
Abstract
To examine the impacts while taking cubic PbTiO3 with various Sr concentrations, a thorough theoretical analysis is conducted. This study evaluates the effects of adding Sr to the ab-initio method using the generalized gradient approximation (GGA), which is used in the Cambridge serial total energy package (CASTEP), on the structural, total, and partial density of states (TDOS/PDOS), modification in the electronic bandgap, and optical properties. All of the PbTiO3 structural properties are in good agreement with previously published values. For 100% Sr substitution, a change in the perfect cubic structure of SrTiO3 is seen. The indirect bandgap of pure PbTiO3 is found to be 1.655 eV. Due to the non-linear band gap dependency on the composition of the material, a reduction in bandgap is recorded by increasing the Sr percentage. As the Sr atoms in PbTiO3 are repeatedly added, the contribution of these orbitals in creating energy states increases. Numerous optical properties have been compared throughout a range of Sr concentrations, and the results show that SrTiO3 exhibits modified optical behaviour and a lower refractive index than PbTiO3. It is also shown how these optical characteristics change with Sr content. We suggest using this SrTiO3 cubic phase as a possible material for optoelectronic applications.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
The ABO3 perovskite materials have a vast range of applications such as infrared sensors, piezoelectric actuators, fuel cells, hard disks, capacitors, ultrasonic transducers and ferroelectricity etc. The name perovskite was first coined by Gustav in 1839, for CaTiO3 (Gillani et al. 2019; Rizwan et al. 2019a; Rizwan et al. 2021a; Kuma et al. 2019). They are also being prepared in the laboratory by experimental techniques such as; sol–gel (Chilibon et al. 2012), co-precipitation (Murugan et al. 2006) and solid-state reaction (Kambale et al. 2014). Lead titanate PbTiO3 (PTO) is studied extensively because it is a visible light photocatalyst so it is used as an oxidizer and has the ability of spontaneous polarization (Mete et al. 2019). They are also used in the manufacturing of detectors, nonvolatile memories and sensors (Mete et al. 2019; Meštrić et al. 2005). Since PTO has ferroelectric material so it has a large band gap and also has a wide absorption spectrum in the UV region (Lemziouka et al. 2021). Lead-based materials have a wide range of applications but these compounds like PTO have adverse effects on the environment and climate (Rizwan et al. 2021b).
At present, there is a big need for alternatives to lead compounds like PTO that have the properties like it but without the involvement of unsafe lead Pb. In this work, the material is made to a deficit in lead by replacing it with Sr atoms and inclusion of strontium modifies its optical and electronic properties a slight increment in band gap energies leads the material towards the higher energy spectrum. Ti and O have covalent interaction which is responsible for the ferroelectric nature of lead titanate (Stashans et al. 2002). PbTiO3 at 490 °C exhibits the phase transformation from tetragonal space group (P4mm) ferroelectric to cubic (Pm-3 m) (Kumar, et al. 2000; Shirane et al. 1956). A detailed explanation of cubic phase of lead titanate (PbTiO3) is given by Jiang et al. (2006). The electrical and optical properties of PbTiO3 have been discussed experimentally by Piskunov et al. (2004), Wang et al. (2014). However, currently, these properties set off a strong interest towards theoretical work (Kuma et al. 2019). In this work, the electrical, structural and optical properties of pure and Sr-doped PbTiO3 have been studied by using GGA-PBE functional. The direct band gap is observed in pure and all doped forms (Pb0.89Sr0.11TiO3, Pb0.67Sr0.33TiO3, Pb0.45Sr0.55TiO3) so there are fewer energy losses as due to phonons further, a prime expansion in band gap values is also observed.
Therefore, the doped-PTO is more useful since it has wider band gaps and as a result its photo-activity lies in visible region as well as in UV region. Hence, it has the ability to utilized the energy of the both regions of solar spectrum which makes it more functional. The present studies also reveals that doped PTO has less reflectivity which as a result its absorption increases so it can be a good photo-catalyst. Elastic properties discloses its stiffness as well as brittleness.
2 Computational details
The first principal calculation predicts results without performing any experiment and plays a vital role in exploring new material applications. In this work, Material studio-based Cambridge Serial Total Energy Package (CASTEP) was used to design and calculate structural, optical, elastic and electronic properties of pure and Sr-doped PbTiO3.
