
Abstract
Plasma-catalysis has attracted significant interest in recent years as an alternative for the direct upgrading of methane into higher-value products. Plasma-catalysis systems can enable the electrification of chemical processes; however, they are highly complex with many previous studies even reporting negative impacts on methane conversion. The present work focuses on the non-oxidative plasma-catalysis of pure methane in a Dielectric Barrier Discharge (DBD) reactor at atmospheric pressure and with no external heating. A range of transition and noble metals (Ni, Fe, Rh, Pt, Pd) supported on γ-Al2O3 are studied, complemented by plasma-only and support-only experiments. All reactor packings are investigated either with pure methane or co-feeding of helium or argon to assess the role of noble gases in enhancing methane activation via energy transfer mechanisms. Electrical diagnostics and charge characteristics from Lissajous plots, and electron temperature and collision rates calculations via BOLSIG+ are used to support the findings with the aim of elucidating the impact of both active metal and noble gas on the reaction pathways and activity. The optimal combination of Pd catalyst and Ar co-feeding achieves a substantial improvement over non-catalytic pure methane results, with C2+ yield rising from 30% to almost 45% at a concurrent reduction of energy cost from 2.4 to 1.7 \(\:\text{M}\text{J}\:{\text{m}\text{o}\text{l}}_{\text{C}{\text{H}}_{4}}^{-1}\) and from 9 to 4.7 \(\:\text{M}\text{J}\:\text{m}\text{o}{\text{l}}_{{\text{C}}_{2+}}^{-1}\). Pd, along with Pt, further displayed the lowest coke deposition rates among all packings with overall stable product composition during testing.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The depletion of fossil sources necessitates the development of technologies for the sustainable production of carbon-based chemicals. Upgrading methane into value-added compounds has been extensively investigated given the molecule’s ready availability from natural and shale gas [1, 2], and renewable biogas [3]. The use of plasmas for methane’s activation has attracted significant interest over recent years due to growing environmental and energy security concerns [4]. Plasmas, enabling the activation of methane even at ambient conditions, appear to have the potential to drive the electrification of the chemical industry, replacing the need for thermal energy supply [5]. With almost instantaneous transient response and inherent modularity, plasmas are particularly well suited for decentralised, renewable energy-driven deployment. In these processes, the electric field accelerates the naturally present electrons to collide with methane molecules and form a range of reactive species including ions, excited states, and radicals. Radical coupling reactions within the plasma lead to the formation of higher hydrocarbon species [4, 6], while catalytic reactions, either between two adsorbed species (Langmuir − Hinshelwood mechanism,) or between a gas phase species and an adsorbed one (Eley − Rideal mechanism,) can steer selectivity towards desired higher hydrocarbons [4, 6,7,8].
Plasma discharges can be operated under different degrees of deviation from thermal equilibrium; however, the low gas bulk temperature and the ease of reactor assembly of Dielectric Barrier Discharges (DBD) has made them particularly suitable for the study of plasma-catalysis systems [8,9,10,11,12]. Despite this, plasma-catalysis systems are highly complex, with the plasma affecting the catalyst and vice versa [6, 13,14,15]. The catalyst, acting as a dielectric or a conductor, can be responsible for local field enhancement and surface discharge formation, while the plasma can increase the adsorption probability of species [16, 17], lower the activation barrier of surface processes, and modify the catalyst surface area and functionality [18, 19].
As elaborated in the multiple reviews published, the dry reforming of methane via plasma-catalysis in DBD reactors has been extensively studied [5, 7, 20,21,22,23]; however, the non-oxidative upgrading has received comparatively less attention and with often conflicting findings reported [4, 24]. In an early study, Indarto reported a slight increase in CH4 conversion in the presence of a mixed oxide Zn and Cr catalyst compared to plasma-only results, however the higher production of hydrogen over hydrocarbons indicated that the catalyst promoted the formation of coke [25]. Using a CH4/Ar/H2 mixture as feed and externally heating at 240 °C, Górska et al. reported a 20% increase of C2 selectivity with a Cu/ZnO/Al2O3 catalyst versus no packing at a minimal change of conversion [26]. Contrarily, Kasinathan et al. using a 10% CH4 in Ar mixture, clearly observed an increase in the conversion of methane and selectivity to C3 hydrocarbons by the addition of MgO/Al2O3 or TiO2/Al2O3 compared to Al2O3 or plasma alone [27]. Jo et al. comparing the use of Al2O3 alone and a Pt/Al2O3 catalyst, observed in the presence of the catalyst a decrease in methane conversion and a shift in the selectivity of C2 products from unsaturated species to ethane [28]. Taheraslani and Gardeniers made similar observations when comparing Al2O3 with Pd/Al2O3 catalysts, with higher loadings of Pd promoting the production of C2 and C3 alkanes but also lowering the conversion of methane [29]. Pd further led to reduced carbon deposits, overall improving C2 yields and energy efficiency [29]. Pd/Al2O3 catalysts were also investigated by García-Moncada et al., who specifically studied the effect of the thickness of the catalyst layer in structured reactors in the presence of argon [30, 31]. Pd was shown to suppress the formation of carbon, linked to the effective hydrogenation of acetylene on the metal [30], the latter further enhanced by mild heating at 75 °C [31]. In the study by Liu et al. comparing single-atom and nanoparticle Pt catalysts supported on CeO2, the abundance of low coordination sites on the single-atom case was found to promote C–H bond scission and hinder unselective coke formation pathways [32]. Recently, in the work of Nguyen et al. Pd-Ga bimetallic catalysts supported on 13X zeolite achieved higher CH4 conversion and hydrocarbons yield compared to either Pd or Ga monometallic catalysts, suggesting that synergisms between the two metals limited the total dehydrogenation of CH4 [33]. Evidently, most recent works have focused on the use of Pt or Pd based catalysts with an exception being the study by Ghanbari et al. over a Ni–K2O/Al2O3 catalyst [34]. Nonetheless, the very high specific energy inputs and catalyst loadings used, in combination with no hydrocarbons formation being reported, suggested that the main reaction active in that work was methane cracking to carbon and hydrogen. Of particular note is the work of Lee et al., who combined a range of in situ/operando spectroscopic techniques to study plasma–surface interactions leading to carbon deposition over Ni during non-oxidative coupling of methane [35].
Given the lower breakdown voltage of noble gases in comparison to methane [36], a range of studies has further investigated specifically their introduction in methane plasmas to reduce energy requirements and improve discharge homogeneity [37,38,39,40,41,42]. Jo et al. studied the impact of Ar, He and Ne [43], and Kr and Xe [44] in double-sided dielectric barrier discharge reactors, whereas different dielectrics in methane plasmas diluted in Ar were tested by Kundu et al. [45]. The cracking of methane diluted in Ar in pulsed DBD was studied by Barni et al. [46], while He excitation and its impact on methane pyrolysis in a radio-frequency plasma was investigated by Sun and Chen [47]. Finally, Huang et al. analysed the non-catalytic activation of methane in the presence of Ar at low temperatures using nanosecond pulsed plasmas [48]. A common finding in these works was that methane activation is enhanced due to Penning ionisation and dissociation pathways that are induced by electronically excited states of the noble gases.
Previous studies have been inconclusive on whether clear catalytic effects exist in the non-oxidative coupling on methane (NOCM) plasma catalysis [28], in cases reporting even negative impacts on methane conversion. The diverse conditions and designs used, including catalyst pellet size and supports of different permittivity, feed mixture composition, and electrode configurations all impact the plasma properties, discharge mode, and electric field distribution to varying degrees. Negative effects induced by the catalyst presence have been specifically linked to the support dielectric constant affecting the discharge mode and consequently the strength of the electric field and the distance between plasma species and catalyst surface. Metal particles also have been reported to affect the formation of discharge streamers with effects being dependant on the metal. The size of the catalyst pellets further can affect the electric field intensity but also the number of electron collisions with walls. Finally, the wide range of species present within a plasma can contribute to the surface kinetics possibly negatively as, for example, hydrogen radicals promoting the reverse reaction of methyl species to methane. The multitude of physical and chemical interactions between the plasma and the catalyst have been elaborated by Loenders et al. [49] and are particularly important to NOCM as both the activation of methane to methyl radicals and the coupling of the latter to ethane can take place within the plasma. Furthermore, most NOCM studies have used specific metal/support combinations (typically Pd or Pt based) making discerning the role of catalysis even more challenging.
The present work focuses on NOCM in a DBD reactor at atmospheric pressure and with no external heating. We methodically aim to elucidate the specific roles of the active metal and the noble gases presence in the reaction pathways, activity, and selectivity of the reaction. A range of transition and noble metals (Ni, Fe, Rh, Pt, Pd) supported on γAl2O3 are used as catalysts, while experiments are further carried out with the plasma only and the support only at equivalent conditions. Pt and Pd are selected due to their established hydrogenation activity and consequent popularity in literature. Ni and Fe are chosen as low-cost metals frequently studied in high temperature reactions, whereas Rh is selected on account of its typically very high activity and stability in methane thermal catalysis [50]. Given the potential of noble gases to enhance the reaction performance, to discern the impact of their presence, all reaction modes and catalysts are studied either with pure methane or co-feeding of helium or argon. The latter are selected as the cheapest and most frequently studied in relevant literature. Electrical diagnostics and BOLSIG+ calculations are used in all cases to consolidate the findings. In addition to the advancements in the methodological evaluation of this reaction and in the understanding of the catalyst and noble gas effects, optimal activity achieved using Pd and argon co-feeding is among the highest reported in literature.
