
Previous studies at the V. V. Zakusov Science Research Institute of Pharmacology created a dimeric dipeptide mimetic of the most exposed fourth loop of nerve growth factor (NGF), bis-(N-monosuccinyl-L-glutamyl-Llysine) hexamethylenediamide (GK-2), which activates specific TrkA receptors and has neuroprotective activity in vitro (at 105 to 109 M) and in vivo (0.1 – 10 mg/kg, i.p., or p.o.). The present report describes the construction and synthesis of a mimetic of the first loop of NGF based on the β-turn (-Lys32-Gly33-Lys34-Glu35-), bis-(N-aminocaproyl-glycyl-L-lysine) hexamethylenediamide (GK-6). The structure of GK-6 preserves the central dipeptide fragment of the β-turn -Gly33-Lys34-, the Lys32 residue being substituted by its bioisostere – a 6-aminocaproic acid residue - and the dimeric structure of NGF being reproduced by dimerization at the C-terminal using a bivalent hexamethylenediamine spacer. Structure-activity relationships of GK-6 were studied by sequential substitution of the side groups of the peptide by hydrogen to produce bis-(N-acetyl-glycyl-Llysine) hexamethylenediamide (GTS-611) and bis-(N-aminocaproyl-glycyl-glycine) hexamethylenediamine (GTS-613). Mimetic GL-6 and its analogs GTS-613, which contains the N-aminocaproyl radical, had neuroprotective effects at concentrations of 106 and 105M in conditions of oxidative stress in HT-22 neuron cultures. Dipeptide GTS-611, in which the aminocaproyl fragment was replaced by an acetyl residue, had no neuroprotective activity in these conditions, pointing to the importance of Lys32 in NGF for this property. In contrast to the loop 4 mimetic GK-2, the loop 1 mimetics GK-6 and GTS-611 showed differentiation-inducing activity on PC12 cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Previous studies at the V. V. Zakusov Science Research Institute of Pharmacology created a dimeric dipeptide mimetic based on the structure of the β-turn of the most exposed loop 4 of nerve growth factor (NGF), i.e., bis-(N-monosuccinate-L-glutamyl-L-lysine) hexamethylenediamide (GK-2) [1 – 5]. GK-2 was shown to be able to activate TrkA receptors and the PI3/Akt signal pathway [6, 7]. In vitro experiments showed that micronanomolar concentrations of GK-2 displayed the neuroprotective activity intrinsic to NGF [8]. In in vivo experiments, the neuroprotective properties of GK-2 were demonstrated in experimental models of Alzheimer’s disease, Parkinson’s disease, and ischemic cerebral stroke [7]. In contrast to full-size NGF, GK-2 had no differentiation- inducing activity [1].
We report here the preparation of three mimetics of another loop of NGF, loop 1. Their neuroprotective and differentiation- inducing activities were studied in vitro. Construction of the novel mimetics was based on published data on the crystal structure of the NGF homodimer (pdb ID: 1btg) [9]. Loop 1 can be seen, like loop 4, to be exposed, which may make a contribution to the binding of the neurotrophin to TrkA receptors. The basis for modeling was the sequence of the most exposed part of NGF loop 1, i.e., its β-turn (-Lys32-Gly33-Lys34-Glu35-). The structure of the mimetic preserved the central dipeptide fragment of the β-turn (Gly33-Lys34) which, on the basis of geometrical considerations, may penetrate more deeply into the receptor binding zone and be most completely recognized by it. The preceding amino acid residue, Lys32, was substituted by its bioisostere -a 6-aminocaproic acid residue. Based on data on the interactions of neurotrophins with Trk receptors in the dimeric form, the two mimetics of the β-turn were dimerized headto-head with a hexamethylenediamine spacer. This yielded the dimeric dipeptide NGF loop 1 mimetic GK-6, i.e., bis-(N-aminocaproyl-glycyl-L-lysine) hexamethylenediamide. The effects of the nature of the N-acyl and side radical of lysine on the activity of GK-6 were studied by constructing two analogs of GK-6 (Fig. 1): bis-(N-acetyl-glycyl-L-lysine) hexamethylenediamide (GTS-611) and bis-(N-aminocaproyl-glycyl-glycine) hexamethylenediamine (GTS-613). The neuroprotective activities of these compounds were studied in vitro on HT-22 neuronal cultures in conditions of oxidative stress induced by H2O2. In addition, the differentiation-inducing effects of mimetics GK-6 and GTS-611 were studied in PC12 cells.

Construction of NGF mimetics GK-6, GTS-611, and GTS-613.
EXPERIMENTAL CHEMICAL SECTION
These studies used commercially available L-amino acids and there derivatives from Sigma and Fluka. Melting temperatures were determined on an Optimelt MPA100 instrument (Stanford Research Systems, USA) in open capillaries without correction. 1H and 13C NMR spectra were recorded on the ppm scale using a Bruker Fourier 300 HD spectrometer (300 and 75 MHz respectively) in DMSO-d6 solution with tetramethylsilane (0 ppm) as the internal standard. Signal assignments were made on the basis of analysis of one-dimensional and two-dimensional homonuclear 1H-1H COSY spectra and heteronuclear 1H-13C COSY spectra (HSQC and HMBC).
Specific optical rotation was recorded on an ADP 440 automatic polarimeter (Bellingham + Stanley Ltd., UK). TLC was run on glass DC Kieselgel 60 G/F254 plates (Merck, Germany) in the following solvent systems: chloroform and methanol, 6:1 (A), chloroform and acetone, 2:1 (B); chloroform, methanol, water, and acetic acid, 15:10:2:3 (C), chloroform, methanol, water, and acetic acid, 8:10:2:3 (D), n-butanol, acetic acid, water, 3:1:1 (E), chloroform, methanol, water, acetic acid, 10:15:2:3 (F), benzene and methanol (I), and dioxane and water, 9:1 (K).
