
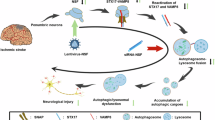
Abstract
CCT2 is a eukaryotic chaperonin TCP-1 ring complex subunit that mediates protein folding, autophagosome incorporation, and protein aggregation. In this study, we investigated the effects of CCT on oxidative and ischemic damage using in vitro and in vivo experimental models. The Tat-CCT2 fusion protein was efficiently delivered into HT22 cells in a concentration- and time-dependent manner, and the delivered protein was gradually degraded in HT22 cells. Incubation with Tat-CCT2 significantly ameliorated the 200 µM hydrogen peroxide (H2O2)-induced reduction in cell viability in a concentration-dependent manner, and 8 µM Tat-CCT2 treatment significantly alleviated H2O2-induced DNA fragmentation and reactive oxygen species formation in HT22 cells. In gerbils, CCT2 protein was efficiently delivered into pyramidal cells in CA1 region by intraperitoneally injecting 0.5 mg/kg Tat-CCT2, as opposed to control CCT2. In addition, treatment with 0.2 or 0.5 mg/kg Tat-CCT2 mitigated ischemia-induced hyperlocomotive activity 1 d after ischemia and confirmed the neuroprotective effects by NeuN immunohistochemistry in the hippocampal CA1 region 4 d after ischemia. Tat-CCT2 treatment significantly reduced the ischemia-induced activation of astrocytes and microglia in the hippocampal CA1 region 4 d after ischemia. Furthermore, treatment with 0.2 or 0.5 mg/kg Tat-CCT2 facilitated ischemia-induced autophagic activity and ameliorated ischemia-induced autophagic initiation in the hippocampus 1 d after ischemia based on western blotting for LC3B and Beclin-1, respectively. Levels of p62, an autophagic substrate, significantly increased in the hippocampus following treatment with Tat-CCT2. These results suggested that Tat-CCT2 exerts neuroprotective effects against oxidative stress and ischemic damage by promoting the autophagic removal of damaged proteins or organelles.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Stroke is a life-threatening disorder that causes a reduction in the quality of life due to permanent disability [1, 2]. Among all strokes, ischemia is the most prevalent (87%), and the absolute number of patients with ischemia has increased by 70%, although the age-standardized incidence rate has decreased [3]. Mongolian gerbils are used as animal models for transient forebrain ischemia because of the incomplete development of their posterior communicating arteries [4, 5]. Obstruction of cerebral vessels and recanalization lead to significant changes in the morphology of neurons and glial cells in the hippocampus, thalamus, and cerebral cortex [6,7,8]. Many mechanisms of neuronal damage have been proposed, including excitotoxicity, oxidative stress, inflammation, and autophagy [9,10,11,12]; however, these mechanisms interact with each other. The removal of protein aggregates, such as ubiquitinated proteins and damaged organelles, is important for reducing neuronal damage after ischemia [12, 13].
With the massive aggregation of proteins and damaged organelles after ischemia, chaperones directly bind to misfolded proteins to counteract aggregation [14, 15]. Among the various chaperones, the eukaryotic chaperonin TCP-1 ring complex (TRiC) is responsible for the folding of approximately 10% of cytosolic protein substrates [16, 17] and consists of eight subunits (CCT1–8) [18]. Among these, CCT2 is closely associated with autophagosome incorporation and clearance of aggregated proteins [19]. Extracellular vesicles of mesenchymal stem cells derived from umbilical cord blood (UC-MSC-EVs) have high CCT2 content, and treatment with UC-MSC-EVs can significantly ameliorate liver injury 6 h after ischemia [20]. In contrast, knockdown of CCTs in UC-MSC-EVs decreases their hepatoprotective effects against ischemic damage [20]. However, no comprehensive study has been conducted to elucidate the effects of CCT2 on ischemic brain damage.
The Tat peptide, derived from the transactivator of the human immunodeficiency virus transcription, is a cell-penetrating peptide that is widely used for the delivery of large and small molecules into brain cells because the blood-brain barrier prevents molecules in the circulating blood from crossing into neurons [21, 22]. In a previous study, we observed that the Tat-fusion protein was efficiently delivered into the HT22 hippocampal cell line and the gerbil hippocampus [23].