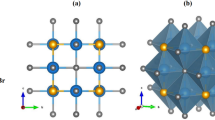
PbTiO3 belongs to space group (Pm-3m) and consists of a perfect cubic structure above 4900 C temperatures. In PbTiO3, Lead (Pb) is placed at corners (0, 0, 0), Titanium (Ti) at body centered (0.5, 0.5, 0.5) and Oxygen (O) at face centre (0.5,0.5,0). A supercell of 2 × 2 × 1 containing 42 atoms was constructed to study the Sr-doping effect in PbTiO3 as shown in Fig. 1. For pure PbTiO3, valance band electronic configuration of Pb, Ti and O are: [Xe], 6s2, 4f1, 5d1, 6p2, [Ar]: 3d2, 4s2 and [He]: 2s2, 2p4 respectively.

A unit cell of PbTiO3 where Pb atoms are at coroner, Ti at body center and Oxygen atoms are at face centered position
Generalized gradient approximation (GGA) proposed by Perdew Burke–Erzerhoff (PBE) or (GGA-PBE) was implemented for electron–electron interaction, while the plane wave pseudo potential approach was implemented for electron–ion interactions. Pseudo potential approach enhances the calculation speed with improved efficiency. Geometry optimization was performed to obtain ground state energy structure and refined atomic position and lattice parameters by considering the following parameters: Cut-off energy 489.8 eV, energy convergence precision 10–5 eV/atom, Residual forces per atom < 2 meV/Å.
3 Results and discussion
3.1 Structural Properties
To calculate the structural, electronic and optical properties of pure and Sr-doped PbTiO3, geometry optimizations are performed on each supercell which relaxes PbTiO3 crystal lattice from external stress. Structural optimization is done with Birch Murnaghan’s equation of state (Birch 1947) to predict structural stability of PbTiO3 and dopants by achieving lowest or ground state energies. Volume optimization curves or energy vs volume (E–V) curves are shown in Fig. 2. The calculated lattice parameters for pure PbTiO3 are a = b = c = 3.975 Å (lattice volume 62.807 Å) which are in good agreement with published experimental and theoretical lattice parameters as shown in Table 1. The difference between published experimental and theoretical values for pure PTO and this study is very small which shows the accuracy of our calculations. The same procedure was adopted for Sr-doped PbTiO3 and calculated lattice parameters are also given in Table 1. It is worth noting that lattice parameters decrease by increasing the Sr-dopants and pure STO calculated results are also in good agreement with studied values. The small difference may be due to the different radii of Pb and Sr.

The total energy as a function of volume for a unit cell of Sr-PbTiO3 fitted to Birch Murnaghan’s equation of state
3.2 Elastic properties
Elastic properties are important parameters of any material and can provide useful information and mechanical stability, bonding characteristics between neighbouring plates, ductility, brittleness, anisotropy and stiffness. The calculated elastic properties of PbTiO3 and Sr-doped PbTiO3 are given in Table 2. For a cubic system, three elastic constants C11, C12 and C44 should satisfy Born’s stability criteria (Ullah et al. 2022a).
From Table 2, we can see that PbTiO3 and Sr-doped PbTiO3 are mechanically stable as Born’s stability criterion given in equation is well satisfied.
The values of Share modulus (G), Young modulus (E), Bulk modulus (B), and Poison ratio (ν), for the perovskite compounds, are calculated by the following relations (Ullah et al. 2022b).
Brittleness and ductility of any material can be categorised by the B/G ratio. According to Pugh (Rizwan et al. 2022), if B/G > 1.75 then the material is considered ductile and if B/G < 1.75 then the material is considered Fragile or Brittle. As shown in Table 2, PbTiO3 and Sr-doped PbTiO3 are categorized as brittle because the value of B/G is less than 1.75 for all dopants. The shear modulus (G) and bulk modulus (B) are used to measure the hardness of the material, the larger the value of G stiffer the material (Ilyas et al. 2022). From Table 2, it can be seen that by increasing the Sr- dopant level in PbTiO3 the values of G increase and it becomes stiffer than pure one which also fulfils the requirement of this work.