Amine-containing compounds were detected with ninhydrin, compounds containing amide groups were detected with the chlorotoluidine test, compounds with open carboxyl groups were detected with bromocresol green, and compounds containing aromatic groups were detected in UV light.
DMF was purified by redistillation over ninhydrin. Diethyl ether was stored over solid NaOH. Ethyl acetate, dichloromethane, chloroform, benzene, acetone, hexane, petroleum ether, methanol, and ethanol (all reagent grade) were used without additional purification. Compounds obtained previously in our laboratory were: Z-Gly-OH (compound 1), Z-Gly-OSu (2), (Z-Gly-LLys(Boc)-NH-)2(CH2)6(8), (H-Gly-L-Lys-(Boc)-NH-)2-(CH2)6(9) [17]; Z-L-Lys(Boc)-OSu (5), (Z-L-Lys(Boc)-NH-)2(CH2)6(6), (H-L-Lys(Boc)-NH-)2(CH2)6(7) [18].
Synthesis of Starting Compounds
N- tert -Butyloxycarbonyl-6-aminohexanoic acid, (Boc-NH-(CH 2 ) 5 -COOH) (3), was prepared as described in [11]. A solution of 5.72 g of NaHCO3 in 57.2 ml of water and 115 ml of isopropyl alcohol were added sequentially to a solution of 15.00 g (114.4 mmol) of 6-aminocaproic acid in 115 ml of 1 M NaOH with mixing. Di-tert-butylpyrocarbonate (35 ml 139.5 mmol) was then added to the reaction mix in portions over 20 min at room temperature and the resulting reaction was mixed for 18 h. The reaction was treated with 150 ml of water and extracted with 2 x 110ml of petroleum ether (to remove excess di-tert-butylpyrocarbonate). The aqueous solution was acidified with 1 M HCl at 0 – 5°C to pH ~3.0 and product was extracted with 3 × 200 ml of ethyl acetate. The ethyl acetate solution was washed with 200 ml of water and 190 ml of saturated NaCl solution, dried over Na2SO4, and evaporated. This yielded 21.82 g (~82%) of product as a yellow material, which was dried in vacuo and used without further purification. Rf 0.74 (A), Rf 0.65 (B), Rf 0.79 (ethyl acetate). The 1H NMR spectrum, DMSO-d6, δ, ppm, was: 1.19 – 1.26 (m, 2H, CγH2 Aca), 1.30 – 1.36 (m, 11H, CδH2 Aca, -O(CH3)3), 1.42 – 1.52 (m, 2H, CβH2 Asa), 2.17 (t, J 7.4 Hz, 2H, CαH2Aca), 2.87 (m, J 5.4 Hz, 2H, CεH2 Aca), 6.78 (t, 3J 5.40 Hz, 1H, HN-(CH2)5-CO Aca), 12.00 (s, 1H, COOH).
tert -Butyloxycarbonyl-6-aminohexanoic acid N-oxysuccinimide ester, (Boc-NH-(CH 2 ) 5 -COOSu) (4). N-hydroxysuccinimide (11.94 g, 0.1037 mol) and 80 ml of ethyl acetate were sequentially added to a solution of 21.82 g (0.0943 mol) of Boc-Aca-OH in 150 ml of ethyl acetate with mixing. The reaction mix was cooled to 0 – 5°C and 22.38 g (0.1084 mol) of DCHC in 120 ml of ethyl acetate was added. The reaction was mixed with cooling for 1.5 h and at room temperature for 20 h. Oxalic acid (1.26 g) was then added (to remove excess DCHC) and after 1 h the precipitate of DCHM was collected by filtration and washed on the filter with 100 ml of ethyl acetate. The filtrate was evaporated in vacuo at 45°C. The resulting oil was triturated under diethyl ether and held overnight in a refrigerator; the resulting precipitate was collected by filtration, thoroughly squeezed, and dried over CaCl2 in a desiccator. The yield was 27.7 g (~90%) as a white crystalline substance. Rf 0.88 (A), Rf 0.82 (ethyl acetate); Tm 81 – 85°C. The 1H NMR spectrum, DMSO-d6, δ, ppm was: 1.31 – 1.37 (m, 13H, CγH2 CδH2 Aca, -O(CH3)3), 1.56 – 1.65 (m, 2H, CβH2 Aca), 2.65 (t, J 7.4 Hz, 2H, CαH2 Aca), 2.80 (s, 4H, -CH2-CH2-OSu), 2.89 (m, J 5.4 Hz, 2H, CεH 2 Aca), 6.79 (t, J 5.4 Hz, 1H, NH Aca).
Acetic acid N-oxysuccinimide ester (Ac-OSu) was prepared as described in [19] from 13.4 g (0.1164 mol) of N-hydroxysuccinimide with a yield of 96%. Rf 0.85 (A), Rf 0.56 (ethyl acetate), Rf 0.70 (K); Tm was 130 – 134°C. The 1H NMR spectrum, DMSO-d6, δ, ppm was: 2.34 (s, 3H, CH3CO-), 2.80 (m, 4H, -CH2CH2-OSu). Published data [19]: Tm 133 – 134°C.
N- tert -Butyloxycarbonyl-glycine (Boc-Gly-OH) (12) was prepared as described in [11] with a yield of 81%. Rf 0.75 (C), Rf 0.91 (E), Rf 0.25 (B); Tm 88 – 91°C. Published data [20]: Tm 87 – 88°C.