Methodology
Experimental Setup and Analysis
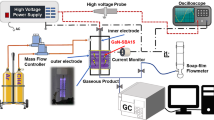
The experiments were carried out in a DBD plasma reactor setup at atmospheric pressure (Fig. 1) using a High Voltage (HV) AC power supply (PVM500-2500). A steel foil wrapped around a quartz tube (9 mm O.D., 1 mm wall thickness) acted as the HV electrode defining a cylindrical discharge area of 6 cm length. The ground electrode consisted of a stainless-steel wire (1 mm O.D.) placed inside of a second quartz tube (3 mm O.D., 0.9 mm wall thickness), resulting in a double dielectric barrier configuration with a gas gap of 2 mm. The voltage and charge data were sampled using a HV probe (Cal Test CT4028) and a differential probe (Micsig DP20003) that measured the voltage across a 955 pF ceramic capacitor placed to the ground line. Both probes were connected to a digital oscilloscope (Tektronix TDS2012B). The inlet gas composition and flow rate were controlled by a series of mass flow controllers (Bronkhorst EL-Flow) connected to the reactor. For plasma only experiments, methane flowrate was varied from 10 to 120 Nml min− 1 (99.9% CH4), which for the given reactor geometry amounted to a residence time variation from 11.3 s to 0.9 s. All plasma-catalysis experiments were performed using a constant flow rate of methane of 25 Nml min − 1 with focus placed on the impact of catalyst and packing within the plasma zone. Given the particle size used (350–500 μm, see section “Catalyst preparation and characterisation”) and assuming a bed porosity of 0.4, this flow corresponded to a residence time of 1.8 s. For the noble gas co-feeding experiments an additional flow of 75 Nml min− 1 He or Ar was used, decreasing the residence to 0.45 s. Further details on the rationale for flowrates selected during the different sets of experiments are provided in the respective results sections. The applied power was modified by manipulating the high voltage output of the power supply. The frequency in all experiments was kept constant and equal to the resonant frequency of the circuit, at a value of approximately 25 kHz. Gas products analysis took place using a HP 5890 Gas Chromatograph (GC) equipped with a Flame Ionisation Detector (FID) and a HayeSep T column that separated the C1-C6 hydrocarbons.

Schematic of dielectric barrier discharge reactor setup
Following ignition, the plasma was allowed to homogenize for 15 min to ensure it is “fully bridged” across the discharge zone [51] and that the charge is evenly distributed [28]. A minimum of two consecutive GC samples were collected at each condition across a time frame of 60 min to ensure the stable operation and lack of pronounced deactivation. Thermogravimetric analysis (TGA) of spent catalyst samples was performed to quantify the amount of carbon deposited on the catalyst (see section “Catalyst preparation and characterisation” for details).
The results presented in following sections, are expressed in terms of methane conversion (\(\:{X}_{C{H}_{4}}\)), product selectivities (\(\:{S}_{i}\)) and carbon balance closure (\(\:{C}_{Bal}\)):
where \(\:{F}_{i}^{In/Out}\) is the inlet/outlet molar flow of compound \(\:i\) (mol s-1), \(\:{w}_{{C}_{TGA}}\) the weight of carbon deposited on the catalytic bed determined by TGA analysis (g), and \(\:{t}_{exp}\) the duration of the experiment (s). In the case of non-catalytic experiments, any solid carbon formed was not possible to be quantified and is, hence, implicitly included in \(\:{S}_{{C}_{unknown}}\). As the FID GC used did not allow the direct quantification of hydrogen, its flowrate and selectivity were determined based on the detected hydrocarbon products and stoichiometric reactions of the form \(\:xC{H}_{4}\to\:{C}_{x}{H}_{y}+\frac{4x-y}{2}{H}_{2}\), according to Eqs. (6) and (7):
Electrical Diagnostics
The discharge power, \(\:P\) (W), was estimated from the voltage, \(\:V\) (V), and charge, \(\:Q\) (C), signals by integration of the area of the Q-V Lissajous plot [51, 52]. The specific energy input, \(\:SEI\) (kJ l− 1), was estimated based on the discharge power and inlet flowrate, \(\:{Q}_{In}\) (Nml min− 1).
The gap, \(\:{C}_{g}\), and dielectric, \(\:{C}_{d}\), capacitances (F) were determined using tangential lines on the Lissajous plot, considering an equivalent electrical circuit comprising series capacitances for the gap and dielectric (Fig. 2), as described in detail by Peeters and Butterworth [51]. In short, during “plasma-off” phases of an AC cycle in a DBD cell, no charge is transferred between the electrodes, hence the slope of lines AB and CD is equal to \(\:{C}_{cell}\). The cell capacitance comprises of the gap and dielectric capacitances and is related to them through the standard formula for total capacitance of capacitances in series:
During “plasma-on” phases, plasma breakdown occurs so the slope of lines BC and DA is equal to the dielectric capacitance (\(\:{C}_{d}\)) for a “fully bridged gap”. Having determined \(\:{C}_{cell}\) and \(\:{C}_{d}\) from the Lissajous plot, Eq. (9) is used to obtain \(\:{C}_{g}\). Given non-idealities of the Lissajous plot during experimentation, especially in the presence of a catalyst, the dielectric capacitance should be considered an effective capacitance, \(\:{C}_{eff}\), while the gap capacitance accounts for the contribution of the integrated gas and solid present in the region between dielectrics [53, 54]. In the following, \(\:{C}_{eff}\) is used instead of \(\:{C}_{d}\) to reflect this.

Simple equivalent electrical circuit of a DBD comprising of dielectric and gap capacitances in series and corresponding ideal Q-V Lissajous plot indicating slopes used for the estimation of DBD capacitances. Other quantities annotated are discussed in the main text
The minimum voltage, \(\:{U}_{min}\:\), (voltage at zero charge on the Lissajous plot) was used to estimate the breakdown voltage of the plasma using Eq. (10) [53, 55, 56].
The electric field strength, \(\:E\) (V m− 1), and the reduced electric field strength, \(\:E/N\) (V m2), were estimated from the gas gap, \(\:{d}_{gap}\) (m), and the breakdown voltage using Eqs. (11) and (12), respectively [57]. The number density, \(\:{n}_{0}\) (m− 3), was estimated using the ideal gas law at atmospheric pressure and the reactor temperature measured using an IR thermometer (Fluke 62 MAX) immediately after switching the HV power off to avoid any interference with emission from the plasma.
The electron temperature, \(\:{T}_{e}\) (eV), and electron mobility, \(\:{\mu\:}_{e}\) (m2 V− 1 s− 1), were calculated using the BOLSIG+ software (12/2019 version) [58], which solves the Boltzmann equation for electrons in weakly ionized gases. Inputs to BOLSIG+ comprised the gas temperature, gas concentration, reduced electric field strength (determined above), and relevant collisional cross sections. BOLSIG+ calculations were carried out using both the inlet and outlet reactor concentrations. The works of Maitre et al. and cited sources therein were used to retrieve detailed partial cross sections for C1, C2 and C3 species [59], and Ar and He [60]. Due to lack of detailed data in databases for higher hydrocarbons, total cross sections were obtained from literature for butane, butylene and isobutane [61], and the C5 and C6 lumps [62].
The electron density, \(\:{n}_{e}\) (m− 3), was calculated using Eq. (13) [63,64,65]:
where \(\:e\) is the elementary charge in C, and \(\:{J}_{c}^{e}\) the conduction current (A). The latter was estimated according to Eq. (14) from the stored net charge \(\:\varDelta\:Q\) (C) (approximated as \(\:\varDelta\:Q=2{Q}_{min}\), with \(\:{Q}_{min}\) being the charge at zero voltage on the Lissajous plot), discharge area \(\:A\) (m2) and the period \(\:T\) (s). The discharge area was approximated as the cylindrical area defined by the external diameter of the internal dielectric tube (3 mm) and the length of the discharge zone (6 cm).
The energy cost, \(\:EC\) (MJ mol− 1), for the conversion of methane and production of hydrocarbons and hydrogen were estimated using Eq. (15) to (17).
Catalyst Preparation and Characterisation
A range of catalysts were procured from Alfa Aesar in powder form, namely Pd/Al2O3, Pt/Al2O3, Rh/Al2O3, all with a 5 wt% metal loading. These catalysts were pelletized, crushed, and sieved to a particle size of 350–500 μm. Additionally, two catalysts, a 5 wt% Ni/Al2O3 and a 5 wt% Fe/Al2O3, were prepared using a standard wet impregnation method. The metal precursors (Ni(NO3)2·6H2O or Fe(NO3)3·9H2O, both from Merck) were dissolved in deionized water to obtain the desired metal loading. Support γ-Al2O3 (Alfa Aesar) was added to the solution and the resulting mixture was stirred for 4 h at 80 °C. The remaining liquid water was evaporated, and the resulting slurry dried in an oven overnight at 110 °C. The resulting solid was pelletized, crushed, and sieved to a size of 350–500 μm. The home-made pelletized catalysts were calcined with air at 500 °C for 4 h to remove the nitrates from the precursors, and then allowed to cool and stored. The home-made calcined catalysts were reduced thermally at 600 °C under 100 Nml min− 1 flow of 5% H2/N2 for 1 h to obtain the reduced metal state. The commercial catalysts were procured in reduced state.
0.5 g of each catalyst were mixed with 0.5 g of γ-Al2O3 particles of same particle size and were loaded to the reactor. The catalyst bed was suspended by quartz wool plugs at both sides, ensuring that the quartz wool was located outside of the HV electrode zone to minimise any effect on plasma chemistry [66]. Before reaction, all catalysts were reduced in situ in a 5% H2/He plasma and a power of 17 W for 1 h to ensure that the active metals are in a fully reduced state, as partial oxidation could have taken place during storage or loading in the reactor. Equivalent experiments with an empty reactor tube, and experiments with the reactor loaded with only γ-Al2O3 particles were also conducted for comparison using the same power and flowrate as that of the plasma-catalysis experiments.
Temperature Programmed Reduction (TPR) analysis of fresh oxidised samples of the home-made catalysts as obtained after calcination was carried out using a TPDRO 1100 instrument with a TCD detector and a trap bed placed before the detector to remove the moisture. Samples were heated from 20 °C to 600 oC with a 10 oC min− 1 temperature ramp, using a 5% H2/N2 flow. XRD patterns of the same samples were obtained at room temperature on a Panalytical powder diffractometer over an angular range of 5o < 2θ < 80o with a step-size of 0.02o. Spent catalyst samples were examined by TGA using a Mettler Toledo TGA/DSC 3 + analyser to determine the amount of deposited carbon. Samples were heated from 20 °C to 800 °C under a flow of air at 10 Nml min− 1 followed by a holding time of 30 min at 800 °C.
