind of clinical investigation. These comparative nonclinical investigations should have as their primary objective the identification of any differences between the Reference Biologic and the Biosimilar. The design of the research that is not done in humans can change depending on the clinical parameters, which can include the therapeutic index as well as the type and quantity of indications that are applied. The nonclinical summary needs to provide complete backing for the approach that was selected. Unless there is a compelling reason not to, nonclinical studies ought to be carried out utilizing both the Reference Biologic and the final version of the Biosimilar that is designed for clinical use. The biosimilar medication should have the same dosage form, dose, strength, and method of administration as the reference biologic, and any differences between the two should be explained. In order to conduct a nonclinical evaluation, the following investigations are required:
Studies on Pharmacodynamics
-
i
Studies conducted in vitro: To evaluate the comparability of biosimilar and reference biologics, cell-based in vitro bioassays (such as cell cytotoxicity, growth assays, neutralizing, and receptor binding assays) should be used.
-
ii
Studies conducted in vivo: Clinically relevant efficacy and potency activity of reference biologic, if correctly reflect by invitro assay, in such condition in vivo evaluation of biological/pharmacodynamic activity may not be required. In cases where in-vitro assays are unable to properly represent efficacy and potency, in vivo studies should be conducted.
9 Toxicological Studies
In vivo toxicity studies must include at least one repeat-dose toxicity trial in a pharmacologically relevant species using the intended route of administration. The applicant must provide a scientific justification for the choice of animal model(s) to be used, based on data from the scientific papers. Toxicology studies, with RCGM approval, must be performed using rodents or nonrodents. The only route of administration would be the one scheduled per schedule Y, regardless of whether the animal model used is pharmacologically relevant or not. Normally, the study would last at least 28 days, including a recovery period of 14 days. However, the time frame may change from case to case based on the dose and other factors (Table 1).
The procedures and study reports should include thorough descriptions of the following toxicity testing phases:
-
Measures done prior to euthanasia, such as weighing the patient or drawing blood.
-
The immediate aftermath of euthanasia, the necropsy, a thorough account, the weights of the organs, and the removal of organ samples for histopathology. Biochemical components, equipment, and English and metric phrases.
-
Haematology test methodology and factors (automated or manual). The use of statistical methods.
-
Bone marrow was either extracted, subjected to a smear analysis, or subjected to histopathology examination. Candidates should consider the following things in instances of histopathological observations:
-
It is necessary to note every observation that is believed to differ from the described normal histology, as well as how frequently it occurs in each group.
-
The protocol should include the recommended course of action in the event of premature death or morbidity.
-
If an animal's organs were not all examined, such as when only 4 of 5 livers were examined in 5 animals, the cause for this omission should be noted.
10 Immunogenicity
It is typically required to use a multi-tiered approach that includes immunoassays for screening and confirmatory purposes that identify binding ADAs (Anti-Drug Antibodies), followed by assays that estimate ADA magnitude and neutralization potential. Deviations from this approach must be justified.
The advantages and drawbacks of the assays and formats used today, as well as how to interpret the findings, have all been thoroughly reviewed. The method for testing antibodies and the assays chosen must be supported by the vendor. Assay control, validation and the establishment of cut-off lines for separating samples that contain antibodies from those that do not should both receive careful consideration. The pharmacological target and any remaining drug in the sample are two factors that could potentially interact with the matrix components. Corrective actions should be taken to lessen this influence. For instance, measures like providing time for the drug to be cleared from the circulation prior to sampling or incorporating steps for dissociating immune complexes and/or removing the drug can be used for drug interference (which frequently happens with samples taken from patients given mAbs). It is important to take precautions to make sure the application of such strategies does not jeopardize ADA diagnosis or patient care.
Comparative immunogenicity testing should be carried out when necessary using the same assay design and sampling frequency. Antibody testing is carried out using the therapeutic administered to the patient in order to evaluate immunogenicity in the development of novel drugs. Applying this idea to biosimilars makes it extremely difficult to create screening assays with comparable sensitivity for the two patient groups (biosimilar and RP) within the same trial. As a result, in the case of biosimilars, relative immunogenicity is frequently evaluated using a single assay that uses the biosimilar's drug component as the antigen for sample testing for both groups. All antibodies produced against the copycat can be found using this method. The manufacturer must show that the method(s) used are appropriate and provide evidence that they measure ADA to the RP and to the biosimilar to a comparable degree.