In the current study, we investigated the effects of the Tat-CCT2 fusion protein on hydrogen peroxide (H2O2)-induced oxidative damage in HT22 cells and ischemia-induced hippocampal damage in gerbils. In addition, we examined the neuroprotective mechanisms of Tat-CCT2 against autophagy-induced ischemic damage in the hippocampus.
Experimental Procedures
Tat-CCT2 Synthesis
To facilitate the delivery of CCT2 into cells and the blood-brain barrier, CCT2 protein was synthesized using the pET15b vector (Novagen, Merck Millipore, Darmstadt, Germany) with human CCT2 cDNA and a histidine-tag (His-tag) with or without the Tat peptide, as shown in Fig. 1A. As described previously [23], Escherichia coli BL21 cells were cultivated in broth media after transformation with Tat-CCT2 or CCT2 plasmids. Protein expression was induced by treatment with 0.5 mM isopropyl-β-D-thiogalactoside (Duchefa, Haarlem, the Netherlands) at 37 °C for 6 h. Proteins were purified using a Ni2+-nitrilotriacetic acid Sepharose affinity column and PD-10 column chromatography. Finally, purified proteins were confirmed for the presence of His-tags through western blotting, as previously described [23, 24].
Delivery of Tat-CCT2 into HT22 Cells
The concentration- and time-dependent delivery of Tat-CCT2 and CCT2 proteins was assessed by western blot analysis of His-tags in HT22 cells. Tat-CCT2 or CCT (0–8.0 µM) was incubated for 1 h, or 8.0 µM of protein was added to HT22 cells for various incubation times (0–60 min). To examine the intracellular degradation of proteins, 8.0 µM of protein was incubated for a longer period (0–60 h). Thereafter, the cells were harvested and subjected to western blotting for detecting His-tags, as previously described [23, 24].
The delivery of Tat-CCT2 and CCT was morphologically confirmed by immunocytochemical staining of His-tags in HT22 cells. Briefly, HT22 cells cultured on coverslips were incubated with 8.0 µM of proteins for 1 h and fixed with 4% paraformaldehyde at 25 °C for 5 min. Fixed cells were sequentially incubated with rabbit anti-His-tag (1:1000, Novus Biologicals, Centennial, CO, USA) and Alexa Fluor 488-conjugated anti-rabbit IgG (1:1,000, Invitrogen, Carlsbad, CA, USA) with 1 µg/mL 4,6-diamidino-2-phenylindole (DAPI; Roche Applied Science, Mannheim, Germany).
Effects of Tat-CCT2 and CCT2 against oxidative stress induced by H2O2 in HT22 cells.
Oxidative damage was induced by incubation with 200 µM H2O2 for 1 h, and Tat-CCT2 or CCT (0–8.0 µM) was treated simultaneously with H2O2 to validate the optimal concentration of protein to exhibit neuroprotective effects against oxidative stress. Thereafter, the culture media were harvested, incubated with water-soluble tetrazolium salt (WST-1) reagent for 1 h, and the formazan dye formed was quantified using an enzyme-linked immunosorbent assay (ELISA) plate reader (Fluoroskan Ascent, Labsystems Multiskan MCC/340, Helsinki, Finland). To visualize the surviving cells after H2O2 treatment, HT22 cells cultured on coverslips were incubated with 8 µM Tat-CCT2 or CCT1 with 200 µM H2O2 for 1 h and then incubated with 1 µmol/L 5-carboxyfluorescein diacetate, acetoxymethyl ester (5- CFDA AM, Invitrogen, Carlsbad, CA, USA) with 1 µg/mL DAPI in phosphate-buffered saline (PBS) at 37 °C for 30 min.
To determine DNA damage and reactive oxygen species (ROS) formation, HT22 cells cultured on coverslips were incubated with 8 µM Tat-CCT2 or CCT1 and 200 µM H2O2 for 1 h. Thereafter, cells were washed with 4% paraformaldehyde or 50 mM PBS for terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate-biotin nick end labeling (TUNEL) and 2,7-dichlorofluorescein diacetate (DCF-DA) staining, respectively. Cells were exposed to 20 µM DCF-DA for 30 min to visualize ROS formation, and DAPI staining was performed according to the manufacturer’s instructions, as previously described [23, 24].
