
Abstract
This study investigates the influence of hydrostatic pressure on structural, electronic, mechanical and optical properties of Sr3PX3 (X = Cl and Br) compounds, by using the first-principles density functional theory (DFT) within the pressure range of 0–30 GPa with a span of 10 GPa. For Sr₃PCl₃ and Sr3PBr3, the dynamical stability is confirmed by the fact that the phonon dispersion curves do not contain imaginary modes. Pressure-induced band gap alterations in Sr3PCl3 and Sr3PBr3 reveal semiconducting behavior: GGA measurements show a decrease from 1.70 eV and 1.55 eV at ambient pressure to 0.22 eV and 0.21 eV at 30 GPa; TB-mBJ results show a decrease from 2.73 eV and 2.40 to 1.07 eV and 0.92 eV. This supports their inverse relationship with pressure. The values of Debye and melting temperatures support their high-temperature applications. Effective mass also shows an inverse relationship with induced pressure. The bond length, lattice parameters, and cell volume reduces with pressure. They exhibit ductility, which is further enhanced by the applied pressure. These materials emerge as promising candidates for flexible optoelectronic devices. Optical properties like absorption coefficients, reflectivity, and dielectric functions were observed and found to be significantly influenced by applied pressure. The absorption spectra exhibit a significant redshift with increasing pressure, indicating enhanced potential for optoelectronic applications. Our detailed investigation sheds light on the tunability of Sr3PX3 (X = Cl and Br) properties under pressure, showcasing their potential for cutting-edge applications in optoelectronics and photovoltaics.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
The fastest-growing photovoltaic materials are hybrid organic–inorganic halides at the moment, which are used for solar cells (Huang et al. 2020; Liu et al. 2020; Seo et al. 2016). For their simple crystal structure and a variety of characteristics, including resistivity, piezoelectricity, ferroelectricity, and superconductivity, perovskite oxides are highly valued in the field of material research (Butt et al. 2021; Kouchaksaraie 2011; Verma and Jindal 2009). Perovskites are exceptional because of their broad variety of uses in the most recent developments and their special capacity to undergo phase changes (Khan et al. 2023; Wu et al. 2021). Particularly, lead halide perovskites gained a lot recognition in photovoltaics (PV) field because of their affordable price, appropriate band gaps, high photoinduced carrier mobility (Wehrenfennig et al. 2014; Zhumekenov et al. 2016), and effective visible light absorption (Kojima et al. 2009; Lee et al. 2012; Stoumpos et al. 2013). Lead halide perovskites show great properties but contain toxicity (Liu et al. 2019b; Yang et al. 2020). In addition, regarding lead toxicity, perovskite solar cell manufacture and disposal create significant problems, which limits the sustainability of the technology and puts human and environmental health at risk (Ren et al. 2022). Because of this, researchers have consequently paid close attention to lead-free halide perovskites (Lan et al. 2021; Moghe et al. 2016).
These Lead-free materials are getting a lot of attention because of their remarkable optoelectronic qualities, Conductivity, mechanical stability, absorption coefficient, and tunable band gap properties (Yin et al. 2014; Zhang et al. 2016). In contrast to lead (Pb)-containing PV cells, the bulk of perovskites have more encouraging qualities and are typically more ecologically harmless (Babayigit et al. 2016; Eperon et al. 2015). A broad range of characteristics, including conductivity, insulation, semi-conduction, and superconductivity, are present in perovskite materials (Bhalla et al. 2000; Kanhere and Chen 2014). As a result, perovskites are acknowledged by many as a highly significant class of materials that comprise various crystal shapes (Filip and Giustino 2018). Even though the method of turning electricity into light is the opposite of that of turning light into electricity, the first research on converting electricity into light was published several decades before the initial study on halide perovskites being utilized as photovoltaic sun absorbers (Jiang et al. 2019). Right now, solar technologies are replacing conventional energy sources. Over the past ten years, perovskite materials have seen improvements that have accelerated development (Jung and Park 2015). It’s also making advances in the green solar consumers (Ali et al. 2023; Dandia et al. 2020; Liu et al. 2019a; Tian et al. 2019). The ABX3 group of perovskite materials has garnered huge attention, wherein Sn2+, Ge2+, Eu2+, Dy2+, and Yb2+ have been explored as substitutes for the hazardous Pb2+, resulting in exceptional optoelectronic properties to hold significant value for the perovskite solar cell field (Marshall et al. 2016; Noel et al. 2014; Zhou et al. 2018). The variation of elemental constituents (A, B, or X) within the ABX3 configuration can also be noticed as provoking a form of internal pressure due to the alteration in elemental size, and thus, an overall grasp of the impact of structural deviation demands this influence to be compared precisely.
Recent years have seen a variety of research projects that have used strain engineering to enhance particular material qualities. (Feng and Zhang 2021) identified the stable compounds of high efficiency on A3MX3 structure (the following: A = Ca, Mg, Ba, Sr; M = N, P, As, Sb and X = F, Cl, Br, I) and found Ba3PI3, Ba3AsI3, and Ba3SbI3 stable PV material which showed up to 25.9% efficiency. (Zhou et al. 2016) worked on MAPbI3 by applying pressure, gaining a higher band gap energy, more carrier mobility, and better absorption coefficients for the material that can raise its potential for use in photovoltaics. (Islam et al. 2024) worked with Ca3PCl3 perovskite by applying biaxial strain. Where in the absorption spectra, tensile strain leads to a blue shift (toward greater energy) while compressive strain leads to a redshift (toward lower energy). (Faridi et al. 2018) showed the reverse relationship of pressure with anisotropy and the brittle to ductile shift under hydrostatic pressure of KNbO3. A strontium-based research by (Algahtani et al. 2024) on Sr3AsX3 (X = F and Br) showed high optical absorption within the range of 2–10 eV. The band gap obtained by HSE was quite high around 2.5 eV. Working with A3BX3 (A = Ca, Sr, B = P, As, X = I, Br), which consists of 6 compounds in total, (Ghosh et al. 2024b) discovered that the HSE approach shows a larger band gap than the GGA method. Later, (Ghosh et al. 2023) reduced the band gap of Sr3AsI3 by applying strain. In addition, (Ghosh et al. 2024a) worked with Sr3PBr3 on strain-induced changes and studied structural, electronic, mechanical, and optical properties with FP-DFT, where it exhibits remarkable light absorption within the visible light range.
This study comprehensively investigates the structural, electronic, mechanical, and optical characteristics of Sr3PX3 (X = Cl, Br) under various pressures utilizing density functional theory (DFT). According to earlier studies, no research was done on the pressure-induced characteristics of Sr3PBr3. Therefore, a thorough examination of the physical properties of Sr3PCl3 and Sr3PBr3 is important with pressure-induced changes to be able to obtain a comprehensive understanding of the behaviors in such systems, particularly with regard to their prospective application sectors. The most trustworthy theoretical framework for examining the chemical and physical properties of materials is density functional theory (DFT)-based first-principles technique. (Hossain et al. 2021b; Hossain et al. 2021a; Hossain et al. 2020; Rasheduzzaman et al. 2021). Energy gap modification and structural features are conducted in order to clarify its electronic properties. Optical properties which also changed due to increasing pressure. The red shifting properties of Sr3PX3 (X = Cl, Br) absorption spectra are investigated in relation to the changes in peak positions of the dielectric maxima under various pressure levels. From our investigation, this compound’s optoelectronic characteristics are changed in pressure conditions, for that these materials are suitable for use in optoelectronic devices applications.
2 Computational details
The Sr3PX3 (X = Cl, Br) compound was examined by ab initio computations, which were carried out utilizing the Quantum ESPRESSO (QE) package’s Density Functional Theory (DFT) method (Giannozzi et al. 2009). Self-consistent computations were conducted to solve the Kohn–Sham equations (Kohn and Sham 1965) utilizing the Perdew-Burke-Ernzerhof (GGA-PBE) exchange-correlational functional in the Generalized Gradient Approximation (Perdew et al. 1996) along the ultra-soft pseudo-potentials (Perdew et al. 1992; Vanderbilt 1990). A plane-wave basis was set with a 40 (Ry) kinetic energy cut-off, 440 (Ry) charge density cut-off, and 1.0 × 10− 8 convergence threshold was used to attain convergence. The lattice parameters used for the Sr3PCl3 and Sr3PBr3 structures were 6.05 Å and 6.23 Å, respectively. To enhance the crystal structure and in order to get the lowest energy state feasible, we applied the Broyden–Fletcher–Goldfarb–Shanno (BFGS) optimization method. (Fischer and Almlof 2002) alongside a force convergence threshold of 1 × 10− 3. In addition, the lattice optimization was performed using the vc-relax method with a k-point value of 8 × 8 × 8. GGA-PBE approximations were used to do electronic band structure computations. k-points dimension of 8 × 8 × 8 for self-consistent field (SCF) calculations and 12 × 12 × 12 for non-self-consistent field (NSCF) calculations are utilized. First-order time-dependent perturbation theory was applied to analyze the optical properties of the material formations and assess their dynamic stability (Karsch et al. 1997; Langhoff et al. 1972). For subsequent calculations, we applied the Tran-Blaha modified Becke-Johnson (TB-mBJ) approximation (Tran and Blaha 2009) in WIEN2K code (Blaha et al. 2020), which is specifically developed to enhance the accuracy of electronic band gap predictions, particularly for semiconductors and insulators. To achieve both charge and energy convergence, we utilized a linearized augmented plane-wave basis set with lmax = 10 and RMTKmax = 7 (where Kmax represents the maximum K-value). After achieving dynamic stability, the intricate dielectric functions of Sr3PX3 (X = Cl, Br) were calculated to study their optical properties, which are affected by the photon energy. The optical properties obtained by utilizing the complex dielectric function, were denoted as \(\varepsilon (\omega ) = {\varepsilon }_{1}(\omega )+i{\varepsilon }_{2}(\omega )\), which consists of \({\varepsilon }_{1}(\omega )\) (real part) and \({\varepsilon }_{2}(\omega )\) (imaginary part). The THERMO-PW approach was used to calculate the mechanical characteristics by assessing the elastic constant. The ELATE tool (Gaillac et al. 2016) is used to plot 3D illustration of anisotropy.
3 Results and discussion
3.1 Structural properties
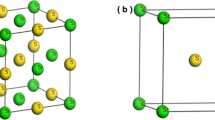
The Sr3PX3 (X = Cl, Br) compounds belong to the cubic space group Pm3m (#221) (Johnsson and Lemmens 2005). Both compounds crystalize in a cubic structure, as demonstrated in Fig. 1a and b. The unit cells of each compound consist of 7 atoms. The Sr, P, and Cl/Br atoms are at in the following Wyckoff positions: Sr atom at (0.5, 0, 0), P atoms at (0 0, 0), and X (Cl/Br) atoms at (0, 0.5, 0.5) (Zhao et al. 2018a). Table 1 covers the Lattice constant (a0), Unit cell optimum volume (V0), Bulk modulus (B), Pressure derivative of bulk modulus (\({B}{\prime}\)), Ground state energy (E0), and Bandgap (Eg). The energy deviation was determined by using the Murnaghan equation (Murnaghan 1944):