Results and Discussion
Non-Catalytic Results
Preliminary non-catalytic non-oxidative methane coupling experiments, in the absence of any solid packing in the reactor, took place to establish baseline reactivity at our conditions. The chromatographic analysis method during this set of experiments provided detection of C1-C3 compounds but was later improved to allow the separation of up to C6 hydrocarbons. As discussed in the following sections, C4+ species comprise only a small fraction of the total distribution of products, hence the impact of not quantifying these in the non-catalytic experimental trends should be considered minor.
Variation of Applied Power
The effect of \(\:SEI\) on CH4 conversion and products selectivities, achieved through variation of applied power, \(\:P\), from 10 to 26 kW (\(\:SEI\) from 13 to 21 kJ l− 1), is presented in Fig. 3. For powers lower than 10 W conversion was negligible, with formed products below the GC detection limits. Increasing \(\:SEI\) led to a roughly linear rise of CH4 conversion, in line with previous experimental and modelling studies [67,68,69]. Applied powers higher than 26 kW led to deviations from this linearity, which, given the constant frequency applied, could indicate that at these conditions charge transfer per voltage cycle was negatively affected, further impacting electron impact dissociation rates [70]. It is further possible that reverse pathways were promoted within the plasma or immediately after the discharge zone at higher applied powers that negatively affected conversion.

CH4 conversion and product selectivities vs. SEI varying applied power in non-catalytic non-oxidative methane coupling. Flowrate: 50 Nml min-1 (100% CH4), Residence time: 2.2 s, Voltage: 8–10 kV (amplitude)
Products comprised primarily of saturated hydrocarbons (C2H6 and C3H8). C3 species are presented lumped as separation of C3H8 and C3H6 was not always attainable. Nonetheless, it is noted that C3H8 comprised in all cases the bulk of this lump. Increasing \(\:SEI\) had a limited effect on product selectivities, with that of C2H6 showing initially a small rise, subsequently stabilising, while, conversely, those of C2H4 and C2H2 at first decreasing before exhibiting a small increase. Given the minor changes in hydrocarbons selectivities, the selectivity to H2 also remained relatively stable at values slightly over 30%. Selectivity trends are consistent with prior works [9] and the primary activation mechanism of CH4 being through electron impact reactions [59]. Simulations have estimated at 300 K reaction (18) to be most probable (79%), followed by (19) (15%) and (20) (5%) [71].
Subsequent recombination reactions in the plasma lead to higher hydrocarbons’ formation, with the high C2H6 selectivity indicating that methyl radical coupling (21) is pronounced [9, 59, 72].
The high C3 selectivity further suggests that electron impact reactions with C2H6 lead to its dehydrogenation and further recombination with CH3 radicals according to reactions (22) and (23).
Unsaturated C2 hydrocarbons form from either electron impact reactions of C2H6 and C2H5 or directly from coupling of methylidene and methylidyne radicals. The overall lower selectivity of C2H4 and particularly C2H2 indicates the low participation of these pathways at our conditions. Considering the higher threshold energies of the electron impact reactions involved in the formation of CHx radicals versus those for C2Hy radicals, the slightly increased selectivity of C2 species observed at the highest \(\:SEI\) tested suggests a promotion of the CHx-based pathways at higher applied powers.
The selectivity to unknown carbon products, \(\:{S}_{{C}_{unknown}}\), ranges from approximately 17% to below 8% as \(\:SEI\) increases. These unknown products are primarily suggested to be C4 species, which were not possible to be identified by GC in these experiments. In the work of Xu and Tu [9], substantial production of butane was reported, suggested to originate from the direct coupling of C2H5 radicals:
C2H5 radicals are expected to be at high densities at our conditions, considering the high concentration of C2H6 across the range of conditions, supporting the possibility of C4 production, as will also be demostrated in subsequent sections. Alternatively, C4 production could also be taking place via coupling reaction (25).
Given the high concentration of propane, the density of C3H7 radicals would be similarly high, while the rates of reactions (24) and (25) at our experimental conditions are comparable at approximately 107 m3 mol− 1 s− 1 [73].
Nonetheless, the carbon lack can also partially be attributed to the inability to quantify precisely products at the very low conversions observed at low \(\:SEI\). The carbon balance closure being above 98% in these experiments, despite the missing carbon and the almost linear decrease of the latter as conversion increases, further supports this argument.
Variation of Feed Flowrate
Figure 4 presents the effect of \(\:SEI\) on conversion and product selectivities following a variation of CH4 inlet flowrate from 10 to 120 Nml min− 1 at constant applied power. For these experiments, the amplitude of the applied voltage was maintained at 10 kV, resulting in an applied power of approximately 35 W and an \(\:SEI\) that ranged from 17 to 212 kJ l− 1 as the flowrate decreased. The substantial and relatively abrupt rise in the selectivity of unknown hydrocarbons at \(\:SEI\) values above 50 kJ l− 1 is noteworthy and will be elaborated below. At lower \(\:SEI\) values, results are in line with the trends discussed in relation to Fig. 3, with C2H6 and C3 (primarily C3H8) again being the dominant products. The selectivity of C2H6 in this low \(\:SEI\) range is higher than that of the lumped C3; however, as \(\:SEI\) increases the lumped C3 selectivity becomes the largest again. These trends further support the previously discussed mechanism of C3H8 formation primarily taking place via methyl addition to C2H5, the latter formed from the dehydrogenation of C2H6 via electron impacts. The rise in the selectivity of unknown hydrocarbons as \(\:SEI\) increases is accompanied by an overall decrease in the selectivity of all C2 and C3 products, consistent with the stipulated formation of unidentified C4 hydrocarbons via reactions (24) and (25). It is noteworthy that the abrupt rise in the selectivity of unknown hydrocarbons at \(\:SEI\) above 50 kJ l− 1 is accompanied by a similarly sharp drop in the selectivity of C2H6, in agreement with these pathways, and particularly that based on C2H5 coupling reaction (24), being active. In these experiments, the \(\:SEI\) reaches values much higher to those obtained in Fig. 3, in further support of the proposed mechanism, as C2H5 radicals’ production via electron impacts would be promoted at higher \(\:SEI\). The findings are in further agreement with those reported in the works of Xu and Tu [9] and Lü and Li [74] during similar residence time variation experiments. The selectivity to H2, as it is linked to the identified hydrocarbons, follows the inverse trend of \(\:{S}_{{C}_{unknown}}\) decreasing with increasing \(\:SEI\) from values close to 40% to slightly below 20%. Soot-like deposits on the reactor walls were also observed at the higher \(\:SEI\) values tested, indicating that a fraction of carbon imbalance is due to the formation of carbonaceous material either due to complete methane decomposition or due to polymerisation of higher hydrocarbons.

CH4 conversion and product selectivities vs. SEI varying flowrate in non-catalytic non-oxidative methane coupling. Flowrate: 10–120 Nml min-1 (100% CH4), Residence time: 11.3–0.9 s, Voltage: 10 kV (amplitude)
Catalytic Results
The catalytic experiments were performed at a fixed \(\:SEI\) of ≈ 42 kJ l− 1 using constant power (17.6 W), flowrate (25 Nml min− 1), and frequency (≈ 25 kHz), to allow for discriminating the impact of the catalyst and packing materials placed within the plasma zone. The frequency was again the resonant value of the circuit, while the \(\:SEI\) was set at approximately the middle of the range studied in relevant literature (typically from 1 to 100 kJ l− 1) [4]. The flowrate was selected to maintain a similar residence time to that of the non-catalytic runs and achieve a measurable conversion at relatively low selectivity to unknown carbon products. Specifically, for the flowrate variation non-catalytic experiments (Fig. 4), at the applied power of 35 W, an \(\:SEI\) of 42 kJ l− 1 was obtained at a CH4 flow of 50 Nml min− 1, which further corresponded to a residence time of 2.2 s for the empty tube. For the catalytic experiments, assuming a bed porosity of 0.4, the applied power of 17.6 W and CH4 flow of 25 Nml min− 1 corresponded to a similar residence time of 1.8 s. For this set of experiments, improved GC analysis allowed the detection of up to C6 hydrocarbons, significantly reducing the amount of unidentified products.
The reducibility of the home-made calcined catalysts, Ni/Al2O3 and Fe/Al2O3, was investigated by TPR analysis, and the profiles are presented in Figure S1 in the Supplementary Information (SI). Regarding the Ni catalyst, two H2 consumption peaks are observed. The first peak at 320 oC is attributed to the reduction of NiO, while the second peak at 600 oC is ascribed to the reduction of Ni species strongly interacting with the support. For the Fe catalyst, a single reduction peak at 440 oC is visible, indicating the presence of iron oxide at one oxidation state. The diffractograms of the home-made calcined catalysts are shown in Figure S2 in the SI. For the Fe sample, the hematite α-Fe2O3 structure is identified at 35.6° 2θ, while for the Ni sample the peak at 37.4° 2θ indicates the NiO phase. In both cases, the peaks ascribed to Fe or Ni crystalline phases are hard to detect suggesting the good dispersion of the latter over the γ-Al2O3 support. Peaks characteristic of the γ-Al2O3 support are visible at 37°, 46° and 67° 2θ on both samples.