The fluorescent structures were visualized using a fluorescence microscope (Nikon Eclipse 80i, Tokyo, Japan), and the total fluorescence intensity was measured using an ELISA plate reader (Fluoroskan Ascent, Labsystems Multiskan MCC/340, Helsinki, Finland), as previously described [23, 24].
Ethical Statement
Male Mongolian gerbils (Meriones unguiculatus) were obtained from Japan SLC, Inc. (Shizuoka, Japan). The experimental animal study protocol was approved by the Institutional Animal Care and Use Committee of the Seoul National University (SNU-200313-2-4). The experimental procedures were conducted according to the ARRIVE 2.0 guidelines [25] and the guidelines of the American Veterinary Medical Association and our institution.
Delivery of Tat-CCT2 into Gerbil hippocampus
To visualize the delivery of Tat-CCT2 and CCT2 proteins into the hippocampus, animals (n = 5 in each group) were intraperitoneally injected with Tat peptide, 0.5 mg/kg Tat-CCT2 or CCT2, and anesthetized with 5% isoflurane (Baxtor, Deerfield, IL, USA). Transcardiac perfusion was performed with 0.9% physiological saline and 4% paraformaldehyde 2 h after protein treatment. The brains were then removed, post-fixed with the same fixative at 25 °C for 12 h, and treated with a 30% sucrose solution. Thereafter, tissue Sect. (30 μm thickness) were prepared between 1.4 and 2.0 mm caudal to the bregma using a sliding microtome (HM430, Thermo Scientific, Waltham, MA) with a freezing stage [26]. Five Sect. (90-µm apart from each other) were selected and sequentially incubated with rabbit anti-His-tag (1:1000, Novus Biologicals) and Cy3-conjugated goat anti-rabbit IgG (1:500, Jackson Immuno Research Inc., PA, USA).
Effects of Tat-CCT2 and CCT2 against neuronal death induced by ischemia in gerbils.
To observe ischemia-induced hyperlocomotive activity, neuronal death, and glial activation in the hippocampus, animals (n = 5 in each group) were anesthetized with 3.0% isoflurane (Baxter) mixed with nitrous oxide and oxygen. A ventral neck incision was made and both the common carotid arteries were exposed. The arteries were occluded for 5 min with aneurysm clips, and body temperature was controlled using a rectal-probe thermostat blanket and an infrared radiation lamp. The retinal artery was observed using an ophthalmoscope (HEINE K180®, Heine Optotechnik, Herrsching, Germany) to confirm the occlusion and reperfusion of the common carotid artery. Immediately after reperfusion, the gerbils were intraperitoneally injected with the Tat peptide, CCT2 (0.5 mg/kg), and Tat-CCT2 (0.2 or 0.5 mg/kg). Sham-operated animals were used as controls.
Spontaneous motor activity was tracked because hyperlocomotive activity was induced 1 d after ischemia due to functional deficits in the hippocampal CA1 region, as opposed to other brain regions [27, 28] in a sound-proof Plexiglas cage (25 × 20 × 12 cm) using a digital camera system (Basler, Ahrensburg, Germany), and the distance traveled over time was analyzed using XT14 software (Ethovision, Wageningen, Netherlands).
After 4 d of ischemia, the animals were euthanized, and neuronal survival and glial activation were confirmed by immunohistochemical staining for neuronal nuclei (NeuN), glial fibrillary acidic protein (GFAP), and ionized calcium-binding adapter molecule 1 (Iba-1) in the hippocampus. Tissue processing and immunohistochemical staining were conducted as described above (Delivery of Tat-CCT2 into the gerbil hippocampus). Four Sect. (120-µm apart from each other) were selected and incubated with primary antibodies, such as mouse anti-NeuN (1:1000; EMD Millipore, Temecula, CA), rabbit anti-GFAP (1:1000; EMD Millipore), and rabbit anti-Iba-1 (1:500; Wako, Osaka, Japan), at 25 °C for 12 h. Thereafter, the sections were treated with biotinylated goat anti-rabbit IgG (1:200, Vector, Burlingame, CA) and peroxidase-conjugated streptavidin (1:200, Vector) at 25 °C for 2 h. Immunoreactive signals were visualized using 3,3-diaminobenzidine tetrachloride (Sigma, St. Louis, MO, USA).