2D and 3D crystal structure of cubic Sr3PX3 (X = Cl, Br) unit cell
The energy levels found in the above Murnaghan equation with respect to different volumes are illustrated in Fig. 2a and b. Both compounds are found to have negative ground state energy, but Sr3PBr3 has significantly lower ground state energy compared to Sr3PCl3 indicating a more stable structure. The graphical representation of this effect is shown in Fig. 3a and b. It also shows a lattice parameter of 6.05 Å for Sr3PCl3 and 6.21 Å for Sr3Br3, which aligns with the previous work by (Ghosh et al. 2024a) which was 6.61 Å. A slight difference is notable here, it could have been due to the use of HSE method in previous research.

Volume optimization curves for Sr3PX3 (X = Cl, Br)

Optimized lattice constant and volume for Sr3PX3 (X = Cl, Br) as a function of pressure
As per Table 2, the volume of unit cells decreases with pressure which means the bond lengths also decrease. This leads to the narrowing of bandgap and overlapping conditions between atoms (Xiong et al. 2017). The band gap narrowing is expected to improve the optoelectronic properties of materials for better performance in optoelectronic devices. It was also noted that increased pressure resulted in a reduction in the volume of the unit cells for various materials including cubic FrQCl3 (Hosen et al. 2024a), trigonal and monoclinic MgSiO3 (Gao et al. 2020), cubic RbCaBr3 (Hosen et al. 2024c), and high-entropy alloys (Ahmad et al. 2017).
3.2 Phonon stability
Phonon dispersion analysis allows us to understand the vibrational properties of a material, which in turn influence its thermal and electrical behavior. The dynamic stability of Sr3PCl3 and Sr3PBr3 is assessed by constructing a 2 × 2 × 2 supercell, enabling a detailed analysis of the materials’ vibrational properties and structural stability. Using this larger supercell allows us to capture a broader range of vibrational modes and atomic interactions, leading to a more precise evaluation of the material’s dynamic behavior. Figure 4 show the phonon dispersion curves for Sr3PCl3 and Sr3PBr3. Both material’s phonon dispersion curves have no negative frequencies, confirming their dynamical stability (Jehan et al. 2023; Khattak et al. 2023). With no imaginary frequencies in the Brillouin zone, Sr3PCl3 and Sr3PBr3 remain stable despite minor perturbations, verifying their cubic structures. The dynamic stability of Sr3PBr3 has also been confirmed in prior research through the analysis of phonon dispersion frequencies (Ghosh et al. 2024a). Comparing Sr3PCl3 and Sr3PBr3 reveals similar phonon activity, with modest frequency shifts due to Cl and Br atom’s mass differences, impacting material vibrations. Understanding the phonon behavior of Sr3PCl3 and Sr3PBr3 is crucial for understanding their vibrational spectra and thermal and electrical characteristics.

Phonon dispersion spectrum of Sr3PX3 (X = Cl, Br)
3.3 Electronic properties
Investigating the electronic characteristics of Sr3PX3 (X = Cl, Br) with applied pressure is the key to understanding its future use. Employing Density Functional Theory (DFT) computations via Quantum Espresso, we scrutinize its band configuration, bandgap, electron charge density, density of states (Partial and total), and effective mass. These examinations are important in understanding their electronic behavior and integrating them into the various technologies. One of the characteristics of semiconductor materials is that adjusting the band gap is possible to enable the manipulation of electron flow and is suitable for various uses.
In this research, bandgap (Eg) values for Sr3PX3 (X = Cl, Br) were obtained using both the GGA-PBE and TB-mBJ approximations, with TB-mBJ providing more accurate results. Sr3PCl3 exhibits a bandgap (Eg) of 1.7 eV under zero pressure, which greatly decreases to 0.22 eV under a pressure of 30 GPa, illustrated in Fig. 5. Similarly, Sr3PBr3 shows a reduction in bandgap (Eg) from 1.55 eV without pressure to 0.21 eV at 30 GPa, illustrated in Fig. 6. These values were obtained using GGA-PBE approximation. The initial value matches with the previous research on Sr3PBr3 by (Ghosh et al. 2024a) at zero strain, which was 1.528 eV, which justified the accuracy of our work. Using TB-mBJ, Sr3PCl3 shows a bandgap of 2.73 eV at zero pressure, decreasing to 1.07 eV at 30 GPa, while Sr3PBr3 shows a reduction from 2.40 to 0.92 eV under the same conditions. The corresponding band structures are illustrated in Figs. 7 and 8. The relation of band gap for both compounds with applied pressure is illustrated in Fig. 9. This similar type of reduction of Eg with applied pressure is found in similar types of single-halides. The pressure-induced fluctuations in the bandgap (Eg) of Sr3PCl3 and Sr3PBr3 make them well-suited for pressure-sensing applications, where changes in the Eg can serve as a reliable indicator of pressure levels. Furthermore, the adjustable nature of the Eg renders these substances immensely appealing for optoelectronic gadgets, presenting prospects for crafting devices with customized optical and electronic characteristics. Our findings align with previous research, highlighting the consistent correlation between applied pressure and bandgap reduction in semiconductor materials.