Poisson’s ratio for most metals and alloys varies from 0.25 to 0.5 (Ilyas et al. 2022). The obtained values for Poisson’s ratio of PbTiO3 and Sr-doped PbTiO3 are within limits which indicates the interatomic forces are central and nature of PbTiO3 and dopants is ductile. Cauchy’s pressure can explain the nature of the material as either brittle or ductile, if Cauchy pressure is positive then material is ductile and otherwise brittle nature (Ilyas et al. 2022). Poisson’s ratio and Cauchy pressure also confirm similar behaviour of the materials.
Trends of mechanical properties are shown in Fig. 3. Variation in C11, C12, C44, B, G, and E for pure and Sr-doped PbTiO3 is seen as Sr concentration increases. An almost similar trend in all presented quantities can be seen with increasing Sr. dopants.

Elastic behaviour of PbTiO3 against St Concentrations (a) Elastic constants C11, C12, C44 (b) Elastic moduli i.e. Bulk modulus, Shear modulus and Young’s modulus
3.3 Electronic properties
The possible energy bands available for electrons and energy gaps which are regions that cannot be occupied by electrons both are described after the study of band structure of a material. The electronic band gap is measured by measuring the difference in energy between the maxima of valence band and minimum conduction band. There are two types of energy band gaps first one is the indirect band gap lies between two dissimilar symmetry points and further it involves a transition of an electron from valence to conduction band accompanied by phonons and a remarkable part of energy is dissipated via these transitions and efficiency of photonic devices based on SrTiO3 while the second type of band gap is known as direct band gap which lies between similar symmetry points along the vertical symmetry line and magnifies the optical properties and photo-conductivity by performing the radiative recombination of electron and a hole (Rizwan et al. 2019c).
A band gap is a key element to describe electron transition in semiconductors and insulators. The electronic band structures in the Brillouin zone are plotted using GGA-PBE approximations for both pure and Sr-doped PBTiO3 as shown in Fig. 4. The minima of the conduction band (MCB) and maxima of valance band (MVB) are separated by direct band gap for pure (Niu et al. 2015) and Sr-doped PbTiO3 respectively and direct band gap makes it more efficient because of no loss of energy due to phonons.

Band structures of pristine and Sr-PbTiO3 calculated with GGA-PBE
Pure PTO band structure is shown in Fig. 4a, in which Fermi-level exists at 0.0 eV which shows p-type behaviour of the material. Valance band (VB) maxima lie at G and conduction band (CB) minima exist at G which shows the G-G indirect nature of band which is 1.655 eV and which has a strong agreement with previous theoretical and experimental values given in Table 2. The same behaviour was observed for all Sr-doped PTO. as shown in Fig. 4 there is noticeable increment has been observed in band gap energy values for Pb0.89Sr0.11TiO3, Pb0.67Sr0.33TiO3, Pb0.45Sr0.55TiO3 and this expansion in band gap increases by adding the more Sr atoms in pure PTO but there is no change is noticed in nature of band gaps that are all the versions have direct band gaps which are responsible for fewer energy losses and hens, our material is efficient and its photo-activity now has a wide range on the electromagnetic spectrum in visible regions and also in UV regions. Direct band gap G–G is larger than indirect band gap but these both values are less than experimental values the only cause of this variation is p-d orbital repulsion (Gillani et al. 2020).
Density of states (DOS) is significant to explain electronic, optical and elastic properties of any material and describes energy distribution of all regions (Rizwan et al. 2019a). The total density of states DOS and PDOS are shown in Fig. 5. They both explain the contributions of different overlapped orbitals in the formation of maximum valence band and minimum conduction bands from Fig. 5, It is noted that there is a maximum contribution of the p-orbitals density of states in the formation of maximum valence band (MVB) near the fermi level which lies at 0 eV. The second most contribution to the structure of the MVB is the d-density of states graph shows its amplitude is very small as compared to the p-density of states. The third overlapping orbital is the s-orbital which is also contributing and can be seen in the graph near the fermi energy level on the left side it is noted that it has smallest amplitude so it shows the minimum involvement in development of MVB. In pure PbTiO3, Lead (Pb) has a small contribution in both valance and conduction bands (i.e. VB and CB), while Titanium (Ti) and Oxygen (O) have prime contributions in VB and CB respectively. In Sr-doped PbTiO3, Strontium (Sr) Lead (Pb) has a major contribution in both valance and conduction bands.