Tert -Butyloxycarbonyl-glycine acid N-oxysuccinimide ester (Boc-Gly-OSu) (13) was prepared as described in [18] with a yield of 81%. Rf 0.76 (C), Rf 0.75 (B), Rf 0.81 (ethyl acetate); Tm 131 – 134°C. Published data [21]: Tm 155°C.
Synthesis of Bis-(6-aminocaproyl-glycyl-L-lysine) Hexamethylenediamide, GK-6
Bis-(N- tert -butyloxycarbonyl-6-aminocaproyl-glycyl-Ne- tert -butyloxycarbonyl-lysine hexamethylenediamide (Boc-HN-(CH 2 ) 5 -CO-Gly-L-Lys(Boc)-NH-)2(CH 2 ) 6 (10). A solution of 4.73 g (14.4 mol) of Boc-Aca-OSu in 45 ml of DMF was added to a solution of 4.50 g (6.6 mmol) of (H-Gly-L-Lys-(Boc)-NH-)2(CH2)6 (9) in 40 ml of DMF and the reaction was mixed for 12 h at room temperature, after which 0.16 ml of N,N-dimethyl-1-aminopropane (DMAPA) was added and mixed for 30 min. The reaction mix was diluted with 200 ml of ethyl acetate and 150 ml of water and the aqueous layer was extracted with 200 ml of ethyl acetate. The ethyl acetate solution was washed with 100 ml of water and evaporated, and the residue was taken up in 200 ml of diethyl ether and held overnight in a refrigerator; the precipitate was collected by filtration and washed with diethyl ether (30 ml). The material was dried in a desiccator over CaCl2 and the yield was 6.2 g (~85%) of chromatographically homogeneous product as a white crystalline substance. Rf 0.90 (C), Rf 0.81 (E), Rf 0.6 (A); Tm was 148 – 155°C. The 1H NMR spectrum, DMSO-d6, δ, ppm was: 1.23, 1.37, 1.45 – 1.61 (four m, 64H, 2 CβH2 CγH2 CαH2 Lys,-HN-CH2-(CH2)4-CH2-NH-, 2 CαH2 CβH2 CγH2 Aca, 4-O(CH3)3, Boc), 2.11 (t, J 6.8 Hz, 4H, 2 CαH2 Aca), 2.87 (m, 8H, 2 CεH2 Lys, 2 CεH2 Aca), 3.02 (m, 4H, HN-CH2-(CH2)4-CH2-NH-), 3.68 (broad s, 4H, 2 CH2 Gly), 4.15 (m, 2H, 2 CαH Lys), 6.75 (broad s, 4H, 2 NεH Lys and 2 NH Aca), 7.87 – 7.90 (m, 2H, 2 NH Lys, -HN-(CH2)6-NH-), 8.05 (broad s, 2H, 2 NH Gly).
Bis-(N-6-aminocaproyl-glycyl-L-lysine) hexamethylenediamide acetate, 4CH 3 COOH·(Aca-Gly-L-Lys-NH-) 2 (GK-6). Compound 10 (6.2 g, 5.6 mmol) was treated with 100 ml of 100% TFA and after 1 h the reaction mix was evaporated; the residue was triturated with 200 ml of diethyl ether. Solvent was decanted and the resulting product was dissolved in 500 ml of water and purified on a column containing 100 ml of SP-Sephadex in a 0.1 > 0.6 M gradient of pyridine acetate buffer supplemented with ammonia. The corresponding fractions (monitored by TLC) were collected and evaporated, redistilled with isopropanol, and the resulting precipitate was dried in a vacuum desiccator over CaCl2 to yield 3.6 g (90%) of final product as a solid white substance. Rf 0.02 (C), Rf 0.08 (E), Rf 0.50 (F); \( {\left[\upalpha \right]}_D^{20} \) was-23.34° (c, 1; water). The 1H NMR spectrum, DMSO-d6, δ, ppm was: 1.23, 1.27, 1.37, 1.48, and 1.61 (five m, 32H, 2 CβH2 CγH2 CαH2 Lys, -HN-CH2-(CH2)4-CH2-NH-, 2 CαH2 CβH2 CγH2 Aca), 1.73 (s, 12H, 4CH3 Ac), 2.12 (t, J 7.2 Hz, 4H, 2 CαH2 Aca), 2.64 (m, 8H, 2 CεH2 2 Lys, 2 CεH2 Aca), 3.01 (m, 4H, HN-CH2-(CH2)4-CH2-NH-), 3.67 (broad s, 4H, 2 CH2 Gly), 4.16 (m, 2H, 2 CαH Lys), 8.08 (t, J 5.3 Hz, 2H, -HN-(CH2)6-NH-), 8.18 (d, J 8.0 Hz, 2H, 2 NH Lys), 8.46 (t, J 5.6 Hz, 2H, 2 NH Gly). 2N+H3 Lys and 2N+H3 Aca exchanged with solvent H2O.