Band structure of Sr3PCl3 at various applied pressures with GGA-PBA approximation

Band structure of Sr3PBr3 at various applied pressures with GGA-PBA approximation

Band structure of Sr3PCl3 at various applied pressures with TB-mBJ approximation

Band structure of Sr3PBr3 at various applied pressures with TB-mBJ approximation

Variation of band gap of Sr3PX3 (X = Cl, Br) at various applied pressures
Density of states (DOS) sheds light on the material’s electronic and optical behavior by providing essential insights into the elemental contributions and bonding properties. Our research indicates that the halide elements (Br-4p and Cl-3p), Strontium (4p), and phosphorus (P-3p) play critical roles in influencing the electronic characteristics of the material because of their high density near the Fermi level illustrated in Figs. 10 and 11 respectively. The presence of partially filled valence bands in the DOS around the Fermi level indicates that halides with strontium and phosphorus orbitals are involved in charge transport activities. The valance band’s primary source of contribution is P-3p orbital, minor contributions from Sr-5s and Sr-4p orbitals and in the conduction band major contribution comes from Sr-4p orbitals along with minor contributions from other orbitals. The involvement of Strontium (Sr-4p and Sr-5s) orbitals in both valance and conduction bands plays a crucial role in the electronic arrangement. The peaks of Sr-4p and P-3p shift towards the fermi level while pressure is applied due to the hybridization with Cl-3p (Br-4p), supports the reduction of bandgap (Eg). Total Density of States (TDOS) gives a complete idea about the distribution of electronic states. In our investigation (as depicted in Fig. 12), a decline was noted in TDOS for both compounds when subjected to applied pressure. This decrease stems from the alteration in the Eg and structural compression induced by pressure which justifies the findings of PDOS. Grasping these variations yields noteworthy discoveries on the electronic properties and features of the material, crucial for enhancing its efficacy across diverse applications.

The partial density of states of Sr3PCl3 at various pressures

The partial density of states of Sr3PBr3 at various pressures

The total density of states of Sr3PX3 (X = Cl, Br) at different pressures
The charge density describes the distribution of electronic charges within a given system. It helps to provide an initial prediction on the most dominant type of bonding present in the system. Electron charge density for both compounds at 0 GPa and 30 Gpa, is illustrated in Fig. 13. From the electron charge density, we can observe the real potential positions of the electrons. The right bar indicates the scale of the electron charge density, blue for low and red for high. Both compounds were found to have spherical forms under all pressures. This is an indication that the bond within the materials is highly likely to be mostly ionic. However, a further analysis should be conducted, such as by using Bader charge analysis (Pitriana et al. 2019), if one wants to investigate the degree of the charge transfer between anions and cations in the materials. This is needed before a final conclusion is drawn about the type of bond present in the materials. With increased pressure, the density of electron charge did increase slightly, but not sufficiently to generate any hybridization or bonding.

Electron charge density distribution for Sr3PX3 at 0 and 30 GPa pressure
Finally, effective mass was calculated to understand the charge carriers’ motion under impact of pressure, depicted in Fig. 14 and Table 3. It helps to understand carrier mobility, Band Structure Analysis and transport properties. Effective mass is calculated using this equation (Zhao et al. 2018b),

Effective masses of Sr3PX3 (X = Cl, Br) at different pressures
The effective mass was performed by taking the second derivative of the energy dispersion relation (E-k curve) with respect to momentum (k). The analyzed data showed an inverse relation for both me and mh with pressure. As we know carrier mobility has an inverse relationship with effective mass. While applying pressure, the effective mass of the electrons and holes was reduced, indicating high carrier mobility and charge carrier acceleration, which is advantageous in electronic and optoelectronic applications.
3.4 Mechanical properties
Elastic constants are crucial in establishing the mechanical characteristics of materials, since they dictate the response of a crystal to external forces (Husain et al. 2023a; Rahman et al. 2020). For cubic crystal materials, only three elastic constants are independent which are: C11, C12, and C44 (Husain et al. 2023b). These three independent elastic constants of a compound are bound to follow a set of criteria if the compound is to be mechanically stable. These criteria, also known as Born stability criteria (Born 1940), are:
Sr3PX3 follows all four of these criteria. The Cauchy pressure (\({\text{C}}_{\text{p}}\)), crystal stiffness (\({\text{C}}_{\text{s}}\)) and Kleinman parameters (\(\zeta\)) can be found using the three elastic constants. The equations from which they can be found are:
Cauchy pressure gives an idea of a compound’s brittleness/ductility. Positive value means that the compound is ductile (Feng and Cui 2014). We can come to the conclusion whether Sr3PX3 is ductile or brittle once we also overviewed the Poisson’s and Pugh’s ratios. The ability of a compound to withstand shear deformation when shear stress is applied in the [\(1\overline{1}0\)] direction on the (110) plane is the crystal stiffness which increases with pressure as shown in Table 4. The value of Kleinman parameter can be between 0 and 1. Values closer to 0 indicate an insignificant contribution to bond bending and the near 1 value indicates marginal contribution to bond stretching (Kleinman 1962). Pressure also has an effect as per Table 4. Here, the values also show almost similar results for Sr3PBr3 as (Ghosh et al. 2024a) found.
Table 5 consists of the following mechanical properties alongside the effects of pressure on them:
bulk modulus (B), shear modulus (G), Young’s modulus (Y), Pugh’s ratio (B/G), Poisson’s ratio(ν), Machinability index (µM), Hardness factor (Hv). It is possible to estimate Bulk Modulus and Shear Modulus using the Voigt-Reuss-Hill method, where the Voigt’s and Reuss’s approximation of bulk moduli and then shear moduli are averaged (Gueddouh et al. 2016; Kleinman 1962):
Here, \({\text{B}}_{\text{V}}\) is the Voigt’s bulk modulus, \({\text{B}}_{\text{R}}\) is the Reuss’ bulk modulus and B is the Hill’s bulk modulus or simply bulk modulus.
Similarly, \({\text{G}}_{\text{V}}\) is the Voigt’s shear modulus, \({\text{G}}_{\text{R}}\) is the Reuss’ shear modulus and G is the Hill’s shear modulus or simply shear modulus.
Bulk and shear moduli indicate resistance to fracture and ability to resist shear stress respectively. It can be concluded from Table 5 that Sr3PCl3 has higher fracture resistance and plastically deforms better than Sr3PBr3 as Sr3PCl3 has high bulk modulus and shear modulus (30.42 GPa and 22.05 GPa compared to 28.38 GPa and 19.71 GPa of Sr3PBr3 at ambient pressure). This comparison stays the same at higher pressure. Based on the bulk and shear modulus values, Young’s modulus can be calculated. The correlation among shear modulus, bulk modulus, and Young’s modulus is (Wang et al. 2015):
Young’s modulus of Sr3PCl3 is higher than Sr3PBr3 across all pressure points. At 0 GPa, Young’s modulus of Sr3PCl3 is 53.27 GPa and Sr3PBr3 is 48.01 GPa which means Sr3PCl3 stiffer compared to Sr3PBr3.
Machinability indicates how easy it is for a material to be cut (machined). High machinability also means less tool wear. Machinability can be measured by a machinability index (µM) (Sun et al. 2005):
Both compounds have low machinability. However, the machinability index grows rapidly with increasing pressure. At 30 GPa pressure, the machinability index of Sr3PCl3 becomes 34.01 and the machinability index of Sr3PBr3 becomes 45.34. To note, increasing pressure not only increases mechanical performance of these materials but also increases their machinability.
Hardness can be used to understand a materials’ elastic and plastic properties. Hard materials tend to be brittle in nature but also more wear resistant. Hardness can be measured from following equation (Al-Fahdi et al. 2021):
The hardness for Sr3PCl3 was found to be 5.38 GPa and for Sr3PBr3 it is 4.47 GPa. Sr3PCl3 was harder compared to Sr3PBr3 at ambient pressure but increasing pressure reduces the hardness rapidly for both compounds.
Poisson and Pugh’s ratio are two important parameters to determine a material’s brittleness/ductility alongside Cauchy pressure. Poisson’s ratio can be determined by the following equation:
Pugh’s ratio is a simple analogy of shear and bulk moduli (B/G). There is a critical value of 0.26 for Poisson’s ratio and 1.75 for Pugh’s ratio (Al-Fahdi et al. 2021; Pugh 1954; Vaitheeswaran et al. 2007). When the value exceeds the critical threshold, the material exhibits ductility, while falling below it results in brittleness (Ayaz et al. 2021; Khattak et al. 2022). Poisson’s ratio and Pugh’s ratio Sr3PCl3 and Sr3PBr3 is illustrated in Fig. 15 with variation of pressure. Both Sr3PCl3 and Sr3PBr3 compounds were found to be brittle in normal condition. At 10 GPa pressure, they become ductile, and as the pressure rises, so does their ductility. A similar transformation has also been observed in compounds like Sr₃AsCl₃ and Sr₃SbCl₃ (Hosen 2024).