The potency assay of the product is typically the foundation for neutralization assays that represent the mechanism of action. In situations where the therapeutic binds to a soluble ligand and blocks its biological action, non-cell ligand-based assays are pertinent. The use of functional cell-based bioassays is advised for products with a high risk (for instance, those with non-redundant endogenous homologs and those for which effector functions are crucial). Regulatory officials may be consulted when guidance is required regarding the need for a neutralization assay and the best format to use (cell-based, ligand-based, or based on enzyme activity).
If deemed clinically relevant or in unique circumstances (such as the occurrence of anaphylaxis or the use of specific assay formats), further characterization of antibodies (for example, isotype) should be carried out while taking into consideration the immunogenicity profile of the RP. It is doubtful that the biosimilar would elicit an IgE response, for instance, if the same expression system is used as in the RP. In instances where technical issues with the initial assay occurred, it will be necessary to store patient samples for later testing under the proper storage conditions.
11 Application Data Requirements for Clinical Trials
According to the CDSCO industry guidelines from 2008, the applicant must submit an application for the conduct of clinical trials in addition to the data that was given in the nonclinical application (Lemery et al. 2017). The provided quality data must show that all KQAs are tightly controlled and there is no variation in the CQAs in order to initiate with the clinical evaluation.
Pharmacokinetic (PK) profile studies
The PK findings should be able to support the ensuing Phase III clinical development since the purported biosimilar would be proven to be similar to the Reference product. A pharmacokinetic study of the biosimilar may be conducted after the thorough characterization and comparability checks on quality attributes have been completed in a suitable number of:
Individuals who are generally recognized as.
-
a.
Healthy (NHV) and Normal/or
-
b.
Unhealthy or Patients.
Priority should be given to the following factors when planning comparative pharmacokinetic studies: the condition and disease to be treated, the route of administration, and the indication. Pharmacokinetics primary and secondary parameters such as clearance, volume of distribution, half-life, and linearity of PK profile.
Design factors that are appropriate include:
-
I.
Comparative, single-dose PK studies
-
II.
Cross over
-
III.
or parallel arm
-
IV.
Multiple dose, Comparative parallel arm steady state.
The research on Normal Healthy Volunteers (NHV) is conducted before the study on the safety and effectiveness of Phase III in a sequential development strategy. A sound rationale should be used when choosing the dosage. On the basis of how perceptive is to differences, the administration's course should be selected. It is crucial to specify and support the comparability limits and have a statistically sound justification for the sample size before starting the pharmacokinetic study. Mostly ELISA, HPLC or LC–MS/MS methods employed for the bioanalysis of biologics. The sensitive bioanalytical method needs to be validated to have acceptable specificity, sensitivity, limit of detection, limit of quantification in addition to adequate an accuracy and precision. It should be able to recognize and monitor the evolution of Biosimilar in a complex biological matrix made up of numerous distinct proteins.
Pharmacodynamics (PD) Study
The pharmacokinetic (PK) studies that were part of the biosimilar demanded that the PD studies also place a priority on comparative analysis. In order to establish the differences between biosimilars and the Reference Biologic, comparative, parallel arm, or cross-over PD studies need to be carried out in the most relevant cohort possible (De Mora 2015) (patients or healthy volunteers). People who are otherwise healthy can develop Parkinson's disease (PD) if a PD marker is present, unless doing so would be unethical due to the possibility for adverse effects and toxicity, such as those caused by oncology drugs. Before beginning the demonstration of similarity in PD parameters, it is essential to establish acceptance ranges and to explain them in a way that is easy to understand. The clinical significance of the surrogate markers being used and their clinical validation are prerequisites for the PD research variables (Barbier et al. 2019). It is possible to combine pharmacodynamic and pharmacokinetic studies; in this situation, the PK/PD relationship needs to be described. A phase III clinical study can be combined with a PK study if there are no PD markers accessible but there is the possibility of conducting PK on patients.
Confirmatory safety and efficacy research
This is necessary in order to eliminate any potential risks that may have been overlooked. A further comparative safety and effectiveness trial is not required except in very specific circumstances, such as when comparing a biosimilar to a Reference Biologic at the analytical, non-clinical, and PK/PD levels and finding no residual uncertainties in the findings of those comparisons.