Methane Conversion and Electrical Properties Over Different Packings
CH4 conversion for all packings tested saw either a decrease in comparison to the empty reactor (for Ni/Al2O3, Rh/Al2O3 and pure Al2O3) or remained largely unchanged at approximately 40% (for Pd/Al2O3, Pt/Al2O3 and Fe/Al2O3) (Fig. 5). The drop in conversion over Rh/Al2O3, a typically high-activity formulation in C1 thermal catalysis, is particularly noted. Electrical diagnostics data (Table 1) further showed that the introduction of packing into the plasma zone led in most cases to a decrease of the peak-to-peak charge, \(\:{Q}_{pk-pk}\), and the net stored charge, \(\:\varDelta\:Q\), indicative of a reduction of the charge deposited into the plasma. For \(\:{Q}_{pk-pk}\), the largest drop is observed over Fe/Al2O3, while the lowest value of \(\:\varDelta\:Q\) is observed for the Rh/Al2O3 catalyst. The effective capacitance, \(\:{C}_{eff}\), for all packings is also lower in comparison to the empty reactor, suggesting again a reduction of the overall discharge area [53, 54]. As discussed by Tu et al. [53], the presence of catalyst pellets leads to a modification of the discharge behavior from microdischarges in the empty case to a combination of surface discharges and possibly weak microdischarges in the packed case. The reduction in \(\:{C}_{eff}\) could imply that the surface discharges are not spreading effectively over the catalyst, possibly focusing between the contact points between particles or metal particles [28]. The latter possibility is further demonstrated by \(\:{C}_{eff}\) being lower for all metal loaded catalysts in comparison to pure Al2O3. The drop in charge and effective capacitance led to a lower electron density as calculated by BOLSIG+, indicating a reduced availability of electrons for impact reactions in the cases of packed reactors. Nonetheless, the reduced electric field strength is seen to increase upon introduction of packing, which in principle should result in more effective plasma generation. As discussed in previous modelling studies, though, this enhancement is usually focused on the contact points between particles rather than affecting the entire discharge volume [53, 75,76,77,78]. The stronger electric field does lead to a higher electron temperature calculated by BOLSIG+ in all packed reactors in comparison to the empty case, which is expected to promote conversion and counterbalance in part the negative effects induced by packing. We note however the work of Gadkari and Gu [76], who via 2D simulations observed that even though the average electric field strength and electron temperature increase in a packed DBD compared to the empty case, the average electron density decreases substantially possibly impeding reactor performance. As evidenced in Table 1, electrical diagnostics data are in excellent agreement with the observations of the authors.

CH4 conversion and product selectivities for various catalyst packings in catalytic non-oxidative methane coupling. Flowrate: 25 Nml min-1 (100% CH4), Residence time: 1.8 s, Power: 17.6 W
The impact of packing on plasma generation is further evident in the Lissajous plots (Fig. 6), whose “almond” shape deviates from that of a parallelogram expected for ideal DBDs and obtained for the case of the empty reactor [51]. This shape, characteristic of the presence of surface discharges, indicates that the plasma is not “fully bridged” during the entire voltage cycle [51], a condition that leads to energy losses. For all packings, similar deviation from the ideal shape is observed, with the small differences in voltage and charge profiles affecting electronic properties in Table 1.

Lissajous plots for various catalyst packings in catalytic non-oxidative methane coupling. Flowrate: 25 Nml min-1 (100% CH4), Residence time: 1.8 s, Power: 17.6 W
Product Selectivities and Energy Cost
Products selectivities vary over the different catalysts tested with the production of light hydrocarbons being enhanced for all catalysts in comparison to that over the empty reactor (Fig. 5). Considering that for all metals the differences in electrical properties were small, the selectivity trends provide evidence of the impact of the active metal phase. The combined selectivity to C2-C3 hydrocarbons with plasma alone and Al2O3 was approximately 50% and 55%, rising to as high as 71% and 76% for the Pd and Pt samples, respectively, the two best performing catalysts in terms of CH4 conversion. Similarly, C4-C6 cumulative selectivity drops from 27% for plasma alone to below 15% for Pd and Pt. These differences are mainly attributed to a large shift in C2H6 selectivity, which approached 60% for Pd and Pt, much higher to the equivalent for an empty reactor (29%) and pure Al2O3 (34%). As discussed in “Non-catalytic results”, the production of C4 and higher hydrocarbons is linked to the coupling of C2Hy and CHx radicals, originating from C2H6 and CH4 electron impact dissociations. The combined reduction in higher hydrocarbons and increased ethane selectivity indicate that Pt and Pd promote the hydrogenation of C2 radicals on the catalyst surface. This is further evidenced in the reduction in the selectivity towards unsaturated hydrocarbons, C2H4 and C2H2, over these catalysts in comparison to the other metals and pure Al2O3 packing. Interestingly, over the empty reactor, selectivity to iso-butane (≈ 6%) and butene (≈ 6%) was higher than that of n-butane (≈ 2%). Over all catalysts, and particularly Pt and Pd, the selectivity to the branched and unsaturated C4 compounds exhibited a measurable drop to ~ 3%, while that to n-butane remained largely unaffected (≈ 1.5%). These observations further demonstrate the promotion of surface hydrogenation of C2 and C3 radicals and/or unsaturated compounds over these two metals. Finally, there is a reduction in the selectivity to unknown carbon, suggested to be long chain hydrocarbons that originate from the further coupling or polymerisation of C2 to C4 species. The selectivity to H2 reaches its highest values in the case of Rh, Pt and Pd due to the low selectivity in unknown hydrocarbons and in the empty reactor due to the low selectivity in C2H6. The decrease in unknown carbon species is also evident from the reduction of coke deposits over Pt and Pd in comparison to all other packings. It is noteworthy that the composition of products remained largely stable across the duration of each experimental run, suggesting an overall dominant effect of the plasma chemistry with specific contributions from the catalyst as discussed. Nonetheless, the formation of carbonaceous deposits, ranging in selectivity from 7% (for Pt) to as high as 20% (for Fe), indicates that the long-term stability of the reactor operation would eventually be impacted. To quantify the level of deactivation over each metal, extended time-on-stream experiments would be required, however overheating issues with the HV AC power supply prevented these from being carried out in this work.
Qualitatively, results are in line with the well documented hydrogenation activity of Pt and Pd and prior works on NOCM [28,29,30], where a significant increase in light hydrocarbons with higher loadings of Pd or Pt was reported. Nonetheless, those works also observed that the introduction of the catalyst into the plasma zone led to mild to moderate decrease in the conversion of CH4. García-Moncada et al. suggested that it is the power that determines CH4 conversion and attributed the decrease in conversion over Pd catalysts versus the empty reactor to the hydrogenation of CH3 radicals back to CH4 [30]. Plasma-catalysis microkinetic simulations over Ni by Maitre et al. further suggested that the catalytic hydrogenation of CH3 radicals negatively affected the conversion of CH4 [79]. The possibility of these pathways cannot be excluded in this work; however, as discussed, differences in electrical properties between packed and blank reactors indicate that plasma generation efficiency is overall negatively affected by the presence of the catalysts even at constant power. The Pd, Pt and Fe catalysts maintained CH4 conversion at the level of the blank reactor suggesting that moderate contributions from the catalysts manage to overcome in part this reduction in plasma generation efficiency. The optimum conversion and selectivity over Pd and Pt resulted in the maximum C2+ hydrocarbon yield of 34% achieved over these catalysts, compared to approximately 30% for the empty reactor (Figure S3 in the SI). The energy cost for methane conversion over Pt and Pd was comparable to that over the empty reactor at approximately 2.4 \(\:\text{M}\text{J}\:{\text{m}\text{o}\text{l}}_{C{H}_{4}}^{-1}\); however, a significantly lower energy cost for the production of C2+ hydrocarbons of 6.5 \(\:\text{M}\text{J}\:{\text{m}\text{o}\text{l}}_{{C}_{2+}}^{-1}\) is achieved over Pt and Pd versus 9 \(\:\text{M}\text{J}\:{\text{m}\text{o}\text{l}}_{{C}_{2+}}^{-1}\) over the empty reactor (Figure S3). On Fe, despite the high conversion, the pronounced selectivity to coke and unknown carbon products resulted in reduced C2+ yield of 25%, with all other catalysts performing even worse on account of, primarily, reduced conversion and, secondly, lower C2+ selectivities.
Co-Feeding of Noble Gases
Noble gases can have a significant effect on plasma generation, electrical properties, and methane conversion acting as energy transfer agents [43, 80]. These benefits can possibly counterbalance the cost of feeding, separating, and recycling them in plasma reactors [80, 81]. As discussed in the Introduction, most relevant works have used high noble gas concentrations, typically above 90%, which would be challenging to apply in a realistic process. In our previous modelling work, energy efficiency enhancements were observed at methane concentration of 25% and below due to electron temperature improvements and in the case of Ar promotion of alternative methane activation pathways (elaborated also below) [60]. Based on these findings, and considering the cost of co-feeding expensive noble gases, the impact of noble gas co-feeding was investigated by carrying out experiments at a 75% dilution of helium or argon at a constant power of 17.6 W and frequency of ≈ 25 kHz. Methane flowrate was maintained at 25 Nml min− 1, as in “Catalytic results”, resulting in a higher total flowrate of 100 Nml min− 1 in these experiments and accordingly a lower \(\:SEI\) of ≈ 10.5 kJ l− 1. The increased flowrate implies that the space velocity is also higher in these experiments, which would in principle affect negatively the performance. Alternatively, the power or total flowrate could have been adjusted so that the \(\:SEI\) or the space velocity are kept equal to that in the previous section, however that would imply a potentially drastic change in the energy provided per methane molecule. By keeping the methane molar flow rate and applied power constant, this ratio (\(\:J\:mo{l}_{C{H}_{4}}^{-1}\)) was also equal across all experiments making possible to evaluate if the addition of noble gases is justifiable from a performance perspective.