Calculated a Poisson’s ratio, b Pugh’s ratio for Sr3PX3 (X = Cl, Br) at various applied pressures
Zener anisotropy factor also known as Zener ratio is the quantitative value that determines a material’s deviation from isotropy and is expressed as,
Zener ratio of 1 indicates a material being perfectly isotropic. While values farther from 1 determine anisotropy of varying magnitude, dependent on the deviation of the value itself. As per Table 5, both of the materials are found to be anisotropic at ambient temperature. Anisotropy increases rapidly when increasing pressure is applied. This can be visualized with different 3D anisotropy graphs in Fig. 16. A shift from the spherical form signifies anisotropy. Poisson’s ratio, Young’s modulus, and shear modulus are all found to be anisotropic in 0 GPa and become extremely anisotropic in 30 GPa,

3D structure of Sr3PX3 (X = Cl, Br) at 0 GPa and 30 GPa
Debye temperature can describe various phenomena related to solid-state physics. It can be determined by the following equation (Sun et al. 2005):
Here, \({\Theta }_{D}\) = Debye temperature, h = Planck’s constant, n = number of atoms in a unit cell, \({\text{k}}_{\text{B}}\) = Boltzmann’s constant, \(\rho \) = density, NA = Avogadro’s number, M = molecular weight, \({v}_{m}\) (m/s) = average sound velocity.
\(v_{m}\) can be determined by following set of equations: (where, \(v_{s} { }\left( {{\text{m}}/{\text{s}}} \right)\) = shear sound velocity and \(v_{l} \left( {{\text{m}}/{\text{s}}} \right)\) = longitudinal sound velocity)
The sound velocities, Debye temperature and melting temperature are listened to Table 6. At 0 GPa pressure, Debye temperature of Sr3PCl3 is 282.07 K and Debye temperature of Sr3PBr3 234.29 K. This value rises for both compounds as pressure increases. Under ambient pressure, the melting points of Sr3PCl3 and Sr3PBr3 are 1004.02 K and 971.13 K, respectively. This suggests both compounds to withstand their structural integrity at high temperatures,
3.5 Optical properties
Understanding a compound’s optical essentials in figuring out its possible applications in photovoltaic and optoelectronic systems (Rasheduzzaman et al. 2021). Researchers have developed an interest in lead-free, non-toxic, narrow band gap halides due to their remarkable optical characteristics. Reducing the band gap can be achieved quickly and simply by applying hydrostatic pressure (Hossain et al. 2021c; Islam et al. 2021a). To get the right details regarding a material’s compatibility and boost device efficiency, optical function research is essential. For that, the optical functions of refractive index, dielectric function, absorption, conduction, extinction coefficient, loss function, and reflectivity of under induced pressures between 0 and 30 GPa for Sr3PX3 (X = Cl, Br) are explored.
The dielectric function is a crucial optical property that regulates the rate of charge-carrier recombination (Liu et al. 2018). It presents an understandable image of how optoelectronic devices function, that is employed to investigate how photons and electrons interact (Murnaghan 1937). Figure 17a and b shows a dielectric constant of Sr3PX3 (X = Cl, Br) which have two parts (Real and Imaginary). Dielectric constant of a material is calculated by this equation,

Measured a dielectric function (real part), b dielectric function (imaginary), c absorption, d conductivity for Sr3PX3 (X = Cl, Br) at 0 GPa and 30 GPa
Here, \(\varepsilon_{1} \left( \omega \right)\) represents real part and \(\varepsilon_{2} \left( \omega \right)\) represent imaginary parts respectively. It is clearly apparent from the part of the dielectric functions that is consistent in between the patterns in the spectra of the two materials. Moreover, both Sr3PCl3 and Sr3PBr3 structures exhibit a higher peak in their dielectric constants, with the shift happening at lower photon energies (redshift) as a result of the bandgap being lower as pressure rises. As a consequence of these transformations, under 30 GPa of pressure, it is expected that the researched compounds will capture visible light energy for photovoltaic conversion, as shown in Fig. 17a, which could potentially enhance the efficiency of solar cells. Imaging systems, structural monitoring, and applications that are sensitive to pressure would benefit greatly from this redshift (Hossain et al. 2021c). The static dielectric function (Fig. 17a) of the studied materials at 30 GPa is considerably higher than that at lower pressure. This implies that the two materials will exhibit their best optoelectronic properties at 30 GPa as materials with larger static dielectric function generally possess better optoelectronic performance (Liu et al. 2018). This is because materials with larger \(\varepsilon_{1} \left( 0 \right)\) values tend to possess a lower charge recombination (Hosen et al. 2024b), making them favorable for optoelectronic applications. As seen from Fig. 17a and b, the two compounds’ dielectric functions’ maximum peaks for the real and imaginary portions at 30 GPa are significantly higher in the visible region than those at lower hydrostatic pressure. These results clearly imply that at 30 GPa, the two materials are predicted to possess the best optical properties at visible energy region. In order to analyze optical transitions between the valence and conduction bands, understanding the imaginary component of the dielectric function \(\varepsilon_{2} \left( \omega \right)\) is crucial. The material’s band gap and optical absorption are also connected to the \(\varepsilon_{2} \left( \omega \right)\). In the visible range, the applied pressure raises \(\varepsilon_{2} \left( \omega \right)\), analogous to optical absorption. The peaks also transport a region with low photon energy (redshift), indicating that these compounds are effective visible light absorbers under pressure.
One of the many optical properties used to evaluate the performance of solar cells is the absorption coefficient (Islam et al. 2021b). The optical absorption coefficient (α) measures the capacity to absorb light energy, which offers key insights into the effectiveness of solar energy transformation (Islam et al. 2021a). Figure 17c shows the absorption coefficient of Sr3PX3 (X = Cl, Br) in different pressure levels between 0 and 20 eV energy levels. From the figures, it is clear that the transfer of charges between VB and CB occurs at the band gap as an outcome of the absorption of ultraviolet radiation at 0 GPa. The primary absorption shifts from the electromagnetic spectrum’s ultraviolet to a visible range in response to pressure increase, which is constant with a band gap variation that is pressure-dependent (Hossain et al. 2021c; Islam et al. 2021a; Kholil and Bhuiyan 2021). This is an indication of this material’s better absorptivity of light.
When light with energy exceeding the band gap hits a material, it can excite electrons within the valence band to conduction band. Therefore, with more holes in the valence band and electrons in the conduction band, the material’s conductivity increases. This can be calculated with the equation (Shah et al. 2023),
The optical conductivity for Sr3PX3 (X = Cl, Br) is illustrated in Fig. 17d, it shows a similar property like other optical properties. Rarely any peaks were found in low-energy regions. There is a distinguishing peak difference between Sr3PCl3 and Sr3PBr3, where Sr3PBr3 shows more conductivity. Moreover, with applied pressure, the conductivity also increased for both compounds with redshift. This further supports our initial proposal for utilizing this pressurized application.
The energy of light reflected from the surface of the Sr3PX3 can be used to determine its surface nature (Roknuzzaman et al. 2017). Figure 18a displays reflectivity, R(ω), which is calculated from dielectric function and shown between the energies of 0 and 20 eV. Reflectivity, R is calculated from this relation (Ayub et al. 2024; Mahmud et al. 2024),