Neuroprotective mechanisms of Tat-CCT2 and CCT2 against ischemia damage in gerbils.
To elucidate the neuroprotective mechanisms of Tat-CCT2 against ischemic damage, the animals were euthanized 6 h or 1 d after ischemia for ELISA and western blotting, respectively, and their brains were immediately removed. Hippocampal homogenates were prepared using RIPA extraction buffer containing 1 mM phenylmethylsulfonyl fluoride, 5 mM NaF, 1 mM Na3VO4, and a protease inhibitor cocktail (Roche, Indianapolis, IN, USA) at 4 °C for 15 min and centrifuged at 12,000 × g at 4 °C for 20 min. Oxidative damage in the hippocampus was assessed using the 4-HNE ELISA kit (MyBioSource, San Diego, CA, USA) and 8-iso-PGF2α ELISA kit (Cayman Chemical Company, Ann Arbor, MI, USA) according to the manufacturer’s instructions. For western blotting, the homogenates were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis and electrotransferred onto polyvinylidene difluoride membranes. Membranes were incubated with primary antibodies, such as rabbit anti-microtubule-associated protein 1 light chain 3B (LC3-II, 1:1000, Cell Signaling, Beverly, MA, USA), rabbit anti-p62 (1:1000, Sigma), rabbit anti-Beclin-1 (1:1000, Cell Signaling), or β-actin (1:1000; Cell Signaling). Thereafter, the membranes were incubated with peroxidase-conjugated anti-rabbit IgG, and the protein bands were visualized using chemiluminescent reagents, as recommended by the manufacturer (Amersham, Franklin Lakes, NJ, USA).
Data Quantification and Analysis
For data quantification, four to five sections (at 90–120 μm intervals) were selected, and the number of NeuN-positive neurons was counted in the stratum pyramidale was counted using ImageJ software version 1.53t (National Institutes of Health, Bethesda, MD, USA). The immunoreactivity of GFAP and Iba-1 was quantified as the optical density, calculated as the sum of the gray scale × pixel number using ImageJ software, as previously described [23, 24]. Protein band intensities were quantified through western blotting using the Image Lab software. The data were normalized to percentile values and compared with those of the control group.
All acquired data were presented as mean ± standard deviation. Differences between means were analyzed using the Student’s t-test and one-way analysis of variance, followed by the Bonferroni’s multiple comparison test. Statistical significance was set at p < 0.05.
Results
Confirmation of the Tat-CCT2 protein and its delivery into HT22 cells.
Overexpressed Tat-CCT2 and CCT2 proteins were validated by Coomassie Brilliant Blue staining and western blotting for His-tags due to their insertion into the pET15b vector. Clear single bands of Tat-CCT2 and CCT2 were detected at approximately 52 kDa and 54 kDa, respectively (Fig. 1A).
The concentration- and time-dependent delivery of Tat-CCT2 and CCT2 was assessed using western blotting for His-tags. Treatment with CCT2 did not result in any clear His-tag bands at any concentration or incubation time after protein treatment (Fig. 1B and C). In contrast, incubation with Tat-CCT2 for 1 h detected a His-tag band, the intensity of which was significantly increased in the group after administration of 4.0 µM Tat-CCT2 or higher (Fig. 1B). In addition, treatment with 8.0 µM Tat-CCT2 significantly increased the His-tag signal intensity at incubation times of 30 min or longer (Fig. 1C).
Degradation of the delivered Tat-CCT2 protein was assessed by western blotting for His-tag. His-tag protein levels significantly increased 1 h after protein treatment; thereafter, the protein level was decreased in a time-dependent manner. His-tag protein levels were significantly higher 24 h after protein treatment in the Tat-CCT2-treated group than in the control group (Fig. 1D).