Methane Conversion Over Different Packings
Figures 7 and 8 present the results with He and Ar dilution, respectively, where noticeable differences are observed in comparison to the pure CH4 results (Fig. 5). For both He and Ar co-feeding, CH4 conversion for the empty reactor is lower than that for pure methane (13.9% for CH4/He and 20.4% for CH4/Ar vs. 39.4% for CH4), which is as expected considering the higher space velocity and lower \(\:SEI\) in the co-feeding experiments. Contrarily, the conversion for pure CH4 that would result at a \(\:SEI\) similar to that of the co-feeding experiments would be much lower than that observed in the co-feeding experiments (below 5% according to Fig. 4), with the much higher molar flowrate of CH4 (100 vs. 25 Nml min− 1) clearly contributing to this difference. The presence of noble gases in the empty reactor does not appear to be able to overcome the large difference in \(\:SEI\) that occurs when maintaining the same CH4 molar flow. Nonetheless, a positive impact on the electrical properties of the discharge is to be noted (Tables 1, 2 and 3), based on the Lissajous plots collected for the co-feeding cases (Figures S6 and S7 in the SI for He and Ar co-feeding, respectively). Higher peak-to-peak charge (215.5 nC for He and 203.2 nC for Ar vs. 184.3 nC for pure CH4), effective capacitance (13.60 pF for He and 13.52 pF for Ar vs. 11.91 pF for pure CH4), inlet electron density (2.19 × 1014 m− 3 for He and 2.52 × 1014 m− 3 for Ar vs. 2.12 × 1014 m− 3 for pure CH4), and inlet/outlet electron temperatures (3.21/2.85 eV for He and 3.48/3.01 eV for Ar vs. 2.26/1.11 eV for pure CH4) values should all increase the successful electron impacts, promoting conversion. Similar observations were made by Jo et al., who reported increased electron temperatures with argon at all reduced electric field strengths and voltages tested, and with helium at low reduced electric field strengths [43]. Moreover, in the same work, higher CH4 conversion was achieved with argon co-feeding compared to with helium, also in agreement with the present study.

CH4 conversion and product selectivities for various catalyst packings in catalytic non-oxidative methane coupling under helium dilution. Flowrate: 100 Nml min-1 (25% CH4, 75% He), Residence time: 0.45 s, Power: 17.6 W

CH4 conversion and product selectivities for various catalyst packings in catalytic non-oxidative methane coupling under argon dilution. Flowrate: 100 Nml min-1 (25% CH4, 75% Ar), Residence time: 0.45 s, Power: 17.6 W
A complex interplay between reactivity and noble gases presence becomes evident from the catalytic results (Figs. 7 and 8). For all packings, a higher conversion in comparison to the empty reactor is observed for both He and Ar co-feeding. This is in stark contrast to the pure CH4 results (Fig. 5), where either a decrease or no measurable difference in conversion was observed for packed cases in comparison to the empty reactor. For He co-feeding (Fig. 7), conversion over each catalyst is still lower than that observed with pure CH4 over the same catalyst, although this should again be largely attributed to the lower \(\:SEI\) used. For Ar co-feeding (Fig. 8), a higher CH4 conversion is obtained for each catalyst in comparison to the equivalent pure CH4 results with the same catalyst, which is particularly noteworthy when the large \(\:SEI\) and residence time differences are accounted for (4 times lower to pure CH4 experiments). Over Pt and Pd, the two best performing catalysts in “Catalytic results”, Ar co-feeding led to an increase of conversion to 51% and 56%, respectively, vs. 38% and 39% for pure CH4, and 32% and 32% for He co-feeding, strongly suggesting that synergistic effects or alternate pathways are active in the presence of argon.
Electrical Diagnostics and Electron Properties
For all catalysts, most electrical properties are either enhanced (\(\:{Q}_{pk-pk}\), \(\:{C}_{eff}\), \(\:{n}_{e,in}\)) or show minor variation (\(\:\varDelta\:Q\), \(\:{n}_{e,out}\)) for both He and Ar co-feeding when compared to the equivalent values for pure CH4 (Tables 1, 2 and 3). The increase in inlet electron temperature is particularly pronounced, rising from 2.66 eV and 2.60 eV, over Pt and Pd, for pure CH4 to 3.79 eV and 3.93 eV for He co-feeding and to 4.15 eV and 4.12 eV for Ar co-feeding. Moreover, it is noteworthy, that during co-feeding the electron temperature decreases a lot less between inlet and outlet conditions in comparison to pure CH4. For the latter, \(\:{T}_{e,out}\) is typically 30% lower than \(\:{T}_{e,in}\), whereas for He and Ar co-feeding this drop ranges from 15 to 20%, indicating that noble gas co-feeding assists in sustaining a high electron energy across the catalyst bed.
Even though the ionization threshold energies of noble gases are higher than methane (He 24.6 eV, Ar 15.8 eV, CH4 12.6 eV), noble gas plasmas have lower breakdown voltages than molecular ones [36, 60]. Indeed, for all packings the breakdown voltage was estimated from the Lissajous plots to be approximately 1.3 kV for pure CH4 results, while for He and Ar co-feeding it averaged at 0.96 and 1.06 kV, respectively (Table S1 in the SI). This behaviour is linked to the small size of the noble gas atoms that result in longer mean free paths and less frequent electron collisions in comparison to a pure methane plasma. Consequently, electrons are accelerated more for a given field, leading to higher electron temperatures [36, 60]. The latter enhancement (in agreement with electron temperature results presented in Tables 1, 2 and 3) is critical to the ability of noble gases to promote CH4 conversion [60]. Argon ionisation threshold energies are lower than those of helium, further justifying the higher electron temperatures calculated for the former (Tables 2 and 3).
Electron Impact Collision Rates, Penning Pathways, and Impact on Catalytic Rates
The impact of these effects is further evident in Fig. 9 that presents collision rates of the electron impact dissociation reactions of CH4 towards CH3, CH2 and CH radicals as calculated with BOLSIG+. Specifically, rates are presented for all cases studied, at both inlet and outlet conditions using the experimental reactor compositions and the electrical diagnostics as presented in Tables 1, 2 and 3. For each set of experiments carried out (pure CH4, CH4/He and CH4/Ar), the differences among catalysts are overall limited, with the main improvement being observed in comparison to the empty reactor case. Comparing, however, the cases of noble gas co-feeding with that of pure methane reveals major enhancements in the collision rates for all catalysts studied. At reactor inlet conditions, there are at least two orders of magnitude higher methane dissociation rates calculated in the presence of Ar or He compared to pure CH4, with the difference increasing to six orders of magnitude at the reactor outlet conditions. Moreover, the favourable electron temperature in noble gas presence enables even the high threshold energy dissociation to methylidyne radicals to proceed, whereas in pure CH4 this reaction is essentially not taking place. An equivalent comparison of BOLSIG+ electron collision rates is presented in the SI in Figure S8 for CH4 excitations and in Figure S9 for CH4 ionizations. For the low threshold energy vibrational excitations to \({\rm{C}}{{\rm{H}}_{\rm{4}}}{\rm{(\nu 24)}}\) and \({\rm{C}}{{\rm{H}}_{\rm{4}}}{\rm{(\nu 13)}}\) the differences for all catalysts and feed compositions are minor. For the high threshold energy processes, though, namely electronic excitation to \({\rm{CH}}_{\rm{4}}^{\rm{*}}{\rm{(7}}{\rm{.9eV)}}\) and ionizations to \(\:\text{C}{\text{H}}_{\text{4}}^{\text{+}}\), \(\:\text{C}{\text{H}}_{\text{3}}^{\text{+}}\), \(\:\text{C}{\text{H}}_{\text{2}}^{\text{+}}\) the collision rates in noble gas presence are substantially larger than those in pure CH4. The promotion of the electronic excitation is of notice as the pathway has been identified in plasma-kinetic simulations to be contributing as high as 70% of methane’s conversion towards CH3 formation via the self-dissociation of \({\rm{CH}}_{\rm{4}}^{\rm{*}}{\rm{(7}}{\rm{.9eV)}}\) [59].

BOLSIG+ reaction rates for major electron impact CH4 dissociation reactions calculated as described in “Electrical diagnostics” using the electrical diagnostics data in Tables 1, 2 and 3 and the inlet or outlet reactor composition from experiments using various catalyst packings in catalytic non-oxidative methane coupling at flowrate of 25 Nml min-1 (100% CH4) or 100 Nml min-1 (25% CH4, 75% Ar/He) and power of 17.6 W
Electrical diagnostics and BOLSIG+ results underline the beneficial effects on discharge formation due to noble gas presence. A further contributing factor is the ability of electronically excited states of noble gas species to act as collision partners transferring energy to CH4 according to Penning dissociation and ionisation reactions, where the multiple possible processes are presented in equations (26) and (27), respectively [43, 82,83,84].
These processes, to the degree that have been documented in literature, are characterised by rate constants that are typically two orders of magnitude higher for argon in comparison to those for helium [37, 42]. In the modelling work of Maitre et al., it was found that Penning dissociation channels had a measurable contribution to methane conversion in CH4-Ar mixtures when CH4 concentration was below 50% [60]. This was primarily due to the lower electronic excitation threshold energies of argon compared to helium, which resulted to a significant fraction of the electrons’ energy being channelled into the creation of Ar excited states. All these observations are in line with presented experimental results and the overall higher activity observed for Ar co-feeding experiments.
The latter effects cannot however explain the conversion differences observed among catalysts tested, as they pertain mainly to the activation of methane within the plasma and not on the catalyst surface. Vibrationally excited states have been discussed in literature to affect NOCM turnover frequencies depending on the binding strength of the metal [85]. Nonetheless, based on the collision rates calculated in this work (Figure S8), the populations of \({\rm{C}}{{\rm{H}}_{\rm{4}}}{\rm{(\nu 24)}}\) and \({\rm{C}}{{\rm{H}}_{\rm{4}}}{\rm{(\nu 13)}}\) are not expected to be significantly affected by noble gas co-feeding, while during the pure CH4 case there were no metal-dependent positive effects on turnover observed. However, as evidenced from the collision rates in Fig. 9, Ar or He co-feeding promotes the rates of electron impact CH4 dissociations, which would enhance the population of CHx radicals in noble gas presence. Microkinetic simulations have further demonstrated that plasma-produced radicals can significantly affect surface chemistry during NOCM [79] and CH4 partial oxidation [86] with effects expected to be metal-dependent. Alternatively, collision induced surface reactions is a mechanism proposed in literature wherein impinging noble gas atoms provide the energy required to overcome surface reaction energy barriers [19]. The latter process has been observed both in relation to the dissociative adsorption of CH4 on Ni(111) aided by Ar presence [19, 87, 88], but also for the formation of CO2 from co-adsorbed CO and O2 on Pt(111) aided by Xe atoms [89, 90]. Hence, co-feeding of He or Ar is suggested to be assisting both in the activation of methane on the catalyst surface but also in the promotion of subsequent surface reactions of CHx species either by enhancing the population of the latter or by altering the energetics of their pathways.