Measured a reflectivity, b extinction coefficient, c refractive index and d loss function for Sr3PX3 (X = Cl, Br) at 0 GPa and 30 GPa
From the figures, we see that the visible range reflectivity (R) of Sr3PX3 (X = Cl, Br) increases significantly when pressure rises from 0 to 30 GPa. Plots of ε2(ω) demonstrate that the highest reflectivity values correlate to the energy range where materials absorb the most. The higher reflectivity value when stressed in high-energy zones suggests the material under the study would be more efficient in preventing solar heating.
Figure 18b shows the extinction coefficient of Sr3PX3 (X = Cl, Br) which is highly associated with the static part, \(\varepsilon_{1} \left( \omega \right)\) of dielectric function and expressed as (Mahmud et al. 2024; Shah et al. 2023),
With increasing pressure extinction coefficient curve goes to the higher energy regions (redshift). Raising extinction coefficient means raising the rate at which a substance absorbs or scatters light as it passes through. This can result in diminished light transmission through the substance, causing objects to appear darker or less visible. In practice, a larger extinction coefficient might reduce the visibility or transparency of materials such as optics and environmental monitoring.
Figure 18c displays refractive index, n(ω) of Sr3PX3. This equation gives the refractive index (n) (Ayub et al. 2024; Mahmud et al. 2024),
From the figures it shows that, as pressure rises, n(0) also begins to ascend, which suits both compounds for optoelectronic application. The refractive index is an important metric for measuring the degree of light refraction, especially in photoelectric applications. Photons are slowed by electron interactions while entering a substance, resulting in a refractive index of n(ω) is more than one. The more photons that are slowed down as they pass through a substance, the higher its refractive index.
The electron loss function is the measure of the energy that electrons lose when moving through a dielectric substrate. When photons are produced that have greater energy than the bandgap of the substance, energy is dissipated in Sr3PX3 (X = Cl, Br), as shown by the peak in the plot of L(ω) in Fig. 18d. The following formula is used to calculate the electron loss function (Mahmud et al. 2024; Shah et al. 2023),
L(\(\omega )=\) \(\tau (\frac{-1}{\varepsilon \left(\omega \right)})\)
This figure shows that the energy range of \(8 \sim 10\text{ eV} \), where L(ω) peaks regarding the cubic structure of Sr3PX3. For optical photon spectra and infrared spectra, Since L(ω) peaks appear infrequently below 2 eV, Sr3PX3 could serve as an efficient optical absorption layer. The results show that a significant redshift is caused by increased pressure, indicating an enhancement in optical loss and photon energy for all structures. When developing and optimizing these materials for designated purpose, the loss function of Sr3PX3 should be carefully taken into account as it plays a significant influence on its overall performance.
4 Conclusion
Both compounds maintain mechanical stability across all pressures, as indicated by the Born stability analysis. They also maintain dynamic stability, as evidenced by the positive values in the phonon dispersion curves. The size of Sr3PCl3 exceeds that of Sr3PBr3 due to the larger size of bromine. As pressure is applied, the volume, lattice parameter, and bond length decrease for both compounds. As for electronic properties, both Sr3PCl3 and Sr3PBr3 showed pretty much similar properties. The band gap measured at 30 GPa was 0.22 eV and 0.21 eV using GGA-PBE, while with TB-mBJ, it was 1.07 eV and 0.92 eV, respectively, indicating a relatively low value at this pressure. The density of states revealed a decrease in energy levels with pressure. Effective mass of me and mh also decreased with increased pressure. No hybridization or overlapping were found from the electron charge density. Bulk modulus, Young’s modulus, shear modulus, Poisson’s ratio, and Pugh’s ratio demonstrate improvements for Sr3PCl3 However, the machinability was found to be better for Sr3PBr3. Increased pressure shifted the brittle nature of both compounds to ductile which further increases with applied pressure. The Debye temperature and melting temperatures justify their application in a harsh temperature environment. For optical properties, a red-shift was observed as the spectral lines shifted towards the lower energy side. Absorption increased with the rise in pressure, while reflection decreased in the visible range, indicating favorable characteristics for optoelectronic applications. In terms of the loss function, there was no increase observed until a certain threshold. This research aligns with previous research on Sr3PBr3, with the only notable difference being a slight variation in the lattice parameter. The objective of reducing the band gap and improving optoelectronic and other properties through induced pressure has been achieved successfully. Since this research is simulation-based, there may be some irregularities and discrepancies compared to real-life applications.
Data availability
No datasets were generated or analysed during the current study.
References
Ahmad, A.S., Su, Y., Liu, S.Y., Ståhl, K., Wu, Y.D., Hui, X.D., Ruett, U., Gutowski, O., Glazyrin, K., Liermann, H.P., Franz, H., Wang, H., Wang, X.D., Cao, Q.P., Zhang, D.X., Jiang, J.Z.: Structural stability of high entropy alloys under pressure and temperature. J. Appl. Phys. 121, 235901 (2017). https://doi.org/10.1063/1.4984796
Al-Fahdi, M., Rodriguez, A., Ouyang, T., Hu, M.: High-Throughput computation of new carbon allotropes with diverse hybridization and ultrahigh hardness. Crystals 11, 783 (2021). https://doi.org/10.3390/cryst11070783
Algahtani, A., RehmanLiaqat, F.M., Juraev, N., Khan, I., Alsuhaibani, A.M., AbdullahTirth, V., Refat, M.S., Zaman, A.: Probing the physical properties of Sr3AsX3 (X = F and Br) perovskite compounds for prospective solar cell applications employing the DFT framework. Inorganic Chem. Commun. 162, 112186 (2024). https://doi.org/10.1016/j.inoche.2024.112186
Ali, A.H., Ahmed, A.M., Abdel-Khaliek, A.A., Abd El Khalik, S., Abass, S.M., Shaban, M., Rabia, M.: Preparation of inorganic lead-free CuO/Cs2SnCl6-KI perovskite for green hydrogen production from wastewater by using solar energy. J. Photochem. Photobiol. a: Chem. 445, 115102 (2023). https://doi.org/10.1016/j.jphotochem.2023.115102
Ayaz, U., Shazia, A., Husain, M., Rahman, N., Bonyah, E.: Ab initio investigation of structural, electronic, magnetic, elastic, and optical properties of Cs-based chloro-perovskites CsXCl3 (X = Be and Rh). AIP Adv. 11, 105215 (2021). https://doi.org/10.1063/5.0065663
Ayub, G., Rahman, N., Husain, M., Sohail, M., Khan, R., Sfina, N., Elhadi, M., Azzouz-Rached, A., Alotaibi, A.: Tailoring the structural, elastic, electronic, and optical properties of Cs2ScCuX6 (X = Cl and F) double perovskite compounds via density functional theory (DFT). J. Phys. Chem. Solids 188, 111942 (2024). https://doi.org/10.1016/j.jpcs.2024.111942
Babayigit, A., Duy Thanh, D., Ethirajan, A., Manca, J., Muller, M., Boyen, H.-G., Conings, B.: Assessing the toxicity of Pb- and Sn-based perovskite solar cells in model organism Danio rerio. Sci. Rep. 6, 18721 (2016). https://doi.org/10.1038/srep18721
Bhalla, A.S., Guo, R., Roy, R.: The perovskite structure—a review of its role in ceramic science and technology. Mater. Res. Innovations 4, 3–26 (2000). https://doi.org/10.1007/s100190000062