Effect of Sr on the total density of states (TDOS) of PbTiO3 near Fermi level and Depiction of change in partial density of states with the change in Sr- PbTiO3
Figure 5 shows that the maximum valence band MVB has been majorly contributed by O-2p orbitals but there is also involvement of Pb-6s orbitals while Ti-3d orbitals play a major role than Pb-6p orbitals in the bottom of conduction band near the Fermi level. The lowest end of conduction band consists of t2g (dxy, dyz and dxz) three-fold and eg (3dx2 − y2 and 3dz2) two-fold degenerate states and eg interacts with O and Pb also have a bond with O which reflects a covalence between O-2p and Pb-6s states (Lv et al. 2011). Similar behaviour has been observed for all doped forms but there is a small shifting of orbitals towards the Fermi level. Therefore, it concludes that band structure of the pure and doped PTO is mainly developed by the O-2p orbitals which form MVB and Ti-3d which is responsible for the formation of MCB.
From Fig. 5 it's clear that there is a very small contribution of the Sr element in formation of MVB but the PDOS and TDOS. The PDOS and TDOS express that by increasing the atoms of Sr in PTO the contribution of p-density of states increases below the fermi energy level and simultaneously d-density of states increases in the bottom of the conduction band. The effect of doping on band gap is responsible for variations in energies that is the computed values of band gaps are 1.655, 1.723, 1.804, 1.812 and 1.822 eV at Pure PTO, Pb0.89Sr0.11TiO3, Pb0.67Sr0.33TiO3, Pb0.45Sr0.55TiO3 as seen from Fig. 4. This implies a fundamental increase in band gap at Pb0.45Sr0.55TiO3 due to upward shifting of the conduction band so it reflects direct relation of band gap energy with dopant level.
As compared to pure the Sr-doped PTO has a wide band gap which has an inverse relation with length of a bond and lattice parameters given in Table 2, as a result, covalent bonding is stronger and it is ascribed that more energy is needed to excite an electron valance to empty conduction band in consequence Sr-doped PTO has more resistivity than pure at a specific temperature and Pb-doped PTO has wide band gap which makes it a good photo-catalyst under visible region as well as in UV region and further presence of Sr atoms makes it more thermally stable.
3.4 Optical properties
The Optical properties give important information about the internal structure of a material. Moreover, the recombination rate and mobility of electrons are associated with optical response (Munir et al. 2021). The optical properties like loss function, refractive index, extinction coefficient, absorption and reflectivity are the functions of frequency or energy of a photon and are studied by analyzing band structure of pure and Sr-doped PbTiO3. The optical responses are determined by complex dielectric functions. ε (ω) = ε1(ω) + i ε2 (ω) this equation is known as the equation of dielectric function where ε1(ω) is the real part of the dielectric function (DF), polarization and capacity of storing electrical energy both are associated with it. Dissipation or loss of energy in a medium is determined by an imaginary part of DF known as ε2 (ω) (Gillani et al. 2021).
The ε1(ω) and ε2(ω) of dielectric function as a function of energy are given in Fig. 6, in a set of energy range 0–45 eV. The value of ε1(ω) for pure material at 0 eV is 8.95 but its value is decreased by increasing the number of doped items i.e. 8.23, 7.51, 6.98 and 6.37 for Pb0.89Sr0.11TiO3, Pb0.67Sr0.33TiO3, Pb0.45Sr0.55TiO3 and SrTiO3 respectively. Maximum peaks of the real part are achieved at 2.70, 2.93, 3.02, 3.06 and 3.09 for pure and Sr-doped PbTiO3 respectively. These peaks are produced by the transition of electrons from the top of the valance to the bottom of the conduction band. A further increase in energy caused to decrease peak sharply and they reached negative values at energies 8.17, 8.63, 8.74, 9.02 and 11.82 for pure and Sr-doped PbTiO3 respectively. The magnitude of polarization is proportional to a real part of DF (ε1) and for Sr-doped PTO the value of ε1 is reduced which imputes that polarization is also dropped. Since ε1 is the function of incident photon’s energy therefore it attains highest value at higher frequencies or energies of incident photon.