Synthesis of Bis-(N-acetyl-glycine-L-lysine), GTS-611
Bis-(N-acetyl-glycyl-Ne- tert -butyloxycarbonyl-L-lysine), (Ac-Gly-L-Lys-(Boc)-NH-) 2 (CH 2 ) 6 (11). AcOSu (1.51 g, 9.59 mmol) was added as one batch to a solution of 3.0 g (4.36 mmol) of (H-Gly-L-Lys-(Boc)-NH-)2(CH2)6(9) in 40 ml of DMF with mixing at 5°C on a magnetic stirrer. The reaction mix was held at this temperature for 1.5 h and then at room temperature for 12 h. DMF was evaporated in vacuo at 40°C and the “gel” residue was supplemented with 40 ml of acetone and held for 1 h to formation of a crystalline precipitate. The acetone solution was decanted and the precipitate was washed with hot acetone 2 x 30 ml with filtration using an attachment for hygroscopic substances, dried using a water flow vacuum pump for 2 h and then in a desiccator over CaCl2, and this procedure yielded 2.3 g (81%) of chromatographically homogeneous product (11) as white crystals with a mild beige tinge. Rf 0.91 (D), Rf 0.82 (I), Rf 0.78 (ethyl acetate); Tm 125 – 139°C. The 1H NMR spectrum, DMSO-d6, δ, ppm was: 1.21, 1.36, and 1.59 (three m, 38H, 2 CγH2 CαH2 CβH2 Lys, -NH-CH2-(CH2)4-CH2-NH-, 2-OC(CH3)3Boc), 1.84 (s, 6H, 2 CH3 Ac), 2.86 (m, 4H, 2 CεH2 Lys), 3.00 (m, 4H, -NH-CH2-(CH2)4-CH2-NH-), 3.69 (broad s, 4H, 2 CH2 Gly), 4.14 (m, 2H, 2 CαH Lys), 6.76 (t, J 5.4 Hz, 2H, 2 NεH Lys), 7.84 (t, J 5.4 Hz 2H,-NH-(CH2)6-NH-), 7.93 (d, J 7.7 Hz, 2H, 2 NH Lys), 8.10 (t, J 5.3 Hz, 2H, 2 NH Gly).
Bis-(N-acetyl-glycyl-L-lysine) hexamethylenediamide ditrifluoroacetate, 2CF 3 COOH·(Ac-Gly-L-Lys-NH-) 2 -(CH 2 ) 6 (GTS-611). A solution of 0.3 g (0.46 mmol) of compound 11 in 10 ml of a mixture of 100% TFA and CH2Cl2 (1:1) was mixed the room temperature for 2 h, after which the reaction mix was evaporated and redistilled with methylene chloride 2 x 15 ml); the residue was triturated under dry diethyl ether with decantation (3 x 20 ml) and was left under diethyl ether (20 ml) for 2 h to form a precipitate. The precipitate was collected by filtration and dried on an attachment for hygroscopic substances, and dried in a desiccator in vacuo over CaCl2 (15) mmHg). This yielded 0.26 g (87%) of chromatographically homogeneous product as a white crystalline substance (Tm not measured, hygroscopic). Rf 0.27 (D). The 1H NMR spectrum, DMSO-d6, δ, ppm was: 1.21, 1.36, 1.49, and 1.64 (four m, 20H, 2 CγH2 CαH2 CβH2 Lys,-NH-CH2-(CH2)4-CH2-NH-), 1.84 (s, 6H, 2 CH3 Ac), 2.74 (m, 4H, 2 CεH2 Lys), 3.01 (m, 4H, -NH-CH2-(CH2)4-CH2-NH-), 3.69 (d, J 4.0 Hz, 4H, 2 CH2 Gly), 4.16 (m, 2H, 2 CαH Lys), 7.74 (broad s, 6H, 2 N+H3 Lys), 7.88 (t, J 7.9 Hz, 2H,-NH-(CH2)6-NH-), 8.01 (d, J 8.0 Hz, 2H, 2 NH Lys), 8.18 (t, J 8.2 Hz, 2H, 2 NH Gly).
Synthesis of Bis-(N-6-aminocaproyl-glycyl-glycine) Hexamethylenediamide, GTS-613
Bis-( tert -butyloxycarbonyl-glycine) hexamethylenediamide (Boc-Gly-NH-) 2 (CH 2 ) 6 (14). A solution of 2.7 g (23.22 mmol) of hexamethylenediamine in 30 ml of DMF was poured into a solution of 13.9 g (51 mmol) of Boc-Gly-OSu (13) in 70 ml of DMF with mixing, resulting in precipitation of a small quantity of grayish material, which dissolved completely after 2.5 h. The reaction was mixed for 4 h at room temperature and was then left overnight without mixing. DMF was evaporated in vacuo at 40°C and 200 ml of distilled water previously warmed to 43 – 45°C was added to the residue, which was then left at room temperature until a precipitate formed. The fully formed precipitate was collected by filtration, washed with water to a neutral reaction followed by 50 ml of hexane, and dried in air. This yielded 9.0 g (90%) of chromatographically homogeneous compound 14 as a white crystalline substance. Rf 0.91 (C), Rf 0.88 (E), Rf 0.13 (B); Tm was 60 – 62°C. The 1H NMR spectrum, DMSO-d6, δ, ppm, was: 1.23 (m, 4H, -NH-(CH2)2-(CH2)2–(CH2)2-NH-), 1.37 (broad s, 22H, -NH-CH2-CH2-(CH2)2-CH2-CH2-NH-, 2 -OC(CH3)3), 3.03 (m, 4H, -NHCH2-(CH2)4-CH2-NH-), 3.48 (d, J 6.0 Hz, 4H, 2 CH2 Gly), 6.86 (t, J 6.0 Hz, 2H, 2NH Gly), 7.68 (t, J 5.1 Hz, 2H,-NH-(CH2)6-NH-).