Blaha P., Schwarz K., Tran F., Laskowski R., Madsen G. K. H., Marks L. D.: WIEN2k: An APW+ lo program for calculating the properties of solids. J. Chem. Phys. 152, 074101 (2020). https://doi.org/10.1063/1.5143061
Born, M.: On the stability of crystal lattices. I. Math. Proc. Cambridge Philos. Soc. 36, 160–172 (1940). https://doi.org/10.1017/S0305004100017138
Butt, M.K., Yaseen, M., Iqbal, J., Altowyan, A.S., Murtaza, A., Iqbal, M., Laref, A.: Structural, electronic, half–metallic ferromagnetic and optical properties of cubic MAlO3 (M=Ce, Pr) perovskites: A DFT study. J. Phys. Chem. Solids 154, 110084 (2021). https://doi.org/10.1016/j.jpcs.2021.110084
Dandia, A., Saini, P., Sharma, R., Parewa, V.: Visible light driven perovskite-based photocatalysts: A new candidate for green organic synthesis by photochemical protocol. Curr. Res.green Sustain. Chem. 3, 100031 (2020). https://doi.org/10.1016/j.crgsc.2020.100031
Eperon, G.E., Habisreutinger, S.N., Leijtens, T., Bruijnaers, B.J., van Franeker, J.J., deQuilettes, D.W., Pathak, S., Sutton, R.J., Grancini, G., Ginger, D.S., Janssen, R.A.J., Petrozza, A., Snaith, H.J.: The importance of moisture in hybrid lead halide perovskite thin film fabrication. ACS Nano 9, 9380–9393 (2015). https://doi.org/10.1021/acsnano.5b03626
Faridi, M.A., Tariq, S., Imran Jamil, M., Batool, A., Nadeem, S., Amin, A.: Pressure induced band-gap tuning in KNbO3 for piezoelectric applications: quantum DFT-GGA approach. Chin. J. Phys. 56, 1481–1487 (2018). https://doi.org/10.1016/j.cjph.2018.06.003
Feng, H.-J., Zhang, Q.: Predicting efficiencies >25% A3MX3 photovoltaic materials and Cu ion implantation modification. Appl. Phys. Lett. 118, 111902 (2021). https://doi.org/10.1063/5.0039936
Feng, W., Cui, S.: Mechanical and electronic properties of Ti2AlN and Ti4AlN3: a first-principles study. Can. J. Phys. 92, 1652–1657 (2014). https://doi.org/10.1139/cjp-2013-0746
Filip, M.R., Giustino, F.: The geometric blueprint of perovskites. Proc. Natl. Acad. Sci. u.s.a. 115, 5397–5402 (2018). https://doi.org/10.1073/pnas.1719179115
Fischer, T.H., Almlof, J.: General methods for geometry and wave function optimization. J. Phys. Chem. (2002). https://doi.org/10.1021/j100203a036
Gaillac, R., Pullumbi, P., Coudert, F.-X.: ELATE: an open-source online application for analysis and visualization of elastic tensors. J. Phys. Condens. Matter 28(27), 275201 (2016). https://doi.org/10.1088/0953-8984/28/27/275201
Gao, J., Zeng, W., Tang, B., Fan, D.-H., Liu, Q.-J., Chang, X.-H., Zhong, M.: Effects of pressure on structural, mechanical, and electronic properties of trigonal and monoclinic MgSiO3. Solid State Sci. 105, 106261 (2020). https://doi.org/10.1016/j.solidstatesciences.2020.106261
Ghosh, A., Ahmed, F., Ferdous, M.J., Juhi, M.M.J., Buian, M.F.I., Miazee, A.A., Sajid, M., Maniruzzaman, M., Tighezza, A.M., Ahmmed, M.F., Islam, M.S.: Strain-induced changes in the electronic, optical and mechanical properties of the inorganic cubic halide perovskite Sr3PBr3 with FP-DFT. J. Phys. Chem. Solids 191, 112053 (2024a). https://doi.org/10.1016/j.jpcs.2024.112053
Ghosh, A., Ferdous Rahman, Md., Kuddus, A., Mohammed, M.K.A., Rasidul Islam, Md., Bhattarai, S., Chaudhry, A.R., Irfan, A.: Investigating of novel inorganic cubic perovskites of A3BX3 (A=Ca, Sr, B P, As, X=I, Br) and their photovoltaic performance with efficiency over 28%. J. Alloys Comp. 986, 174097 (2024b). https://doi.org/10.1016/j.jallcom.2024.174097
Ghosh, A., Rahman, M.F., Islam, M.R., Islam, M.S., Amami, M., Hossain, M.K., Md Ismail, A.B.: Inorganic novel cubic halide perovskite Sr3AsI3: Strain-activated electronic and optical properties. Heliyon 9, e19271 (2023). https://doi.org/10.1016/j.heliyon.2023.e19271
Giannozzi, P., Baroni, S., Bonini, N., Calandra, M., Car, R., Cavazzoni, C., Ceresoli, D., Chiarotti, G.L., Cococcioni, M., Dabo, I., DalCorso, A., de Gironcoli, S., Fabris, S., Fratesi, G., Gebauer, R., Gerstmann, U., Gougoussis, C., Kokalj, A., Lazzeri, M., Martin-Samos, L., Marzari, N., Mauri, F., Mazzarello, R., Paolini, S., Pasquarello, A., Paulatto, L., Sbraccia, C., Scandolo, S., Sclauzero, G., Seitsonen, A.P., Smogunov, A., Umari, P., Wentzcovitch, R.M.: QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 21, 395502 (2009)
Gueddouh, A., Bentria, B., Lefkaier, I.K.: First-principle investigations of structure, elastic and bond hardness of FexB (x=1, 2, 3) under pressure. J. Magn. Magn. Mater. 406, 192–199 (2016). https://doi.org/10.1016/j.jmmm.2016.01.013
Hosen, A.: Investigating the effects of hydrostatic pressure on the physical properties of cubic Sr3BCl3 (B = As, Sb) for improved optoelectronic applications: a DFT study. Heliyon (2024). https://doi.org/10.1016/j.heliyon.2024.e35855
Hosen, A., Hossain, Md.A., Abu-Jafar, M.S., Pingak, R.K., Mousa, A.A.: Unraveling lead-free Fr-based perovskites FrQCl3 (Q = Ca, Sr) and their pressure induced physical properties: DFT analysis for advancing optoelectronic performance. J.phys.chem.solids 193, 112211 (2024a). https://doi.org/10.1016/j.jpcs.2024.112211
Hosen, A., Islam, M.R., Badhan, S.H.: Exploring the influence of pressure-induced semiconductor-to-metal transition on the physical properties of cubic perovskites FrXCl3 (X = Ge and Sn). Heliyon (2024b). https://doi.org/10.1016/j.heliyon.2024.e27581
Hosen, A., Islam, M.R., Park, J.: Pressure-induced band gap shifting from ultra-violet to visible spectrum of non-toxic RbCaBr3 cubic perovskite for enhancing optoelectronic applications. J. Inorg. Organomet. Polym. Mater. 34, 1–10 (2024c)
Hossain, K.M., Hasan, Md.Z., Ali, Md.L.: Narrowing bandgap and enhanced mechanical and optoelectronic properties of perovskite halides: effects of metal doping. AIP Adv. 11, 015052 (2021a). https://doi.org/10.1063/5.0039308
Hossain, K.M., Mitro, S.K., Hossain, Md.A., Modak, J.K., Rasheduzzaman, Md., Hasan, Md.Z.: Influence of antimony on the structural, electronic, mechanical, and anisotropic properties of cubic barium stannate. Mater. Today Commun. 26, 101868 (2021b). https://doi.org/10.1016/j.mtcomm.2020.101868
Hossain, K.M., Zahid Hasan, Md., Lokman Ali, Md.: Understanding the influences of Mg doping on the physical properties of SrMoO3 perovskite. Results Phys. 19, 103337 (2020). https://doi.org/10.1016/j.rinp.2020.103337
Hossain, Md.S., Haque Babu, Md.M., Saha, T., Hossain, Md.S., Podder, J., Rana, Md.S., Barik, A., Rani, P.: Pressure induced semiconductor to metal phase transition in cubic CsSnBr3 perovskite. AIP Adv. 11, 055024 (2021c). https://doi.org/10.1063/5.0048979
Huang, Y., Zhang, T., Wang, J., Xu, S.-G., Zhang, P., Chen, S., Yin, W.-J., Zhang, X., Wei, S.-H.: Design of multifunctional quinternary metal-halide perovskite compounds based on cation-anion co-ordering. Chem. Mater. 32, 5949–5957 (2020). https://doi.org/10.1021/acs.chemmater.0c00674
Husain, M., Rahman, N., Amami, M., Zaman, T., Sohail, M., Khan, R., Khan, A.A., Shah, S.A., KhanSaeedullah, A., ReshakAl-ShaalanAlharthiAlharthyAminTirth, A.H.N.H.S.S.A.M.A.V.: Predicting structural, optoelectronic and mechanical properties of germanium based AGeF3 (A = Ga and In) halides perovskites using the DFT computational approach. Opt. Quant. Electron. 55, 536 (2023a). https://doi.org/10.1007/s11082-023-04796-8