Comparison of optical properties of SrTiO3 and Sr-PbTiO3 (a) Re (DF) (b) Im (DF) (c) Absorption (d) Energy Loss Function (e) Reflectivity and (f) Refractive index (g) Extinction Coefficient
The value of ε2 is zero at 0eV for pure and doped which indicates zero energy loss. Figure 6b graph has the highest peak value of 11.4 at 4.0eV for pure but its value is decreased for doped material. The other prominent peaks are shown at 6.1 eV, 23.1 eV and 36.1 eV. When electrons make transitions from valence to conduction bands, these peaks are generated. In the case of doped material peaks slightly shifts towards higher energies.
The imaginary part ε2(ω) plays an important role in determining the absorptive behavior and band structure of the material. In semiconductor materials, the interband transitions are also indicated with ε2(ω). The ε2(ω) plot has two sharp peaks at 1.7 and 5.1 eV with LDA, 1.9 and 5.3 eV with GGA, and 2.4 and 5.6 eV. The information of both ε1(ω) and ε2(ω) permits the calculation of essential optical properties such as refractive index, absorption coefficient, optical conductivity and reflectivity.
Absorption explains the phenomena in which photons that have enough energies to fill the energy gaps of atoms are absorbed and the other photons are transmitted and remain unaffected. From Fig. 6c at 0 eV, there is no absorption for both pure and doped materials like the imaginary part of DF. The observed major absorption peaks for pure PTO occur at 8.09, 13.73, 22.06 and 36.68 eV respectively. For Pb0.89Sr0.11TiO3, peaks shift to 8.49, 13.26, 23.51 and 36.61 eV respectively, for Pb0.67Sr0.33TiO3, peaks shift to 5.58, 12.46, 23.70 and 36.56 eV, for Pb0.45Sr0.55TiO3, peaks shifts to 8.70, 11.52, 23.67 and 36.63 eV and SrTiO3, major peaks observed at 8.78, 11.39, 23.831 and 36.7 eV respectively.
Pb0.45Sr0.55TiO3 and SrTiO3 show similar behavior recurrent to other optical behavior in the UV region. For 5–10 eV the transitions may be due to O-2p VB states of Ti-3s and 3P states and for higher energies, the peaks may be due to inner-band transitions. The highest peak at 22.06 eV for pure and similar ranges from 23.51 to 23.83 peaks for all dopants shows maximum amount of absorption and doped edges occur at higher energies which determine an increase in band gap as dopants concentration increases.
Energy loss is also a function of energy. When a fast-moving electron interacts with material and collides with other particles then some amount of energy is lost is called energy loss (Piskunov et al. 2004). Energy loss function (ELF) is related inversely to imaginary dielectric function and absorption. When a light incident on material then electrons that are not fixed in lattice sites start oscillating and cause the formation of peaks known as the Plasmon peaks and plasma resonance occurs at a specific frequency called plasma frequency (Gillani et al. 2019; Yinnü et al. 2015) It can be seen from Fig. 6d that plasmon peaks are sharper in case of doped material and for a different amount of doping the positions of peaks are slightly different. The highest peak for PbTiO3 is observed at 25.1eV which shifts at 25.9, 26.3, 26.7 and 27 eV for Pb0.89Sr0.11TiO3, Pb0.67Sr0.33TiO3, Pb0.45Sr0.55TiO3 & SrTiO3 respectively. As compared to the highest peak of imaginary of dielectric function the peaks of ELF are shifted towards the high energies. Sharp peak is observed at lower energies for reflecting spectra which decrease with increasing Sr-dopants. It is clear that reflectivity has a direct relationship with ELF but in converse relation with both absorption and imaginary DF.
The complex refractive index is frequency or energy-dependent and displayed by equation N = n + ik here n(ω) is the real part and imaginary part K(ω) is familiar as the extension coefficient. The value of refractive index n(ω) at 0eV is about 3.0 and the spectrum reflects its maximum value at 2.9eV.
However, a decreasing trend is detected in value of n(ω) with a rising number of doped atoms. Figure 6 shows its value is reduced from 3.0 to 2.0 for SrTiO3 which reveals a net change in the band gap. Also, from the spectrum, it’s clear that value of n(ω) is minimum where absorption has maximum value so they are inverse of each other but the imaginary part K(ω) elucidates the energy losses and is in line with absorption spectrum and imaginary part of DF. All the above consultations depicted that the Sr improves the optical responses of pure PbTiO3.