Product Selectivities and Energy Cost
Selectivities obtained are largely similar between the co-feeding and pure CH4 cases (Figs. 7 and 8). Pt and Pd are again characterised by their hydrogenation activity and coking resistance [30], as evidenced by the high selectivity to ethane (44-59% depending on noble gas and metal) and low amount of carbon deposits. Selectivity to higher hydrocarbons and unknown carbon products is accordingly lower over Pt and Pd. As with the pure CH4 trends, selectivity to H2 is higher in cases that the selectivity to unknown carbon products is lower, namely Pt, Pd and pure Al2O3 for both He and Ar co-feeding. Notably, equivalent selectivity trends are observed for all catalysts regardless of the gas composition (e.g., Fe and Pt have the lowest and highest selectivity to C2H6 for all feed compositions tested), suggesting that the surface reactions proceed in a similar manner over each metal and that methyl radical generation is what is primarily affected by the noble gas presence. Nonetheless, there is a relative increase in unsaturated C2 hydrocarbon production with noble gas co-feeding in comparison to pure CH4 over the same catalysts, evidencing a more effective generation of CH2 and CH radicals which further couple to form C2H4 and C2H2. The latter observation is further in agreement with the higher collision rates for electron impact methane dissociations presented in Fig. 9 and the higher electron temperatures obtained from BOLSIG+. A further exception to these trends is the Rh catalyst with Ar co-feeding which showed a higher conversion compared to Al2O3, in contrast to the conversion order obtained with He co-feeding and pure CH4. This evidences that surface activity was low with pure CH4 and He presence but was able to be promoted due to the enhancement from Ar co-feeding, resulting in the catalyst performing better that the pure support. The selectivity to unknown hydrocarbons is higher in most cases with the Ar co-feeding in comparison to the pure CH4 and He co-feeding cases. These unknown hydrocarbons are most likely long chain molecules deposited on the walls or post-catalyst part of the reactor as coke deposits on the bed are lower with Ar co-feeding in most cases. Moreover, dark yellow waxy residues were observed on the reactor walls during Ar co-feeding, in agreement with Jo et al. who attributed similar formations to carbonaceous deposits [80]. Scarduelli et al. moreover analysed these residues with a range of spectroscopic techniques finding they consisted of a mixture of hydrocarbons, with the most represented moiety being the gem-dimethyl group [91].
The overall effect of noble gases co-feeding on C2+ yield and energy cost in comparison to pure CH4 is shown in Fig. 10, while detailed energy cost and yield data for Ar and He co-feeding are provided in Figures S4 and S5 in the SI. Besides the empty reactor and the pure Al2O3, for each metal catalyst the highest yield is obtained with Ar co-feeding, while He co-feeding results in yields even lower than those of the pure CH4 case. The highest overall yields of 41% and 44% are achieved with Pt and Pd, respectively, during Ar co-feeding, in line with the higher methane conversion and selectivity to hydrocarbons attained with these two catalysts. This optimal performance leads to the lowest C2+ energy costs achieved in this study of 4.8 and 4.7 MJ mol− 1. Considering the lower \(\:SEI\) during noble gases co-feeding, these results clearly underline the beneficial effects of Ar presence in the non-oxidative coupling of methane, which coupled with active Pt or Pd catalysts can substantially enhance the process efficiency. This is further evidenced in Table 4 which compares the optimal results achieved in this study with those reported in equivalent works in literature, namely plasma-catalysis NOCM studies in DBD reactors. In studies where some data were not available, approximate values based on provided information are displayed. For studies that further presented time-on-stream data, the initial (and typically highest) values are used. The energy cost for methane conversion in this work in the presence of Ar over the Pd/Al2O3 catalyst is the second lowest of all studies. Interestingly, only the work of Kasinathan et al. reported a lower energy cost [27], although on an oxide-based catalyst and at a much lower conversion (23% vs. 56%) and higher percentage of argon in the feed stream (90% vs. 75%). Furthermore, the optimal combination of CH4 conversion and C2+ selectivity achieved in the present work for the Pd/Al2O3 catalyst and Ar cofeeding result in the highest reported C2+ yield amongst all the studies.

C2+ energy cost and yield for various catalyst packings in catalytic non-oxidative methane coupling. Flowrate: 25 Nml min-1 (100% CH4) or 100 Nml min-1 (25% CH4, 75% Ar/He), Power: 17.6 W
Conclusions
The non-oxidative coupling of methane was investigated in a DBD reactor over a range of metals and noble gas co-feeding cases, further supported via electrical diagnostics and BOLSIG+ calculations. When pure CH4 was used, and depending on metal, minor (Pt, Pd, and Fe) or negative (Ni, Rh) impact on conversion was observed. All catalysts affected the production of C2 hydrocarbons, with Pt and Pd found the most performant in terms of enhancing C2 selectivity and minimizing carbon deposits. Electrical diagnostics and BOLSIG+ calculations overall revealed similar enhancement of properties, namely an increase of electron temperature but a decrease of deposited charge. Selectivity differences evidenced that the observed improvements over Pt and Pd were driven by the hydrogenation activity of the metals, which impacted primarily the pathways of C2 and C3 radicals. Co-feeding of helium or argon was not able to enhance the performance of plasma only reactors; however substantial conversion and selectivity differences were observed during plasma catalysis. Argon co-feeding specifically was able to enhance the conversion achieved over each catalyst in comparison to the pure CH4 case, despite large differences in \(\:SEI\) and residence time. Electrical diagnostics indicated substantial enhancements in electron temperature induced by noble gas co-feeding, which promoted electron impact dissociations of methane as demonstrated via BOLSIG+ calculations. Optimal performance over argon was attributed to Penning pathways that promote the dissociation and ionization of methane in the plasma phase, while differences among catalysts were ascribed to collision induced surface reactions enhancing catalytic methane and CHx pathways. C2+ yields over 40% were achieved with Pt and Pd during Ar co-feeding at an energy cost of ≈ 4.7 \(\:\text{M}\text{J}\:\text{m}\text{o}{\text{l}}_{{\text{C}}_{2+}}^{-1}\).
Data Availability
The authors declare that the data supporting the findings of this study are available within the paper and its Supplementary Information files. Should any raw data files be needed in another format they are available from the corresponding author upon reasonable request.
References
Zichittella G, Pérez-Ramírez J (2021) Status and prospects of the decentralised valorisation of natural gas into energy and energy carriers. Chem Soc Rev 50:2984–3012. https://doi.org/10.1039/D0CS01506G
Galadima A, Muraza O (2016) Revisiting the oxidative coupling of methane to ethylene in the golden period of shale gas: A review. J Ind Eng Chem 37:1–13. https://doi.org/10.1016/j.jiec.2016.03.027
Zhang K, Zhang F, Wu Y-R (2021) Emerging technologies for conversion of sustainable algal biomass into value-added products: A state-of-the-art review. Sci Total Environ 784:147024. https://doi.org/10.1016/j.scitotenv.2021.147024
Scapinello M, Delikonstantis E, Stefanidis GD (2017) The panorama of plasma-assisted non-oxidative methane reforming. Chem Eng Process Process Intensif 117:120–140. https://doi.org/10.1016/j.cep.2017.03.024
Maslova V, Nastase R, Veryasov G et al (2024) Current status and challenges of plasma and plasma-catalysis for methane coupling: A review. Prog Energy Combust Sci 101:101096. https://doi.org/10.1016/j.pecs.2023.101096
Maitre P-A, Bieniek MS, Kechagiopoulos PN (2020) Plasma-enhanced catalysis for the upgrading of methane: A review of modelling and simulation methods. React Chem Eng 5:814–837. https://doi.org/10.1039/D0RE00024H
Puliyalil H, Lašič Jurković D, Dasireddy VDBC, Likozar B (2018) A review of plasma-assisted catalytic conversion of gaseous carbon dioxide and methane into value-added platform chemicals and fuels. RSC Adv 8:27481–27508. https://doi.org/10.1039/C8RA03146K
Nozaki T, Okazaki K (2013) Non-thermal plasma catalysis of methane: Principles, energy efficiency, and applications. Catal Today 211:29–38. https://doi.org/10.1016/j.cattod.2013.04.002
Xu C, Tu X (2013) Plasma-assisted methane conversion in an atmospheric pressure dielectric barrier discharge reactor. J Energy Chem 22:420–425. https://doi.org/10.1016/S2095-4956(13)60055-8