Bis-glycine ditrifluoroacetate hexamethylenediamide, 2CF 3 COOH·(H-Gly-NH)-2(CH 2 ) 6 (15). A solution of 2.0 g (46.45 mmol) of compound 14 in a mixture of 25 ml of CH2Cl2 and 10 ml of 100% TFA was mixed at room temperature for 2 h, after which the reaction mix was evaporated, redistilled with methylene chloride (2 × 15 ml), and the residue was triturated under dry diethyl ether with decantation (3 × 20 ml) and left under diethyl ether (20 ml) for 2 h to form a precipitate. The precipitate was collected by filtration and dried on an attachment for hygroscopic substances. This yielded 1.9 g (90%) of chromatographically homogeneous product 15 as a white crystalline substance (Tm not measured, hygroscopic). Rf 0.33 (D), Rf 0.28 (E); Tm was 165 – 167.5°C. The 1H NMR spectrum, DMSO-d6, δ, ppm, was: 1.27 (m, 4H, -NH-(CH2)2-(CH2)2-(CH2)2-NH-), 1.41(m, 4H, -NH-CH2-CH2-(CH2)2-CH2-CH2-NH-), 3.10 (m,4 H, -NH-CH2-(CH2)4-CH2-NH-), 3.52 (broad s, 4H, 2 CH2 Gly), 8.10 (broad s, 6H, 2 N+H3 Gly), 8.39 (t, J 5.2 Hz, 2H,-NH-(CH2)6-NH-).
Bis-( tert -butyloxycarbonyl-glycyl-glycine) hexamethylenediamide, (Boc-Gly-Gly-NH 2 (CH 2 ) 6 (16). DIPEA (0.83 ml, 4.8 mmol) was poured into a solution of 1.0 g (2.18 mmol) of compound 15 in 15 ml of DMF and 1.31 g (4.8 mmol) of Boc-Gly-OSu (13) was added with mixing. The reaction was mixed for 4 h at room temperature and then left overnight without mixing. DMF was evaporated in vacuo in a rotary evaporator at a temperature of 53°C and then redistilled with water (3 × 20 ml); 50 ml of water prewarned to 45°C was poured onto the residue and left to form a precipitate. The precipitate was collected by filtration, washed with water to a neutral reaction and then with hexane (20 ml) and acetone (20 ml), and dried in air. This produced 0.97 g (82%) of chromatographically homogeneous product 16 as a white crystalline substance. Rf 0.88 (C), Rf 0.73 (E), Rf 0.36 (A); Tm was 131 – 134°C. The 1H NMR spectrum, DMSO-d6, δ, ppm, was: 1.24 (m, 4H, -NH-(CH2)2-(CH2)2-(CH2)2-NH-), 1.39 (broad s, 22H, -NH-CH2-CH2-(CH2)2-CH2-CH2-NH-, 2 -OC(CH3)3), 3.03 (m, 4 H, -NH-CH2-(CH2)4-CH2-NH-), 3.55 and 3.65 (two d, J 5.8 and 5.6 Hz, 8H, 2 CH2 1Gly and 2 CH2 2Gly), 7.04 and 8.01 (two t, J 5.4, 5.1 Hz, 4H, 2 NH 1Gly and 2 NH 2Gly), 7.68 (t, J 5.1 Hz, 2H,-NH-(CH2)6-NH-).
Bis-(glycyl-glycine) hexamethylenediamide ditrifluoroacetate, 2CF 3 COOH·(H-Gly-Gly-NH-) 2 (CH 2 ) 6 (17). A solution of 2.0 g (46.45 mmol) of compound 16 in a mixture of 30 ml of CH2Cl2 and 15 ml of 100% TFA was mixed for 2 h at room temperature, and the reaction mix was then evaporated, redistilled with methylene chloride (2 × 15 ml); the residue was washed (energetically shaken) with dry diethyl ether with decantation (2 × 15 ml); the diethyl ether residue was evaporated and the residue was supplemented with 35 ml of acetonitrile (which completely dissolved the oil) and held for 30 min; the resulting precipitate was collected by filtration using an attachment for hygroscopic substances. This yielded 1.89 g (90%) of chromatographically homogeneous product 17 as a white crystalline substance. Rf 0.18 (C), Rf 0.14 (E); Tm was 175 – 178°C. The 1H NMR spectrum, DMSO-d6, δ, ppm, was: 1.23 (m, 4H, -NH-(CH2)2-(CH2)2-(CH2)2-NH-), 1.37 (m, 4H, -NH-CH2-CH2-(CH2)2-CH2-CH2-NH-), 3.04 (m, 4 H, -NH-CH2-(CH2)4-CH2-NH-), 3.61 (m, 4H, 2CH2 2Gly), 3.75 (d, J 5.7 Hz, 4H, 2 CH2 1Gly), 7.96 (t, J 5.5 Hz, 2H, -NH-(CH2)6-NH-), 8.11 (broad s, 6H, 2 N+H3 2Gly), 8.64 (t, J 5.7 Hz 2H, NH 1Gly).
Bis-(N-6- tert -butyloxycarbonyl-aminocaproyl-glycylglycine) hexamethylenediamide, Boc-Aca-Gly-Gly-NH-) 2 (CH 2 ) 6 (18). Ditrifluoroacetate 17 (1.89 g, 3.3 mmol) was dissolved in 25 ml of DMF and 1.22 ml (7 mmol) of DIPEA was poured in; the reaction mix became gelatinous and a solution of 2.28 g (7.3 mmol) of Boc-Aca-OSu (4) in 15 ml of DMF was poured in and mixed for 18 h; DMF was evaporated on a rotary evaporator in vacuo using an oil pump at a temperature of 40°C. The residue, a mobile yellow gel, was supplemented with 40 ml of acetonitrile and left overnight to form a precipitate. The precipitate was collected by filtration and washed with 15 ml of acetonitrile and 15 ml of diethyl ether. The yield was 1.77 g (70%) as a white crystalline product. Rf 0.83 (C), Rf 0.11 (A); Tm was 220°C (with degradation). The 1H NMR spectrum, DMSO-d6, δ, ppm, was: 1.22 (m, 8H, -NH-(CH2)2-(CH2)2-(CH2)2-NH-, 2 CγH2 Aca), 1.36 (m, 8H, -NH-CH2-CH2-(CH2)2-CH2-CH2-NH-, 2 CαH2 Aca), 1.47 (m, 4H 2 CβH2 Aca), 2.11 (t, J 5.7 Hz 2H, 2 CαH2 Aca), 2.87 (m, 4H, 2 CεH2 Aca), 3.03 (m, 4 H,-NH-CH2-(CH2)4-CH2-NH-), 3.63 (d, J 6.0 Hz 4H, 2CH2 2Gly), 3.66 (d, J 6.0 Hz, 4H, 2 CH2 1Gly), 6.78 (t, J 5.0 Hz 2H, 2 NH Aca), 7.72 (t, 3J 5.5 Hz, 2H, -NH-(CH2)6-NH-), 8.09 (t, J 5.5 Hz, 2H, 2NH 2Gly), 8.14 (t, J 5.0 Hz 2H, NH 1Gly).