4 Conclusion
DFT has been used to study the structure, elastic, mechanical, electronic, and optical, properties of pure and Sr-PbTiO3. Both end materials were discovered to be stable and cubic. It is demonstrated that the Eg of pure PbTiO3 and the theoretical and experimental values are in good accord. Charge correction prevents gap defect states thanks to Sr inclusion. Sr addition results in the lowest Eg (bottom of CB is shifted to lower energy, while VB edge remains unaffected). After Sr addition, we see that the Fermi level shifts toward VB, mimicking the behavior of a p-type degenerate semiconductor. The enhanced characteristics of Sr-PbTiO3 are confirmed by the computed absorption coefficient. Due to their suitability for water splitting and the fact that they are lead-free, both Sr-induced and pure materials are good choices. The PDOS at the bottom of CB changes greatly after Sr doping, whereas the PDOS at the top of VB also changes significantly. The optical band gap decreased when new Sr DOSs were present relative to pure PbTiO3. Additionally, since the Eg of doped material applies to solar cell applications, it will be an excellent replacement for current materials in optical especially solar cell applications. The n and dielectric constants of Sr-PbTiO3 were changed, and the absorption spectra revealed a pronounced blue shift. Sr-optical PbTiO3's and electronic characteristics underwent significant modification. After Sr substitution, noticeable changes in elastic and mechanical properties are seen. SrTiO3 which has been impacted by Sr is therefore presented as a strong candidate in the field of optoelectronic applications.
Data availability
N/A.
References
Birch, F.: Finite elastic strain of cubic crystals. Phys. Rev. 71, 809–824 (1947)
Chilibon, I., et al.: Ferroelectric ceramics by sol–gel methods and applications: a review. J. Sol-Gel Sci. Technol. 64, 571–611 (2012)
Gillani, S., et al.: Effect of magnesium doping on band gap and optical properties of SrZrO3 perovskite: a first-principles study. Optik 191, 132–138 (2019)
Gillani, S., et al.: First-principles investigation of structural, electronic, optical and thermal properties of Zinc doped SrTiO3. Optik 201, 163481–163490 (2020)
Gillani, S., et al.: A systematic computational study to understand the effect of metals (Mg, Ca, Sr) doping and external isotropic static pressure on phase stability, electronic band structure and optical properties of KNbO3. Mater. Sci. Eng. B 271, 115261–115269 (2021)
Ilyas, I., et al.: Pressure-induced elastic, mechanical and opto-electronic response of RbCdF3: a comprehensive computational approach. J. Phys. Chem. Solids 165, 110642–110649 (2022)
Isah, I., et al.: Effect of pressure on structural, elastic and electronic properties of Perovskite PbTiO3. J. Found. Appl. Phys. 8, 179–190 (2021)
Jiang, L., et al.: Prediction of lattice constant in cubic perovskites. J. Phys. Chem. Solids 67, 1531–1536 (2006)
Kambale, K., et al.: Grain growth kinetics of barium titanate synthesized using conventional solid state reaction route. Ceram. Int. 40, 667–673 (2014)
Kuma, S., et al.: Structural, electronic, lattice dynamic, and elastic properties of SnTiO3 and PbTiO3 using density functional theory. Adv. Condens. Matter Phys. 2019, 3176148–3176160 (2019)
Kumar, M., et al.: Effect of A and B site cations on the dielectric and electrical properties of PbTiO3. Phys. Status Solidi 177, 583–591 (2000)
Lemziouka, H., et al.: Structural, dielectric and optical properties of Cu-doped PbTiO3 ceramics prepared by sol–gel. Mater. Today: Proc. 37, 3940–3945 (2021)
Lv, H., et al.: Density functional theory (DFT) investigation on the structure and electronic properties of the cubic perovskite PbTiO3. Appl. Catalysis A 404, 54–58 (2011)