Toth JR, Shen X, Lacks DJ, Sankaran RM (2018) Reaction conversion for a plasma-based steady-state flow process is independent of reactor volume. Ind Eng Chem Res 57:6048–6056. https://doi.org/10.1021/acs.iecr.7b05091
Wang B, Yan W, Ge W, Duan X (2013) Methane conversion into higher hydrocarbons with dielectric barrier discharge micro-plasma reactor. J Energy Chem 22:876–882. https://doi.org/10.1016/S2095-4956(14)60267-9
Saleem F, Kennedy J, Dahiru UH et al (2019) Methane conversion to H₂ and higher hydrocarbons using non-thermal plasma dielectric barrier discharge reactor. Chem Eng Process - Process Intensif 142:107557. https://doi.org/10.1016/j.cep.2019.107557
Bogaerts A, Zhang Q-Z, Zhang Y-R et al (2019) Burning questions of plasma catalysis: Answers by modeling. Catal Today 337:3–14. https://doi.org/10.1016/j.cattod.2019.04.077
Chen H, Mu Y, Xu S et al (2020) Recent advances in non-thermal plasma (NTP) catalysis towards C1 chemistry. Chin J Chem Eng 28:2010–2021. https://doi.org/10.1016/j.cjche.2020.05.027
Neyts EC, Bogaerts A (2014) Understanding plasma catalysis through modelling and simulation—a review. J Phys Appl Phys 47:224010. https://doi.org/10.1088/0022-3727/47/22/224010
Juurlink LBF, Killelea DR, Utz AL (2009) State-resolved probes of methane dissociation dynamics. Prog Surf Sci 84:69–134. https://doi.org/10.1016/j.progsurf.2009.01.001
Sheng Z, Watanabe Y, Kim H-H et al (2020) Plasma-enabled mode-selective activation of CH4 for dry reforming: First touch on the kinetic analysis. Chem Eng J 399:125751. https://doi.org/10.1016/j.cej.2020.125751
Whitehead JC (2016) Plasma–catalysis: The known knowns, the known unknowns and the unknown unknowns. J Phys Appl Phys 49:243001. https://doi.org/10.1088/0022-3727/49/24/243001
Neyts EC (2016) Plasma-surface interactions in plasma catalysis. Plasma Chem Plasma Process 36:185–212. https://doi.org/10.1007/s11090-015-9662-5
Wang N, Otor HO, Rivera-Castro G, Hicks JC (2024) Plasma catalysis for hydrogen production: A bright future for decarbonization. ACS Catal 14:6749–6798. https://doi.org/10.1021/acscatal.3c05434
Nozaki T, Chen X, Kim D-Y, Zhan C (2023) Combination of DBD and catalysts for CH4 and CO2 conversion: Basics and applications. Plasma Chem Plasma Process 43:1385–1410. https://doi.org/10.1007/s11090-023-10382-3
Lee DH, Kang H, Kim Y et al (2023) Plasma-assisted hydrogen generation: A mechanistic review. Fuel Process Technol 247:107761. https://doi.org/10.1016/j.fuproc.2023.107761
Xu S, Chen H, Fan X (2023) Rational design of catalysts for non-thermal plasma (NTP) catalysis: A reflective review. Catal Today 419:114144. https://doi.org/10.1016/j.cattod.2023.114144
Lamichhane P, Pourali N, Scott L et al (2024) Critical review: ‘Green’ ethylene production through emerging technologies, with a focus on plasma catalysis. Renew Sustain Energy Rev 189:114044. https://doi.org/10.1016/j.rser.2023.114044
Indarto A (2008) Hydrogen production from methane in a dielectric barrier discharge using oxide zinc and chromium as catalyst. J Chin Inst Chem Eng 39:23–28. https://doi.org/10.1016/j.jcice.2007.10.001
Górska A, Krawczyk K, Jodzis S, Schmidt-Szałowski K (2011) Non-oxidative methane coupling using Cu/ZnO/Al₂O₃ catalyst in DBD. Fuel 90:1946–1952. https://doi.org/10.1016/j.fuel.2010.12.023
Kasinathan P, Park S, Choi WC et al (2014) Plasma-enhanced methane direct conversion over particle-size adjusted MOx/Al₂O₃ (M = Ti and Mg) catalysts. Plasma Chem Plasma Process 34:1317–1330. https://doi.org/10.1007/s11090-014-9574-9
Jo S, Kim T, Lee DH et al (2014) Effect of the electric conductivity of a catalyst on methane activation in a dielectric barrier discharge reactor. Plasma Chem Plasma Process 34:175–186. https://doi.org/10.1007/s11090-013-9505-1
Taheraslani M, Gardeniers H (2020) Plasma catalytic conversion of CH4 to alkanes, olefins and H2 in a packed bed DBD reactor. Processes 8:774. https://doi.org/10.3390/pr8070774
García-Moncada N, van Rooij G, Cents T, Lefferts L (2021) Catalyst-assisted DBD plasma for coupling of methane: Minimizing carbon-deposits by structured reactors. Catal Today 369:210–220. https://doi.org/10.1016/j.cattod.2020.04.028
García-Moncada N, Cents T, van Rooij G, Lefferts L (2020) Minimizing carbon deposition in plasma-induced methane coupling with structured hydrogenation catalysts. J Energy Chem 58:271–279. https://doi.org/10.1016/j.jechem.2020.09.006
Liu L, Das S, Zhang Z, Kawi S (2022) Nonoxidative coupling of methane over ceria-supported single-atom Pt catalysts in DBD plasma. ACS Appl Mater Interfaces 14:5363–5375. https://doi.org/10.1021/acsami.1c21550
Nguyen HM, Gorky F, Guthrie S et al (2023) Plasma catalytic non-oxidative methane conversion to hydrogen and value-added hydrocarbons on zeolite 13X. Energy Convers Manag 286:117082. https://doi.org/10.1016/j.enconman.2023.117082
Ghanbari M, Binazadeh M, Zafarnak S et al (2020) Hydrogen production via catalytic pulsed plasma conversion of methane: Effect of Ni–K2O/Al2O3 loading, applied voltage, and argon flow rate. Int J Hydrog Energy 45:13899–13910. https://doi.org/10.1016/j.ijhydene.2020.03.099
Lee G, Go DB, O’Brien CP (2021) Direct observation of plasma-stimulated activation of surface species using multimodal in situ/operando spectroscopy combining polarization-modulation infrared reflection-absorption spectroscopy, optical emission spectroscopy, and mass spectrometry. ACS Appl Mater Interfaces 13:56242–56253. https://doi.org/10.1021/acsami.1c18169
Lieberman MA, Lichtenberg AJ (2005) Principles of plasma discharges and materials processing, 2nd edn
Lefkowitz JK, Guo P, Rousso A, Ju Y (2015) Species and temperature measurements of methane oxidation in a nanosecond repetitively pulsed discharge. Philos Trans R Soc Math Phys Eng Sci 373:20140333. https://doi.org/10.1098/rsta.2014.0333
Mao X, Rousso A, Chen Q, Ju Y (2019) Numerical modeling of ignition enhancement of CH₄/O₂/He mixtures using a hybrid repetitive nanosecond and DC discharge. Proc Combust Inst 37:5545–5552. https://doi.org/10.1016/j.proci.2018.05.106
Starikovskiy A, Aleksandrov N (2013) Plasma-assisted ignition and combustion. Prog Energy Combust Sci 39:61–110. https://doi.org/10.1016/j.pecs.2012.05.003
Kosarev IN, Aleksandrov NL, Kindysheva SV et al (2008) Kinetics of ignition of saturated hydrocarbons by nonequilibrium plasma: CH₄-containing mixtures. Combust Flame 154:569–586. https://doi.org/10.1016/j.combustflame.2008.03.007
Aleksandrov NL, Kindysheva SV, Kukaev EN et al (2009) Simulation of the ignition of a methane-air mixture by a high-voltage nanosecond discharge. Plasma Phys Rep 35:867. https://doi.org/10.1134/S1063780X09100109
Sun W, Uddi M, Won SH et al (2012) Kinetic effects of non-equilibrium plasma-assisted methane oxidation on diffusion flame extinction limits. Combust Flame 159:221–229. https://doi.org/10.1016/j.combustflame.2011.07.008
Jo S, Lee DH, Kang WS, Song Y-H (2013) Methane activation using noble gases in a dielectric barrier discharge reactor. Phys Plasmas 20:083509. https://doi.org/10.1063/1.4818795
Jo S, Lee DH, Kim K-T et al (2014) Methane activation using Kr and Xe in a dielectric barrier discharge reactor. Phys Plasmas 21:103504. https://doi.org/10.1063/1.4897171
Kundu SK, Kennedy EM, Gaikwad VV et al (2012) Experimental investigation of alumina and quartz as dielectrics for a cylindrical double dielectric barrier discharge reactor in argon diluted methane plasma. Chem Eng J 180:178–189. https://doi.org/10.1016/j.cej.2011.11.039
Barni R, Benocci R, Spinicchia N et al (2019) An experimental study of plasma cracking of methane using DBDs aimed at hydrogen production. Plasma Chem Plasma Process 39:241–258. https://doi.org/10.1007/s11090-018-9940-0
Sun J, Chen Q (2019) Kinetic roles of vibrational excitation in RF plasma assisted methane pyrolysis. J Energy Chem 39:188–197. https://doi.org/10.1016/j.jechem.2019.01.028
Huang B, Zhang C, Bai H et al (2020) Energy pooling mechanism for catalyst-free methane activation in nanosecond pulsed non-thermal plasmas. Chem Eng J 396:125185. https://doi.org/10.1016/j.cej.2020.125185
Loenders B, Michiels R, Bogaerts A (2023) Is a catalyst always beneficial in plasma catalysis? Insights from the many physical and chemical interactions. J Energy Chem 85:501–533. https://doi.org/10.1016/j.jechem.2023.06.016
Kechagiopoulos PN, Angeli SD, Lemonidou AA (2017) Low temperature steam reforming of methane: A combined isotopic and microkinetic study. Appl Catal B Environ 205:238–253. https://doi.org/10.1016/j.apcatb.2016.12.033
Peeters F, Butterworth T (2018) Electrical diagnostics of dielectric barrier discharges. In: Nikiforov A, Chen Z (eds) Atmospheric pressure plasma. IntechOpen, Rijeka
Peeters FJJ, van de Sanden MCM (2014) The influence of partial surface discharging on the electrical characterization of DBDs. Plasma Sources Sci Technol 24:015016. https://doi.org/10.1088/0963-0252/24/1/015016
Tu X, Gallon HJ, Twigg MV et al (2011) Dry reforming of methane over a Ni/Al2O3 catalyst in a coaxial dielectric barrier discharge reactor. J Phys Appl Phys 44:274007. https://doi.org/10.1088/0022-3727/44/27/274007