Bis-(N-6-aminocaproyl-glycyl-glycine) hexamethylenediamide ditrifluoroacetate, 2CF 3 COOH·(Aca-Gly-Gly-NH-) 2 (CH 2 ) 6 (GTS-613). A solution of 0.30 g (0.389 mmol) of compound 18 in 5 ml of a mixture of 100% TFA and 15 ml of CH2Cl2 was mixed at room temperature for 2 h; the reaction mix was evaporated and then redistilled with methylene chloride (2 × 15 ml); the residue was triturated under dry diethyl ether with decantation (3 × 20 ml) and left under diethyl ether (20 ml) for 2 h to form a precipitate. The precipitate was collected by filtration and dried on an attachment for hygroscopic substances, and then dried in a desiccator in vacuo over CaCl2 (15 mmHg). The yield was 0.23 g (87%) of chromatographically homogeneous GTS-613 as a white crystalline substance. Rf 0.18 (C), Rf 0.14 (E); Tm was 112 – 120°C (with degradation). The 1H NMR spectrum, DMSO-d6, δ, ppm, was: 1.23 – 1.26 (m, 4H,-NH-(CH2)2-(CH2)2-(CH2)2-NH-), 1.29 – 1.31 (m, 4H, 2 CγH2 Aca), 1.38 (m, 4H, -NH-CH2-CH2-(CH2)2-CH2-CH2-NH-), 1.46 – 1.57 (m, 8H, 2 CαH2 and 2 CβH2 Aca), 2.11 (t, J 7.4 Hz, 4H, 2 CαH Aca), 2.77 (m, 4H, 2 CεH2 Aca), 3.03 (m, 4H, -NHCH2(CH2)4CH2NH-), 3.65 and 3.69 (two d, J 5.8, 5.6 Hz, 8H, 2 CH2 2Gly and 2CH2 1Gly), 7.75 (broad s, 8H, 2 N+H3 Aca, -NH-(CH2)6-NH-), 8.10 and 8.16 (two t, J 5.8, 5.6 Hz, 2 NH 2Gly and 2 NH 1Gly). The 13C NMR spectrum (DMSO-d6), δ, ppm, was: 173.18 (s, 2C, 2CO, Aca), 169.86 and 168.94 (two s, 4C, 2CO 2Gly and 2CO 1Gly), 158.79 (q, 2C, 2JC-F 34.27 Hz, CF3COOH), 116.84 (q, 2C, 1JC-F 295.03 Hz, CF3 COOH), 42.74 and 42.47 (two s, 4C, 2Cα 2Gly and 2Cα 1Gly), 39.21 (s, 2C, 2Cε Aca), 38.94 (s, 2C, 2 C1 of spacer), 35.32 (s, 2C, 2Cδ Aca), 29.46 (s, 2C, C2 of spacer), 27.29 (s, 2C, 2Cδ Aca), 26.51 (s, 2C, 2 C3 of spacer), 25.95 (s, 2C, 2Cγ Aca), 25.01 (s, 2C, 2Cβ Aca).
EXPERIMENTAL BIOLOGICAL SECTION
The neuroprotective activity of compounds was assessed [13] in cultures of immortalized mouse hippocampal cells, line HT-22. Cells were cultured in 96-well plates at a density of 3500 cells/well in DMEM medium (HyClone, USA) containing 5% fetal bovine serum (Gibco, USA) and 2 mM L-glutamate (ICN Pharmaceuticals, USA) and incubated at 37°C in 5% CO2 to formation of a monolayer. Peptides were added 24 h before harmful actions in the concentration range 105 to 108 M.
Oxidative stress was modeled using H2O2 at a final concentration of 1.5 mM. Cells with H2O2 were incubated in a 5% CO2 atmosphere at 37°C for 30 min [22]. Medium was then changed for normal medium and cell viability was assayed after 4 h using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (AppliChem, Panreac, Germany). Optical density was measured on a Multiscan EX spectrophotometer (Thermo) at a wavelength of 600 nm.
Activity in experiments against oxidative stress was computed as
where Dexp is the optical absorption in the experimental conditions, \( {D}_{{\mathrm{H}}_2{\mathrm{O}}_2} \) is the optical absorption in the active control (with H2O2), and Dcontr is the optical absorption in controls (without H2O2).
Statistical analysis was run using the standard program bundle Statistica 6.0 (StatSoft Inc. USA). MTT test results were analyzed using nonparametric statistics and qualitative data were analyzed using the Kruskal-Wallis test followed by the Dunn test (ANOVA). Results were regarded as significant at p ≤ 0.05.