Meštrić, H., et al.: Iron-oxygen vacancy defect centers in PbTiO3: Newman superposition model analysis and density functional calculations. Phys. Rev. B 71, 134109–134119 (2005)
Mete, E., et al.: Double perovskite structure induced by co addition to PbTiO3: insights from dft and experimental solid-state NMR spectroscopy. The J. Phys. Chem. C 123, 27132–2719 (2019)
Munir, J., et al.: An insight into the electronic, optical and transport properties of promising Zintl-phase BaMg2P2. Physica B 618(1), 413181–413187 (2021)
Murugan, A., et al.: Phase evolution of NiTiO3 prepared by coprecipitation method. Mater. Lett. 60, 1791–1792 (2006)
Niu, P., et al.: First-principles study of nitrogen doping and oxygen vacancy in cubic PbTiO3. Comput. Mater. Sci. 98, 10–14 (2015)
Piskunov, S., et al.: Bulk properties and electronic structure of SrTiO3, BaTiO3, PbTiO3 perovskites: an ab initio HF/DFT study. Comput. Mater. Sci. 29, 165–178 (2004)
Rizwan, M., et al.: Band gap modulation effect on electronic and optical properties in PbTiO3 under stress: a DFT study. The Euro. Phys. J. Appl. Phys. 88, 10501–10507 (2019a)
Rizwan, M., et al.: Implementation of magnesium doping in SrTiO3 for correlating electronic, structural and optical properties: a DFT study. Chin. J. Phys. 62, 388–394 (2019b)
Rizwan, M., et al.: Structural, electronic and optical properties of copper-doped SrTiO3 perovskite: a DFT study. Physica B 552, 52–57 (2019c)
Rizwan, M., et al.: Alteration impact of electronic properties of c-SrTiO3 on optical response due to Ca inclusion: a DFT study. Physica B 602, 412558 (2021a)
Rizwan, M., et al.: Putting DFT to trial: For the exploration to correlate structural, electronic and optical properties of M-doped (M= Group I, II, III, XII, XVI) lead free high piezoelectric c-BiAlO3. Mater. Sci. Eng. B 264, 114959–114968 (2021b)
Rizwan, M., et al.: Photocatalytic and optical properties of (Mg: La) CaTiO3: insights from first principles studies. J. Phys. Chem. Solids 169, 110830–110837 (2022)
Shirane, G., et al.: X-ray and neutron diffraction study of ferroelectric PbTiO2. Acta Crystallogr. A 9, 131–140 (1956)
Stashans, A., et al.: Structural properties of PbTiO3 and PbZrxTi1−xO3: a quantum-chemical study. Int. J. Quantum Chem. 87, 145–151 (2002)
Tse, J., et al.: Unraveling the impact of graphene addition to thermoelectric SrTiO3 and La-doped SrTiO3 materials: a density functional theory study. ACS Appl. Mater. Interfaces 13(34), 41303–41314 (2021)
Ullah, H.M.N., et al.: A DFT study of optical, elastic, mechanical, and overall water-splitting photocatalytic properties of pristine and Cd substituted BaZrO3: a lead free environment friendly material. Mater. Sci. Eng. B 286, 116041–116054 (2022a)
Ullah, H.M.N., et al.: A computational study for mechanical, thermoelectric and optoelectronic applications of BiAlO3 under static pressure. J. Phys. Chem. Solids 168, 110819–110830 (2022b)
Wang, L., et al.: First-principles study of tetragonal PbTiO3: phonon and thermal expansion. Mater. Res. Bull. 49, 509–513 (2014)
Yaseen, M., et al.: Investigation of optical and thermoelectric properties of PbTiO3 under pressure. Physica B 615, 412857–412865 (2021)
Yinnü, Z., et al.: Effects of N concentration on electronic and optical properties of N-doped PbTiO3. J. Semicond. 36, 093005–093010 (2015)
Funding
No funding available.
Author information
Authors and Affiliations
Contributions
Dr. HN, Dr, R and Dr. HMN done main calculations. Dr. U, Dr. A and Dr. A helped in writing the manuscript text and preparing figures. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Rizwan, M., Naeem, H., Naeem Ullah, H.M. et al. Fine band gap tuning via Sr incorporated PbTiO3 for optoelectronic application: a DFT study. Opt Quant Electron 56, 122 (2024). https://doi.org/10.1007/s11082-023-05775-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11082-023-05775-9