The delivered Tat-CCT2 protein was visualized by immunohistochemical staining for His-tag. In the control and CCT-treated groups, no His-tag immunoreactivity was observed in HT22 cells, whereas in the Tat-CCT2-treated group, His-tag-immunoreactive structures were mainly identified in the cytoplasm of HT22 cells (Fig. 1E).

Synthesis of Tat-CCT2 and its control protein and confirmation of their delivery into HT22 cells. (A) Tat-CCT2 and its control (CCT2) are synthesized with His-tag and CCT2 with or without the Tat expression vector, respectively. The expressions of Tat-CCT2 and CCT2 proteins are detected by Coomassie Brilliant Blue staining and western blotting for His-tags. (B and C) Concentration- and time-dependent delivery of Tat-CCT2 and CCT2 proteins is confirmed by western blotting for His-tag in HT22 cells. (D) Protein degradation of delivered Tat-CCT2 protein is assessed by western blotting for His-tag in HT22 cells. All assays are performed in triplicates. Data are expressed as mean ± standard deviation and analyzed using the Student’s t-test (n = 5, ap < 0.05, significantly different from the control group). (E) Intracellularly delivered proteins are visualized by immunofluorescent staining for His-tags with DAPI in HT22 cells. Scale bar = 20 μm
Effects of Tat-CCT2 on oxidative stress induced by 200 µM H2O2 in HT22 cells.
Cells that survived after H2O2-induced oxidative damage were determined by a WST-1 assay with incubations of various concentrations (2.0–8.0 µM) of CCT2 and Tat-CCT2 in HT22 cells. Co-treatment with 200 µM H2O2 and vehicle significantly reduced cell viability to 53.3% ± 8.7% of that of the control group. Treatment with CCT2 did not show any significant ameliorative effects on cell survival, whereas Tat-CCT2 treatment prevented the reduction in cell viability induced by H2O2 in a concentration-dependent manner. In particular, cell viability was significantly improved by treatment with 8 µM Tat-CCT2 to 79.6% ± 15.9% of that of the control group, although it remained significantly lower than that of the control group (Fig. 2A).
To visualize surviving HT22 cells, 5-CFDA-AM staining was performed 1 h after treatment with 8.0 µM Tat-CCT2 or CCT1 with 200 µM H2O2. In the control group, many cells were stained with 5-CFDA-AM-based on DAPI staining. In the vehicle- and CCT2-treated groups, fewer 5-CFDA AM-stained cells were identified than in the control group. In addition, the fluorescence intensities in these groups were significantly decreased to 27.1% ± 8.3% and 28.5% ± 9.8% of that of the control group, respectively. In contrast, 5-CFDA-AM-stained cells were abundantly observed in the Tat-CCT2-treated group, and the fluorescence intensity was 84.3% ± 8.5% of that of the control group (Fig. 2B).
ROS formation was visualized by the DCF-DA staining of HT22 cells. In the control group, no DCF-stained cells were detected in the HT22 cells, whereas strong DCF fluorescence was observed in the vehicle-treated group. In this group, DCF fluorescence intensity significantly increased to 810.8% ± 113.3% of that of the control group. In the CCT2-treated group, DCF fluorescence was observed in HT22 cells; however, the fluorescence intensity was significantly lower (536.1% ± 165.6% of that of the control group) than that in the vehicle-treated group. In the Tat-CCT2-treated group, DCF fluorescent structures were faintly detected, and the fluorescence intensity was significantly lower (166.0% ± 33.0% of that of the control group) than that in the vehicle- or CCT2-treated groups (Fig. 2C).
DNA damage was visualized by the TUNEL staining of HT22 cells. TUNEL-positive cells were barely detectable in the control group but were abundant in the vehicle- and CCT2-treated groups. The fluorescence intensity in the vehicle- and CCT2-treated groups significantly increased to 753.8% ± 206.8% and 608.5% ± 98.5% of that of the control group, respectively. No significant differences were observed in the TUNEL fluorescence intensity between the vehicle- and CCT2-treated groups. In the Tat-CCT2-treated group, few TUNEL-positive cells were identified, and the fluorescence intensity was significantly ameliorated to 248.4% ± 62.3% of that of the control group compared to the vehicle- or CCT2-treated groups (Fig. 2D).