Determination of the differentiation-inducing activity of peptide mimetics of nerve growth factor NGF was performed using pheochromocytoma cell line PC12. Cells were cultured in DMEM medium supplemented with 5% fetal bovine serum (FBS) in a CO2 incubator at a temperature of 37°C and 5% CO2 to monolayer formation. Undifferentiated PC12 cells were cultured at a density of 3500 per well in DMEM medium containing 1% FBS. At the moment of sowing, NGF (BD Biodiversity, UK) was added to the culture medium at a final concentration of 100 ng/ml (≈ 109 M) as positive control. This NGF concentration is used in experiments for detection of its neuroprotective and differentiation-inducing activities in PC12 cells [23] and is not cytotoxic [24]. Dipeptides GK-6 and GTS-611 were added to a concentration of 106 M.
Study peptides and NGF were then added to the culture medium every 48 h for six days [25]. Cells were photographed on day 7 using a Nikon Eclipse TS1200-F microscope (Japan) at a magnification of ×100. Cells with processes longer than one cell diameter were regarded as differentiated.
Data were analyzed statistically using the Mann-Whitney U test. Data are presented as m ± s.d. Data were regarded as significant at p ≤ 0.05.
RESULTS AND DISCUSSION
Synthesis of Mimetics of NGF Loop 1 – GK-6, GTS-611, and GTS-613
FK-6 and GTS-611 were synthesized by classical peptide synthesis methods in solution using elongation of the peptide chain at the C terminus using the Z/Boc protective group strategy with N-hydroxysuccinimide-activated esters (scheme 1).

Synthesis of NGF loop 1 dipeptide mimetics GK-6 and GTS-611.
Peptides GK-6 and GTS-611 were synthesized using commercially available Z-L-Lys(Boc)-OH, H-Gly-OH, and 6-aminocaproic acid. Z-protected glycine (1) was prepared by reaction of the sodium salt of glycine with benzyloxycarbonysuccinimide (Z-OSu) in aqueous-acetone solution [10]. Boc-protected aminocaproic acid Boc-Aca-OH was obtained using di-tert-butylpyrocarbonate using the Pozdnev method [11]. N-hydroxysuccinimide esters of Z/Boc-protected lysine (5), Boc-Aca-OH (4), and Z-glycine (2) were synthesized by the Andersen method [12] using N-hydroxysuccinimide and dicyclohexylcarbodiimide (DCHD) at temperatures from 0°C to +5°C. Condensation of the activated ester of protected L-lysine (5) with hexamethylenediamine in DMF at room temperature produced the corresponding (Z-L-Lys(Boc)-NH-)2(CH2)6 hexamethylenediamide with a yield of 93%, which was then Z-deblocked by hydrogenolysis with hydrogen in the presence of 10% Pd/C. The resulting product 7 was condensed in DMF with Z-Gly-OSu (2) to produce (Z-Gly-L-Lys(Boc)-NH-)2(CH2)6 (8) with a yield of 95%. Removal of the Z group by catalytic hydrogenolysis produced bis-dipeptide (H-Gly-L-Lys(Boc)-NH-)2(CH2)6 (9), which was then acylated with Boc-Aca-OSu (4) and Ac-OSu in DMF to convert it to the N-aminocaproyl and N-acetyl derivatives (Boc-Aca-Gly-L-Lys(Boc)-NH-)2(CH2)6(10) and (Aca-Gly-L-Lys(Boc)-NH-)2(CH2)6 (11) with yields of 95% and 81% respectively. Removal of Boc protection from bis-dipeptides 10 and 11 by acidolysis with TFA in dichloromethane produced trifluoroacetates GK-6 and GTS-611. Target peptide GK-6 was converted to the non-salt form by ion exchange chromatography (SP-Sephadex, pyridine-acetate buffer), purified by RP HPLC, and lyophilized. The overall yields of GK-16 and GTS-611 were 61% and 52% respectively. The structures and diastereomeric purities (>98%) of all compounds were confirmed by 1H and 13C NMR spectroscopy.
GTS-613 was synthesized using the Boc protective groups strategy (scheme 2). At the first stage, Boc-Gly-OH (12) was prepared from glycine, followed by preparation of its activated ester 13, which was condensed with hexamethylenediamine to produce bis-glycine 14 with a yield of 90%. Acidolysis with TFA in dichloromethane was then used to prepare bis-glycine trifluoroacetate 15, which was converted to free base using DIPEA and condensed with ester 13 to produce bis-dipeptide 16. Acidolysis of peptide 16 and further acylation with ester 4 produced the N-aminocaproyl derivative of bis-dipeptide 18, and final removal of Boc protection produced target compound GTS-611 as the ditrifluoroacetate with an overall yield in terms of glycine of 24%. Peptide GTS-613 was homogeneous by TLC. The structure and diastereomeric purity (98%) were confirmed by 1H and 13C NMR spectroscopy.

Synthesis of NGF loop 1 dipeptide mimetic GTS-613.
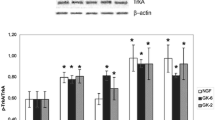
The neuroprotective activity of the peptides synthesized here was studied in a model of oxidative stress using hydrogen peroxide in cultures of immortalized mouse hippocampal cells line HT-22 [13] over the concentration range 10 5 to 10 8 M. Peptides were added 24 h before the harmful agent. The results are presented in Table 1. GK-6 had neuroprotective activity at concentrations to 106 M, increasing cell viability by 66% of the maximum possible. In these conditions, NGF increased viability by 97%. Neuroprotective activity persisted when the lysine residue was replaced with a glycine residue (compound GTS-613). Dipeptide GTS-613 was active to a concentration of 106 M at a level of 44%. At the same time, substitution of the N-aminocaproyl radical with an acetyl radical (compound GTS-611) led to complete loss of activity.
Thus, the nature of the N-terminal substituent playing the role of Lys32 was critical for the neuroprotective activity of the NGF loop 1 mimetic, i.e., dimeric dipeptide GK-6. The C-terminal lysine residue corresponding to Lys34 in the NGF sequence does not play an important role in neuroprotective activity.