Lack of a study on efficacy and safety
If all of the following conditions are met, study can be excluded like, the confirmatory clinical safety and efficacy study:
-
i.
Physicochemical and in vitro methodologies can be used with high confidence to characterize the structural and functional comparability of biosimilars and the Reference Biologic.
-
ii.
The biosimilar is equivalent to the Reference Biologic in all preliminary evaluations.
-
iii.
The applicant supported the efficacy/PD measurements and safety measurements, including meaningful immunogenicity assessments over an appropriate time period, in the PK/PD study, which demonstrated that clinically validated PD markers could be compared (Lemery et al. 2017).
The confirmatory clinical safety and efficacy study cannot be omitted, particularly for high-molecular-weight biologics such as monoclonal antibodies. If the safety and efficacy research is waived based on convincing PK/PD data and comparable quality non-clinical data, all of the approved indications for the reference product may be used.
Information on Safety and Immunogenicity
It is required that a clone go through both a pre-approval safety evaluation as well as a post-approval safety assessment. This provides details about the product's susceptibility to certain conditions. The primary goal of the safety statistics that are required for pre-approval is to guarantee that there will be no unforeseeable problems with the product's safety. In addition to the data that have already been published on the Reference Biologic, are necessary in order to guarantee that there are no unanticipated safety concerns. A comprehensive method must be supplied, in addition to the suggested non-comparative post-marketing study, in order to evaluate the biosimilar's level of safety.
The Extrapolation of Efficacy and Safety Information to Other Indications
The safety and efficacy statistics of a specific clinical indication of a biosimilar may be extrapolated to other clinical indications if the following criteria are met. This is due to the fact that clinical research has been done on the specific clinical indication in issue.
-
Validation and demonstration of quality comparability to Reference Biologic.
-
Comparability with regard to initial assessment in reference to the Reference Biologic has been shown.
-
One indication has shown that the treatment is clinically safe and efficacious.
-
For the treatment of other therapeutic indications, the mechanism of action is unaltered.
-
The involved receptor(s) are identical to those found in other therapeutic applications.
12 Data Requirements for Market Authorization Applications
For market authorization the application must adhere to the guidelines outlined in the CDSCO industry advice document from 2008. Information on comparability of quality must also be provided with the proper justification in cases where commercial manufacturing is conducted at a different scale or using a different process than that used to produce batches for phase III clinical trials. Each of these instances will be handled separately (Requirements and Authorization 2016).
13 Pharmacovigilance Strategy
It is unlikely that the rare adverse events will occur because the clinical studies on Biosimilars that were performed prior to market authorization were so small-scale. Post- market surveillance with comprehensive pharmacovigilance protocol should be designed by the applicant or manufacturer, to evaluate the safety of biosimilar (Baldo et al. 2018). Periodic Safety update report (PSURs), should include regular safety update report which is submitted every six months for the initial two years after the approval of biosimilar. For the later few years the PSURs, annually need to submitted to DCGI office as per the Schedule Y.
14 Post-marketing Analysis (Phase IV Study)
Data collected through a pre-defined single arm study with typically more than 200 evaluable patients and compared to historical data of the Reference Biologic after market approval for additional safety information is used to finally further reduce the residual risk of the biosimilar. If there are no exceptional circumstances, the research ought to be finished no later than two years after receiving either a manufacturing license or a marketing authorization, at the very latest. Because ensuring participant safety is the primary focus of the post-marketing phase IV research, the following factors need to be taken into account when designing the protocol:
-
The top priority is safety.
-
Secondary outcomes include immunogenicity and efficacy.
The findings of post-marketing studies should be submitted to DCGI due to the limited scope of clinical studies on Biosimilars conducted prior to market authorization. Post-market study plans should be included in pharmacovigilance plans, and CDSCO should be updated on the studies.
15 Exceptions
The size of the clinical trial community can be reduced in the case of a biosimilar that can be assessed for rare diseases depending on the rarity, severity, and limited availability of therapeutic alternatives (Table 2).