Husain, M., Rahman, N., Sfina, N., Al-Shaalan, N.H., Alharthi, S., Alharthy, S.A., Amin, M.A., Tirth, V., Khan, R., Sohail, M., Azzouz-Rached, A., Khattak, S.A., Khan, M.Y.: The comparative investigations of structural, optoelectronic, and mechanical properties of AgBeX3 (X = F and Cl) metal halide-perovskites for prospective energy applications utilizing DFT approach. Opt. Quant. Electron. 55, 920 (2023b). https://doi.org/10.1007/s11082-023-05187-9
Islam, M.A., Islam, J., Islam, M.N., Sen, S.K., Hossain, A.K.M.A.: Enhanced ductility and optoelectronic properties of environment-friendly CsGeCl3 under pressure. AIP Adv. 11, 045014 (2021a). https://doi.org/10.1063/5.0048849
Islam, M.A., Rahaman, Md.Z., Sen, S.K.: A comparative study of hydrostatic pressure treated environmentally friendly perovskites CsXBr3 (X = Ge/Sn) for optoelectronic applications. AIP Adv. 11, 075109 (2021b). https://doi.org/10.1063/5.0057287
Islam, Md.R., Zahid, A., Rahman, M.A., Rahman, Md.F., Islam, M.A., Hossain, M.K., Ali, M.A., Iqbal, M.A., Bakhsh, F.I., Ahmad, S.: Tuning the optical, electronic, and mechanical properties of inorganic Ca3PCl3 perovskite via biaxial strain. J.phys. Chem.solids 184, 111722 (2024). https://doi.org/10.1016/j.jpcs.2023.111722
Jehan, A., Husain, M., Tirth, V., Algahtani, A., Uzair, M., Rahman, N., Khan, A., Khan, S.N.: Investigation of the structural, electronic, mechanical, and optical properties of NaXCl3 (X= Be, Mg) using density functional theory. RSC Adv. 13, 28395–28406 (2023)
Jiang, Q., Zhao, Y., Zhang, X., Yang, X., Chen, Y., Chu, Z., Ye, Q., Li, X., Yin, Z., You, J.: Surface passivation of perovskite film for efficient solar cells. Nat. Photonics 13, 460–466 (2019). https://doi.org/10.1038/s41566-019-0398-2
Johnsson, M., Lemmens, P.: Crystallography and Chemistry of Perovskites. (2005). http://arxiv.org/abs/cond-mat/0506606
Jung, H.S., Park, N.-G.: Perovskite Solar Cells: From Materials to Devices. Small 11, 10–25 (2015). https://doi.org/10.1002/smll.201402767
Kanhere, P., Chen, Z.: A review on visible Light active perovskite-based photocatalysts. Molecules 19, 19995–20022 (2014). https://doi.org/10.3390/molecules191219995
Karsch, F., Patkós, A., Petreczky, P.: Screened perturbation theory. Phys. Lett. B 401, 69–73 (1997). https://doi.org/10.1016/S0370-2693(97)00392-4
Khan, S.A., Khan, N.Z., Sohail, M., Runowski, M., Xu, X., Agathopoulos, S.: Recent developments of lead-free halide-perovskite nanocrystals: synthesis strategies, stability, challenges, and potential in optoelectronic applications. Mater. Today Phys. 34, 101079 (2023)
Khattak, S.A., Abohashrh, M., Ahmad, I., Husain, M., Ullah, I., Zulfiqar, S., Rooh, G., Rahman, N., Khan, G., Khan, T., Salman Khan, M., Shah, S.K., Tirth, V.: Investigation of structural, mechanical, optoelectronic, and thermoelectric properties of BaXF3 (X = Co, Ir) fluoro-perovskites: promising materials for optoelectronic and thermoelectric applications. ACS Omega 8, 5274–5284 (2023). https://doi.org/10.1021/acsomega.2c05845
Khattak, S.A., Wabaidur, S.M., Islam, M.A., Husain, M., Ullah, I., Zulfiqar, S., Rooh, G., Rahman, N., Khan, M.S., Khan, G., Khan, T., Ghlamallah, B.: First-principles structural, elastic and optoelectronics study of sodium niobate and tantalate perovskites. Sci. Rep. 12, 21700 (2022). https://doi.org/10.1038/s41598-022-26250-7
Kholil, M.I., Bhuiyan, M.T.H.: Effects of pressure on narrowing the band gap, visible light absorption, and semi-metallic transition of lead-free perovskite CsSnBr3 for optoelectronic applications. J. Phys. Chem. Solids 154, 110083 (2021). https://doi.org/10.1016/j.jpcs.2021.110083
Kleinman, L.: Deformation potentials in silicon I. Uniaxial Strain. Phys. Rev. 128, 2614–2621 (1962). https://doi.org/10.1103/PhysRev.128.2614
Kohn, W., Sham, L.J.: Self-consistent equations including exchange and correlation effects. Phys. Rev. 140, A1133–A1138 (1965). https://doi.org/10.1103/PhysRev.140.A1133
Kojima, A., Teshima, K., Shirai, Y., Miyasaka, T.: Organometal halide perovskites as visible-light sensitizers for photovoltaic cells. J. Am. Chem. Soc. 131, 6050–6051 (2009). https://doi.org/10.1021/ja809598r
Kouchaksaraie, L.S.: Theoretical calculation of electrical and optical properties of BaZrO3. Int. J. Math. Comput. Phys. Electr. Comput. Eng. 5, 1680–1683 (2011)
Lan, Z., Meng, J., Zheng, K., Castelli, I.E.: Exploring the intrinsic point defects in cesium copper halides. J. Phys. Chem. C 125, 1592–1598 (2021). https://doi.org/10.1021/acs.jpcc.0c11216
Langhoff, P.W., Epstein, S.T., Karplus, M.: Aspects of time-dependent perturbation theory. Rev. Mod. Phys. 44, 602–644 (1972). https://doi.org/10.1103/RevModPhys.44.602
Lee, M.M., Teuscher, J., Miyasaka, T., Murakami, T.N., Snaith, H.J.: Efficient hybrid solar cells based on meso-superstructured organometal halide perovskites. Science 338, 643–647 (2012). https://doi.org/10.1126/science.1228604
Liu, J., Li, N., Jia, J., Dong, J., Qiu, Z., Iqbal, S., Cao, B.: Perovskite films grown with green mixed anti-solvent for highly efficient solar cells with enhanced stability. Sol. Energy 181, 285–292 (2019a). https://doi.org/10.1016/j.solener.2019.02.020
Liu, S.-C., Li, Z., Yang, Y., Wang, X., Chen, Y.-X., Xue, D.-J., Hu, J.-S.: Investigation of oxygen passivation for high-performance all-inorganic perovskite solar cells. J. Am. Chem. Soc. 141, 18075–18082 (2019b). https://doi.org/10.1021/jacs.9b07182
Liu, X., Xie, B., Duan, C., Wang, Z., Fan, B., Zhang, K., Lin, B., Colberts, F.J.M., Ma, W., Janssen, R.A.J., Huang, F., Cao, Y.: A high dielectric constant non-fullerene acceptor for efficient bulk-heterojunction organic solar cells. J. Mater. Chem. A 6, 395–403 (2018). https://doi.org/10.1039/C7TA10136H
Liu, Y.-Q., Wei, D., Cui, H.-L., Wang, D.-Q.: Photovoltaic effect related to methylammonium cation orientation and carrier transport properties in high-performance perovskite solar cells. ACS Appl. Mater. Interfaces 12, 3563–3571 (2020). https://doi.org/10.1021/acsami.9b18452
Mahmud, S., Ali, M.A., Hossain, M.M., Uddin, M.M.: DFT aided prediction of phase stability, optoelectronic and thermoelectric properties of A2AuScX6 (A= Cs, Rb; X= Cl, Br, I) double perovskites for energy harvesting technology. Vacuum 221, 112926 (2024)
Marshall, K.P., Walker, M., Walton, R.I., Hatton, R.A.: Enhanced stability and efficiency in hole-transport-layer-free CsSnI3 perovskite photovoltaics. Nat. Energy 1, 16178 (2016). https://doi.org/10.1038/nenergy.2016.178
Moghe, D., Wang, L., Traverse, C.J., Redoute, A., Sponseller, M., Brown, P.R., Bulović, V., Lunt, R.R.: All vapor-deposited lead-free doped CsSnBr3 planar solar cells. Nano Energy 28, 469–474 (2016). https://doi.org/10.1016/j.nanoen.2016.09.009
Murnaghan, F.D.: Finite deformations of an elastic solid. Am. J. Math. 59, 235–260 (1937). https://doi.org/10.2307/2371405
Murnaghan, F.D.: The compressibility of media under extreme pressures. Proc. Natl. Acad. Sci. 30, 244–247 (1944). https://doi.org/10.1073/pnas.30.9.244
Noel, N.K., Stranks, S.D., Abate, A., Wehrenfennig, C., Guarnera, S., Haghighirad, A.-A., Sadhanala, A., Eperon, G.E., Pathak, S.K., Johnston, M.B., Petrozza, A., Herz, L.M., Snaith, H.J.: Lead-free organic–inorganic tin halide perovskites for photovoltaic applications. Energy Environ. Sci. 7, 3061–3068 (2014). https://doi.org/10.1039/C4EE01076K
Perdew, J.P., Burke, K., Ernzerhof, M.: Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996). https://doi.org/10.1103/PhysRevLett.77.3865
Perdew, J.P., Chevary, J.A., Vosko, S.H., Jackson, K.A., Pederson, M.R., Singh, D.J., Fiolhais, C.: Atoms, molecules, solids, and surfaces: applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B 46, 6671–6687 (1992). https://doi.org/10.1103/PhysRevB.46.6671