Differentiation-inducing activity on PC-12 cells of NGF mimetics GK-6 and GTS-611. The ability of peptide mimetics of nerve growth factor to induce differentiation was assessed in PC21 pheochromocytoma cells. These cells are known to contain TrkA and p75 receptors and addition of NGF is known to induce differentiation along the neuronal pathway [14]. Peptides GK-6 and GTC-611 were added to culture medium to a concentration of 106 M. As shown in Fig. 2, NGF induced differentiation of PC12 cells. The differentiating influence of GK-6 was weaker than that of NGF, while that of GTS-611 was even less marked. Mean process length in differentiated cells after addition of NGF was 59.6 ± 11.0 μm, compared with 44.5 ± 8.5 μm with GK-6 and 36.0 ± 6.2 μm after GTS-611 (Table 2).

Differentiation-inducing activity of peptides GK-6 and GTS-611 in rat PC-12 pheochromocytoma cells. Phase contrast, magnification ×100.
Thus, substitution of the N-aminocaproyl radical of GK-6 with an acetyl radical led to loss of the neuroprotective but not the differentiation-inducing activity.
The causation of these relationships between the cellular effects of the compounds synthesized here and their structures could be linked with the specific features of the activation of the post-receptor transduction pathway, examples of which are given in our previous studies [15, 16].
This study was supported in the framework of a state contract (theme No. 0521-2019-0003 “Search for pharmacological means of selective activation of transduction pathways for neurotrophin receptor tyrosine kinase signals as the basis for creating drugs lacking the side effects of native neurotrophins”).
References
T. A. Gudasheva, T. A. Antipova, and S. B. Seredenin, Dokl. Akad. Nauk, 434(4), 549 – 552 (2010).
Russian Federation Patent RF No. 2410392 (2011).
US Patent SShA No. 9683014 B2 (2017).
Korean People’s Republic Patent KNR No. 102365294 B (2016).
European Patent EPV ER No. 2397488 (2019).
T. A. Gudasheva, P. Y. Povarnina, T. A. Antipova, et al., J. Biomed. Sci., 22(1), 1 – 10 (2015).
S. B. Seredenin and T. A. Gudasheva, Zh. Nevrol. Psikhiat. im. S. S. Korsakova, No. 6, 63 – 70 (2015).
T. A. Antipova, T. A. Gudasheva, and S. B. Seredenin, Byull. Éksperim. Biol. Med., 150(11), 537 – 540 (2010).
D. R. Holland, L. S. Cousens, W. Meng, et al., J. Mol. Biol., 239(3), 385 – 400 (1994).
A. Paquet, Can. J. Chem., 60(8), 976 – 980 (1982).
V. F. Pozdnev, Khim. Prirod. Soedin., No. 6, 764 – 767 (1974).
G. W. Anderson and J. E. Zimmerman, J. Am. Chem. Soc., 86(9), 1839 – 1842 (1964).
G. R. Jackson, K. Werrbach-Perez, E. L. Ezell, et al., Brain Res., 592(1 – 2), 239 – 248 (1992).
J. H. Chang, E. Mellon, N. C. Schanen, et al., J. Biol. Chem., 278(44), 42877 – 42885 (2003).
T. A. Gudasheva, P. Povarnina, A. V. Tarasiuk, and S. B. Seredenin, Curr. Pharm. Design., 25, 729 – 737 (2019).
R. U. Ostrovskaya, S. V. Ivanov, T. A. Gudasheva, and S. B. Seredenin, Acta Naturae, 11(1), 48 – 57 (2019).
N. M. Sazonova, A. V. Tarasyuk, Yu. N. Firsova, et al., Khim.- Farm. Zh., 54(2), 24 – 31 (2020); Pharm. Chem. J., 54(2), 126 – 133 (2020); Pharm. Chem. J., 54(2), 126 – 133 (2020).
N. M. Sazonova, A. V. Tarasyuk, D. V. Kurilov, et al., Khim.- Farm. Zh., 49(7), 10 – 19 (2015); Pharm. Chem. J., 49(7), 439 – 448 (2015).
S. M. Andreev, L. A. Pavlova, Yu. A. Davidovich, et al. Izv. Akad. Nauk. SSSR, 1078 – 1081 (1980).
Org. Synth. Coll., No. 7, 70 (1990).
T. Munegumi and T. Yamada, Int. J. Polymer Sci., ID 8364710, 1 – 17 (2017).
K. Riveles, L. Z. Huang, and M. Quik, Neurotoxicology, 29(3), 421 – 427 (2008).
Y. Gong, J. Wu, H. Qiang, et al., BMB Rep., 41(4), 287 – 293 (2008).
D. L. Senger and R. B. Campenot, J. Cell Biol., 138(2), 411 – 412 (1997).
V. Kukhtina, V. Tsetlin, Yu. Utkin, et al., J. Natural Toxins., 10(1), 9 – 16 (2001).
Author information
Authors and Affiliations
Corresponding author
Additional information
This material has in part been published as an academic thesis [1].
Translated from Khimiko-Farmatsevticheskii Zhurnal, Vol. 54, No. 11, pp. 22 – 30, November, 2020.
Rights and permissions
About this article

Cite this article
Tarasyuk, A.V., Sazonova, N.M., Rebeko, A.G. et al. Design and Synthesis of Dipeptide Nerve Growth Factor Loop 1 Mimetics and In Vitro Studies of their Neuroprotective and Differentiation-Inducing Activities. Pharm Chem J 54, 1126–1135 (2021). https://doi.org/10.1007/s11094-021-02330-2
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11094-021-02330-2