Conclusion
Despite the many challenges that must be overcome, research and development of biosimilars persist. Biosimilarity must be established using a personalized strategy across the whole product development process, beginning with the structural and functional assessment and progressing through nonclinical and clinical research. This must be completed before any inferences about biosimilarity may be made. It has become abundantly evident that a new approach is required for the creation of biosimilars. This is especially true when deciding upon CQAs and study outcomes for preclinical and clinical research. The healthcare business faces a difficulty with the launch of biosimilars because of the need to systematically understand and comprehend the scientific basis of resemblance to the reference product. The introduction of biosimilars raises this difficulty. When it comes to generating biosimilars, clinical studies are a blunt instrument, while analytical evaluation is a tool that is substantially more sensitive in determining similarity. This is something that should be made known to the widest possible audience.
References
Administration TG (2018) Biosimilar medicines regulation. http://www.ema.europa.eu/ema/index.jsp?curl=pages/special_topics/document_listing/document_listing_000318.jsp
Anon (2022) Annex 3—Guidelines on evaluation of biosimilars. 977:1–44
Bachu RD, Abou-Dahech M, Balaji S, Boddu SHS, Amos S, Singh V, Babu RJ, Tiwari AK (2022) Oncology biosimilars: new developments and future directions. Cancer Rep. 5(11):1–20. https://doi.org/10.1002/cnr2.1720
Baldo P, Fornasier G, Ciolfi L, Sartor I, Francescon S (2018) Pharmacovigilance in oncology. Int J Clin Pharm 40(4):832–841. https://doi.org/10.1007/s11096-018-0706-9
Barbier L, Declerck P, Simoens S, Neven P, Vulto AG, Huys I (2019) The arrival of biosimilar monoclonal antibodies in oncology: clinical studies for trastuzumab biosimilars. Br J Cancer 121(3):199–210. https://doi.org/10.1038/s41416-019-0480-z
Barbier L, Mbuaki A, Simoens S, Declerck P, Vulto AG, Huys I (2022) Regulatory information and guidance on biosimilars and their use across Europe: a call for strengthened one voice messaging. Front Med 9(March):1–16. https://doi.org/10.3389/fmed.2022.820755
Bielsky MC, Cook A, Wallington A, Exley A, Kauser S, Hay JL, Both L, Brown D (2020) Streamlined approval of biosimilars: moving on from the confirmatory efficacy trial. Drug Discov Today 25(11):1910–1918. https://doi.org/10.1016/j.drudis.2020.09.006
Chadwick L, Zhao S, Mysler E, Moots RJ (2018) Review of biosimilar trials and data on etanercept in rheumatoid arthritis. Curr Rheumatol Rep 20(12). https://doi.org/10.1007/s11926-018-0799-0
De Mora F (2015) Biosimilar: what it is not. Br J Clin Pharmacol 80(5):949–956. https://doi.org/10.1111/bcp.12656
Declerck P, Danesi R, Petersel D, Jacobs I (2017) The language of biosimilars: clarification, definitions, and regulatory aspects. Drugs 77(6):671–677. https://doi.org/10.1007/s40265-017-0717-1
European Medicines Agency (1995) Production and quality control of medicinal products derived by recombinant DNA technology. In: 3AB1a, pp 1–11
Galbraith D (2017) Ich Q5a. In: ICH quality guidelines, pp 311–335. https://doi.org/10.1002/9781118971147.ch10
Humphreys SZ (2022) Real-world evidence of a successful biosimilar adoption program. Future Oncol 18(16):1997–2006. https://doi.org/10.2217/fon-2021-1584
ICH (2010) Quality of biotechnological/biological products: derivation and characterization of cell substrates. Fed Reg 75(180):56928–56935
Ishii-Watabe A, Kuwabara T (2019) Biosimilarity assessment of biosimilar therapeutic monoclonal antibodies. Drug Metab Pharmacokinet 34(1):64–70. https://doi.org/10.1016/j.dmpk.2018.11.004