Francke K-P, Rudolph R, Miessner H (2003) Design and operating characteristics of a simple and reliable DBD reactor for use with atmospheric air. Plasma Chem Plasma Process 23:47–57. https://doi.org/10.1023/A:1022412718224
Kogelschatz U (2003) Dielectric-barrier discharges: Their history, discharge physics, and industrial applications. Plasma Chem Plasma Process 23:1–46. https://doi.org/10.1023/A:1022470901385
Wagner H-E, Brandenburg R, Kozlov KV et al (2003) The barrier discharge: Basic properties and applications to surface treatment. Vacuum 71:417–436. https://doi.org/10.1016/S0042-207X(02)00765-0
Chawdhury P, Bhargavi KVSS, Subrahmanyam C (2021) A single-stage partial oxidation of methane to methanol: A step forward in the synthesis of oxygenates. Sustain Energy Fuels 5:3351–3362. https://doi.org/10.1039/D1SE00557J
Hagelaar GJM, Pitchford LC (2005) Solving the Boltzmann equation to obtain electron transport coefficients and rate coefficients for fluid models. Plasma Sources Sci Technol 14:722–733. https://doi.org/10.1088/0963-0252/14/4/011
Maitre P-A, Bieniek MS, Kechagiopoulos PN (2021) Modelling excited species and their role on kinetic pathways in the non-oxidative coupling of methane by dielectric barrier discharge. Chem Eng Sci 234:116399. https://doi.org/10.1016/j.ces.2020.116399
Maitre P-A, Long J, Bieniek MS et al (2022) Investigating the effects of helium, argon and hydrogen co-feeding on the non-oxidative coupling of methane in a dielectric barrier discharge reactor. Chem Eng Sci 259:117731. https://doi.org/10.1016/j.ces.2022.117731
Floeder K, Fromme D, Raith W et al (1985) Total cross section measurements for positron and electron scattering on hydrocarbons between 5 and 400 eV. J Phys B Mol Phys 18:3347. https://doi.org/10.1088/0022-3700/18/16/019
Szmytkowski C, Stefanowska S, Zawadzki M et al (2015) Electron collisions with methyl-substituted ethylenes: Cross section measurements and calculations for 2-methyl–2-butene and 2,3-dimethyl–2-butene. J Chem Phys 143:064306. https://doi.org/10.1063/1.4927703
Gómez-Ramírez A, Montoro-Damas M, Rodríguez A MA, et al (2017) Improving the pollutant removal efficiency of packed-bed plasma reactors incorporating ferroelectric components. Chem Eng J 314:311–319. https://doi.org/10.1016/j.cej.2016.11.065
Hong J, Aramesh M, Shimoni O et al (2016) Plasma catalytic synthesis of ammonia using functionalized-carbon coatings in an atmospheric-pressure non-equilibrium discharge. Plasma Chem Plasma Process 36:917–940. https://doi.org/10.1007/s11090-016-9711-8
Yun Y (2003) Alternating-current glow and pseudoglow discharges in atmospheric pressure. IEEE Trans Plasma Sci 31:174–176. https://doi.org/10.1109/TPS.2003.808878
Gallon HJ, Tu X, Whitehead JC (2012) Effects of reactor packing materials on H2 production by CO2 reforming of CH4 in a dielectric barrier discharge. Plasma Process Polym 9:90–97. https://doi.org/10.1002/ppap.201100130
Tu X, Whitehead JC (2012) Plasma-catalytic dry reforming of methane in an atmospheric dielectric barrier discharge: Understanding the synergistic effect at low temperature. Appl Catal B Environ 125:439–448. https://doi.org/10.1016/j.apcatb.2012.06.006
Liu SY, Mei DH, Shen Z, Tu X (2014) Nonoxidative conversion of methane in a dielectric barrier discharge reactor: Prediction of reaction performance based on neural network model. J Phys Chem C 118:10686–10693. https://doi.org/10.1021/jp502557s
Chen X, Zhang S, Li S et al (2021) Temperature-independent, nonoxidative methane conversion in nanosecond repetitively pulsed DBD plasma. Sustain Energy Fuels 5:787–800. https://doi.org/10.1039/D0SE01593H
Xu S, Khalaf PI, Martin PA, Whitehead JC (2018) CO2 dissociation in a packed-bed plasma reactor: Effects of operating conditions. Plasma Sources Sci Technol 27:075009. https://doi.org/10.1088/1361-6595/aacd6a
Bie CD, Verheyde B, Martens T et al (2011) Fluid modeling of the conversion of methane into higher hydrocarbons in an atmospheric pressure dielectric barrier discharge. Plasma Process Polym 8:1033–1058. https://doi.org/10.1002/ppap.201100027
Kado S, Urasaki K, Sekine Y et al (2003) Reaction mechanism of methane activation using non-equilibrium pulsed discharge at room temperature. Fuel 82:2291–2297. https://doi.org/10.1016/S0016-2361(03)00163-7
Wang H, You X, Joshi AV et al (2007) USC Mech Version II. High-temperature combustion reaction model of H2/CO/C1-C4 compounds
Lü J, Li Z (2010) Conversion of natural gas to C2 hydrocarbons via cold plasma technology. J Nat Gas Chem 19:375–379. https://doi.org/10.1016/S1003-9953(09)60082-7
Kruszelnicki J, Engeling KW, Foster JE et al (2016) Propagation of negative electrical discharges through 2-dimensional packed bed reactors. J Phys Appl Phys 50:025203. https://doi.org/10.1088/1361-6463/50/2/025203
Gadkari S, Gu S (2018) Influence of catalyst packing configuration on the discharge characteristics of dielectric barrier discharge reactors: A numerical investigation. Phys Plasmas 25:063513. https://doi.org/10.1063/1.5030508
Kang WS, Park JM, Kim Y, Hong SH (2003) Numerical study on influences of barrier arrangements on dielectric barrier discharge characteristics. IEEE Trans Plasma Sci 31:504–510. https://doi.org/10.1109/TPS.2003.815469
Laer KV, Bogaerts A (2016) Fluid modelling of a packed bed dielectric barrier discharge plasma reactor. Plasma Sources Sci Technol 25:015002. https://doi.org/10.1088/0963-0252/25/1/015002
Maitre P-A, Bieniek MS, Kechagiopoulos PN (2022) Plasma-catalysis of nonoxidative methane coupling: A dynamic investigation of plasma and surface microkinetics over Ni(111). J Phys Chem C 126:19987–20003. https://doi.org/10.1021/acs.jpcc.2c03503
Jo S, Lee DH, Song Y-H (2015) Product analysis of methane activation using noble gases in a non-thermal plasma. Chem Eng Sci 130:101–108. https://doi.org/10.1016/j.ces.2015.03.019
Bernardo P, Drioli E, Golemme G (2009) Membrane gas separation: A review/state of the art. Ind Eng Chem Res 48:4638–4663. https://doi.org/10.1021/ie8019032
Zhu W, Inspektor A, Badzian AR et al (1990) Effects of noble gases on diamond deposition from methane-hydrogen microwave plasmas. J Appl Phys 68:1489–1496. https://doi.org/10.1063/1.346679
Balamuta J, Golde MF, Ho Y-S (1983) Product distributions in the reactions of excited noble-gas atoms with hydrogen‐containing compounds. J Chem Phys 79:2822–2830. https://doi.org/10.1063/1.446103
Morgan WL (1992) A critical evaluation of low-energy electron impact cross sections for plasma processing modeling. II: Cl4, SiH4, and CH4. Plasma Chem Plasma Process 12:477–493. https://doi.org/10.1007/BF01447255
Engelmann Y, Mehta P, Neyts EC et al (2020) Predicted influence of plasma activation on nonoxidative coupling of methane on transition metal catalysts. ACS Sustain Chem Eng 8:6043–6054. https://doi.org/10.1021/acssuschemeng.0c00906
Loenders B, Engelmann Y, Bogaerts A (2021) Plasma-catalytic partial oxidation of methane on Pt(111): A microkinetic study on the role of different plasma species. J Phys Chem C 125:2966–2983. https://doi.org/10.1021/acs.jpcc.0c09849
Beckerle JD, Johnson AD, Yang QY, Ceyer ST (1989) Collision induced dissociative chemisorption of CH4 on Ni(111) by inert gas atoms: The mechanism for chemistry with a hammer. J Chem Phys 91:5756–5777. https://doi.org/10.1063/1.457529
Beckerle JD, Yang QY, Johnson AD, Ceyer ST (1987) Collision-induced dissociative chemisorption of adsorbates: Chemistry with a hammer. J Chem Phys 86:7236–7237. https://doi.org/10.1063/1.452325
Vattuone L, Gambardella P, Burghaus U et al (1998) Collision induced desorption and dissociation of O2 chemisorbed on Ag(001). J Chem Phys 109:2490–2502. https://doi.org/10.1063/1.476820
Åkerlund C, Zorić I, Kasemo B (1996) A collision induced reaction: CO2 production on O2 and CO covered Pt(111). J Chem Phys 104:7359–7362. https://doi.org/10.1063/1.471817
Scarduelli G, Guella G, Mancini I et al (2009) Methane oligomerization in a dielectric barrier discharge at atmospheric pressure. Plasma Process Polym 6:27–33. https://doi.org/10.1002/ppap.200800096
Acknowledgements
This study was funded by The Leverhulme Trust (Grant DS-2017-073). James Rogers, a Leverhulme Trust Doctoral Scholar, is part of the 15 PhD scholarships of the “Leverhulme Centre for Doctoral Training in Sustainable Production of Chemicals and Materials” at the University of Aberdeen (Scotland, United Kingdom).
Author information
Authors and Affiliations
Contributions
PNK: Conceptualization, Methodology, Investigation, Writing - Review & Editing, Supervision, Funding acquisition. JR: Investigation, Writing - Original Draft. PAM: Investigation. AJM: Resources, Writing - Review & Editing. MNB: Resources, Writing - Review & Editing.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kechagiopoulos, P.N., Rogers, J., Maitre, PA. et al. Non-Oxidative Coupling of Methane via Plasma-Catalysis Over M/γ-Al2O3 Catalysts (M = Ni, Fe, Rh, Pt and Pd): Impact of Active Metal and Noble Gas Co-Feeding. Plasma Chem Plasma Process (2024). https://doi.org/10.1007/s11090-024-10507-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11090-024-10507-2