Pitriana, P., Wungu, T.D.K., Hidayat, R.: The characteristics of band structures and crystal binding in all-inorganic perovskite APbBr3 studied by the first principle calculations using the density functional theory (DFT) method. Results Phys. 15, 102592 (2019). https://doi.org/10.1016/j.rinp.2019.102592
Pugh, S.F.: XCII. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. London, Edinburgh, and Dublin Philos. Mag. J. Sci. 45, 823–843 (1954). https://doi.org/10.1080/14786440808520496
Rahman, N., Husain, M., Yang, J., Murtaza, G., Sajjad, M., Habib, A., Karim, A., HaqZulfiqar, M.U., Rauf, A.: First principle study of structural, electronic, elastic, and magnetic properties of half-Heusler compounds ScTiX (X= Si, Ge, Pb, In, Sb, and Tl). J. Superconductivity Novel Mag. 33, 3915–3922 (2020)
Rasheduzzaman, Md., Hossain, K.M., Mitro, S.K., Hadi, M.A., Modak, J.K., Hasan, Md.Z.: Structural, mechanical, thermal, and optical properties of inverse-Heusler alloys Cr2CoZ (Z = Al, In): a first-principles investigation. Phys. Lett. A 385, 126967 (2021). https://doi.org/10.1016/j.physleta.2020.126967
Ren, M., Qian, X., Chen, Y., Wang, T., Zhao, Y.: Potential lead toxicity and leakage issues on lead halide perovskite photovoltaics. J. Hazard. Mater. 426, 127848 (2022)
Roknuzzaman, M., Ostrikov, K., Wang, H., Du, A., Tesfamichael, T.: Towards lead-free perovskite photovoltaics and optoelectronics by ab-initio simulations. Sci. Rep. 7, 14025 (2017). https://doi.org/10.1038/s41598-017-13172-y
Seo, J., Noh, J.H., Seok, S.I.: Rational strategies for efficient perovskite solar cells. Acc. Chem. Res. 49, 562–572 (2016). https://doi.org/10.1021/acs.accounts.5b00444
Shah, S.Z.A., Niaz, S., Nasir, T., Sifuna, J.: First principles insight into band gap tuning in bismuth based double perovskites X2NaBiCl6 (X= Cs, Rb, K) for enhanced optoelectronic and thermoelectric properties. Results Chem. 5, 100828 (2023)
Stoumpos, C.C., Malliakas, C.D., Kanatzidis, M.G.: Semiconducting tin and lead iodide perovskites with organic cations: phase transitions, high mobilities, and near-infrared photoluminescent properties. Inorg. Chem. 52, 9019–9038 (2013). https://doi.org/10.1021/ic401215x
Sun, Z., Music, D., Ahuja, R., Schneider, J.M.: Electronic origin of shearing in M2AC (M = Ti, V, Cr, A = Al, Ga). J. Phys. Condens. Matter 17, 7169 (2005). https://doi.org/10.1088/0953-8984/17/46/001
Tian, S., Li, J., Li, S., Bu, T., Mo, Y., Wang, S., Li, W., Huang, F.: A facile green solvent engineering for up-scaling perovskite solar cell modules. Sol. Energy 183, 386–391 (2019). https://doi.org/10.1016/j.solener.2019.03.038
Tran, F., Blaha, P.: Accurate band gaps of semiconductors and insulators with a semilocal exchange-correlation potential. Phys. Rev. Lett. 102, 226401 (2009). https://doi.org/10.1103/PhysRevLett.102.226401
Vaitheeswaran, G., Kanchana, V., Svane, A., Delin, A.: Elastic properties of MgCNi3—a superconducting perovskite. J. Phys. Condens. Matter 19, 326214 (2007). https://doi.org/10.1088/0953-8984/19/32/326214
Vanderbilt, D.: Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 41, 7892–7895 (1990). https://doi.org/10.1103/PhysRevB.41.7892
Verma, A.S., Jindal, V.K.: Lattice constant of cubic perovskites. J. Alloy. Compd. 485, 514–518 (2009)
Wang, L., Kutana, A., Zou, X., Yakobson, B.I.: Electro-mechanical anisotropy of phosphorene. Nanoscale 7, 9746–9751 (2015). https://doi.org/10.1039/C5NR00355E
Wehrenfennig, C., Eperon, G.E., Johnston, M.B., Snaith, H.J., Herz, L.M.: High charge carrier mobilities and lifetimes in organolead trihalide perovskites. Adv. Mater. (Deerfield Beach, Fla.) 26, 1584 (2014)
Wu, T., Qin, Z., Wang, Y., Wu, Y., Chen, W., Zhang, S., Cai, M., Dai, S., Zhang, J., Liu, J., Zhou, Z., Liu, X., Segawa, H., Tan, H., Tang, Q., Fang, J., Li, Y., Ding, L., Ning, Z., Qi, Y., Zhang, Y., Han, L.: The main progress of perovskite solar cells in 2020–2021. Nano-Micro. Lett. 13, 152 (2021). https://doi.org/10.1007/s40820-021-00672-w
Xiong, R., Die, D., Xiao, L., Xu, Y.-G., Shen, X.-Y.: Probing the structural, electronic, and magnetic properties of AgnV (n = 1–12) clusters. Nanoscale Res. Lett. 12, 625 (2017). https://doi.org/10.1186/s11671-017-2394-0
Yang, X., Wang, W., Ran, R., Zhou, W., Shao, Z.: Recent advances in Cs2AgBiBr6-based halide double perovskites as lead-free and inorganic light absorbers for perovskite solar cells. Energy Fuels 34, 10513–10528 (2020). https://doi.org/10.1021/acs.energyfuels.0c02236
Yin, W.-J., Shi, T., Yan, Y.: Unique properties of halide perovskites as possible origins of the superior solar cell performance. Adv. Mater. 26, 4653–4658 (2014). https://doi.org/10.1002/adma.201306281
Zhang, W., Eperon, G.E., Snaith, H.J.: Metal halide perovskites for energy applications. Nat. Energy 1, 1–8 (2016). https://doi.org/10.1038/nenergy.2016.48
Zhao, X., Xiang, C., Huang, M., Ding, M., Jia, C., Jia, C., Sun, L., Sun, L.: Quantum dot solar cells. In: Emerging Photovoltaic Materials, pp. 611–658. John Wiley, Hoboken (2018a)
Zhao, Y.-Q., Wang, X., Liu, B., Yu, Z.-L., He, P.-B., Wan, Q., Cai, M.-Q., Yu, H.-L.: Geometric structure and photovoltaic properties of mixed halide germanium perovskites from theoretical view. Organic Electr. 53, 50–56 (2018b). https://doi.org/10.1016/j.orgel.2017.11.005
Zhou, X., Jankowska, J., Dong, H., Prezhdo, O.V.: Recent theoretical progress in the development of perovskite photovoltaic materials. J. Energy Chem. 27, 637–649 (2018). https://doi.org/10.1016/j.jechem.2017.10.010
Zhou, Y., You, L., Wang, S., Ku, Z., Fan, H., Schmidt, D., Rusydi, A., Chang, L., Wang, L., Ren, P., Chen, L., Yuan, G., Chen, L., Wang, J.: Giant photostriction in organic–inorganic lead halide perovskites. Nat. Commun. 7, 11193 (2016). https://doi.org/10.1038/ncomms11193
Zhumekenov, A.A., Saidaminov, M.I., Haque, M.A., Alarousu, E., Sarmah, S.P., Murali, B., Dursun, I., Miao, X.-H., Abdelhady, A.L., Wu, T., Mohammed, O.F., Bakr, O.M.: Formamidinium lead halide perovskite crystals with unprecedented long carrier dynamics and diffusion length. ACS Energy Lett. 1, 32–37 (2016). https://doi.org/10.1021/acsenergylett.6b00002
Funding
We did not receive any funding for this research.
Author information
Authors and Affiliations
Contributions
S.J.: Investigation, methodology, data curation, writing the original, draft reviewing and editing. M.A.H.: Formal analysis, software, calculation and analysis, writing the original draft, reviewing and editing. M.A.Y.: Data curation, writing the original draft, reviewing and editing. M.M.H.: Data curation, writing the original draft, reviewing and editing. R.K.P.: Data curation, writing the original draft, reviewing and editing. N.F.A.M.: Formal analysis, software, data curation, reviewing and editing. M.S.A.J.: Formal analysis, software, data curation, reviewing and editing. A.A.M.: Formal analysis, software, data curation, reviewing and editing. A.H.: Investigation, conceptualization, software, methodology, data curation, calculation and analysis, writing the original draft, supervision, reviewing and editing.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Joifullah, S., Hossain, M.A., Al Yeamin, M. et al. First-principles investigation of pressure-modulated structural, electronic, mechanical, and optical characteristics of Sr3PX3 (X = Cl, Br) for enhanced optoelectronic application. Opt Quant Electron 56, 1463 (2024). https://doi.org/10.1007/s11082-024-07388-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11082-024-07388-2