Iskit AB (2021) Key concepts in biosimilar medicines: what physicians must know. North Clin Istanb 9(1):86–91. https://doi.org/10.14744/nci.2021.84669
Jimenez AG, Brake B (2011) Biosimilars in the European union—regulatory perspectives. In: ICH GCG ASEAN training workshop
Joshi D, Khursheed R, Gupta S, Wadhwa D, Singh TG, Sharma S, Porwal S, Gauniyal S, Vishwas S, Goyal S, Gupta G, Eri RD, Williams KA, Dua K, Singh SK (2022) Biosimilars in oncology: latest trends and regulatory status. Pharmaceutics 14(12). https://doi.org/10.3390/pharmaceutics14122721
Kang H, Wadhwa M, Knezevic I, Ondari C, Simao M (2023) WHO guidelines on biosimilars: toward improved access to safe and effective products. Ann N Y Acad Sci 96–103. https://doi.org/10.1111/nyas.14965
Kesik-Brodacka M (2018) Progress in biopharmaceutical development. Biotechnol Appl Biochem 65(3):306–322. https://doi.org/10.1002/bab.1617
Kirchhoff CF, Wang XZM, Conlon HD, Anderson S, Ryan AM, Bose A (2017) Biosimilars: key regulatory considerations and similarity assessment tools. Biotechnol Bioeng 114(12):2696–2705. https://doi.org/10.1002/bit.26438
Lemery SJ, Ricci MS, Keegan P, McKee AE, Pazdur R (2017) FDA’s approach to regulating biosimilars. Clin Cancer Res 23(8):1882–1885. https://doi.org/10.1158/1078-0432.CCR-16-1354. Epub 2016 Dec 29. PMID: 28034906
Liu JW, Yang YH, Wu N, Wei JF (2022) Biosimilar monoclonal antibodies in China: a patent review. Bioengineered 13(6):14503–14518. https://doi.org/10.1080/21655979.2022.2090206
Lyu X, Zhao Q, Hui J, Wang T, Lin M, Wang K, Zhang J, Shentu J, Dalby PA, Zhang H, Liu B (2022) The global landscape of approved antibody therapies. Antib Ther 5(4):233–257. https://doi.org/10.1093/abt/tbac021
McKinnon RA, Cook M, Liauw W, Marabani M, Marschner IC, Packer NH, Prins JB (2018) Biosimilarity and interchangeability: principles and evidence: a systematic review. BioDrugs 32(1):27–52. https://doi.org/10.1007/s40259-017-0256-z
Mysler E, Azevedo VF, Danese S, Alvarez D, Iikuni N, Ingram B, Mueller M, Peyrin-Biroulet L (2021) Biosimilar-to-biosimilar switching: what is the rationale and current experience? Drugs 81(16):1859–1879. https://doi.org/10.1007/s40265-021-01610-1
Niazi SK (2022) Molecular biosimilarity—an AI-driven paradigm shift
Requirements R, Authorization M (2016) Guidelines on similar biologics : regulatory requirements for marketing authorization in
Ruppach H (2020) Viral safety for biotherapeutics and biosimilar. Drug Discov Today Technol 37:23–29. https://doi.org/10.1016/j.ddtec.2020.08.001
Schiestl M, Ranganna G, Watson K, Jung B, Roth K, Capsius B, Trieb M, Bias P, Maréchal-Jamil J (2020) The path towards a tailored clinical biosimilar development. BioDrugs 34(3):297–306. https://doi.org/10.1007/s40259-020-00422-1
Wiland P, Batko B, Brzosko M, Kucharz EJ, Samborski W, Swierkot J, Więsik-Szewczyk E, Feldman J (2018) Biosimilar switching—current state of knowledge. Reumatologia 56(4):234–242. https://doi.org/10.5114/reum.2018.77975
Wolff-Holz E, Tiitso K, Vleminckx C, Weise M (2019) Evolution of the EU biosimilar framework: past and future. BioDrugs 33(6):621–634. https://doi.org/10.1007/s40259-019-00377-y
Zhang RM, Puri R, McGill JB (2020) Update on biosimilar insulins: A US perspective. BioDrugs 34(4):505–512. https://doi.org/10.1007/s40259-020-00431-0
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Manwatkar, S., Kumar, B. (2023). Biosimilars: Promising and Rapidly Emerging Biotherapeutics. In: Kulkarni, S., Haghi, A.K., Manwatkar, S. (eds) Novel Technologies in Biosystems, Biomedical & Drug Delivery. Springer, Singapore. https://doi.org/10.1007/978-981-99-5281-6_3
Download citation
DOI: https://doi.org/10.1007/978-981-99-5281-6_3
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-99-5280-9
Online ISBN: 978-981-99-5281-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)