Effects of Tat-CCT2 and its control protein against oxidative stress induced by 200 µM H2O2 in HT22 cells. (A) Concentration-dependent cell viability is determined in HT22 cells using a WST-1 assay 1 h after incubation with protein and hydrogen peroxide (H2O2). (B–D) Visualization of survived cells, ROS formation, and DNA fragmentation are performed by 5-CFDA AM, DCF-DA, and TUNEL staining, respectively, in HT22 cells. Scale bar = 50 μm. All assays are performed in triplicates. Total fluorescence intensity is quantified, and data are analyzed using one-way analysis of variance followed by Tukey’s multiple comparison post hoc test (n = 5 in each experiment; ap < 0.05, significantly different from the control group; bp < 0.05, significantly different from the vehicle group; cp < 0.05, significantly different from the CCT2 group). Data are presented as mean ± standard deviation
Delivery of Tat-CCT2 Protein into hippocampus
The intracellular delivery of Tat-CCT2 and CCT2 proteins into the hippocampus was visualized by immunohistochemical staining for His-tags 2 h after protein treatment. In the control and CCT2-treated groups, no His-tag immunoreactive structures were observed in the hippocampal CA1 region. In the Tat-CCT2-treated group, His-tag immunoreactive structures were abundantly found in various brain regions including hippocampus, cerebral cortex, and thalamus. Especially, His-tag immunoreactive structures were abundantly detected in the hippocampal CA1 region, and His-tag immunoreactivity was significantly increased to 612.1% ± 173.9% of that of the control group (Fig. 3A).
Effects of Tat-CCT2 Against Hippocampal Damage Induced by Transient Forebrain Ischemia in Gerbils
The neuroprotective effects of CCT2 and Tat-CCT2 were evaluated by measuring spontaneous motor activity 1 d after ischemia in gerbils because ischemia-induced hyperlocomotive activity was induced before morphological evidence of neuronal damage (Gerhardt and Boast, 1988). In the vehicle-treated group, animals exhibited hyperlocomotor activity and traveled a longer distance (588.5 m ± 69.8 m) compared to that of the control group (215.9 m ± 36.4 m). The traveled distance in the 0.5 mg/kg CCT2-treated group was comparable to that in the vehicle-treated group. In the 0.2 and 0.5 mg/kg CCT2-treated groups, animals exhibited reduced motor activity and significant decreases in traveled distance (402.5 m ± 113.8 and 289.0 m ± 58.9 m, respectively) compared to those of the vehicle- or CCT2-treated groups (Fig. 3B).
Morphological evidence of neuronal damage in the hippocampus was assessed by immunohistochemical staining for NeuN 4 d after ischemia. In the control group, NeuN-immunoreactive nuclei were abundant in all hippocampal subregions, including CA1. In the vehicle-treated group, NeuN-immunoreactive nuclei were similarly observed in the hippocampus, except in the CA1 region, which demonstrated a significant reduction in NeuN-immunoreactive nuclei to 5.7% ± 1.9% of that in the control group. In the group treated with 0.5 mg/kg CCT2, the distribution pattern of NeuN-immunoreactive neurons was similar to that of the vehicle-treated group, and a higher number of NeuN-immunoreactive neurons (7.0% ± 2.1%) was detected compared to that in the control group. In the group treated with 0.2 mg/kg Tat-CCT2, more NeuN-immunoreactive neurons were observed in the lateral part of the CA1 region, although the medial part exhibited similar neuronal death as seen in vehicle- or CCT2-treated groups. In the group treated with 0.5 mg/kg CCT2, numerous NeuN-immunoreactive neurons were observed in the CA1 region, and a significantly higher number (58.9% ± 7.1% of the control group) of NeuN-immunoreactive neurons was detected compared to the vehicle- or CCT2-treated groups (Fig. 3C).

Delivery of Tat-CCT2 and its control protein into the hippocampus and its effects against transient forebrain ischemia induced by 5 min occlusion of common carotid arteries in gerbils. (A) Protein delivery is assessed by immunohistochemical staining for His-tag 2 h following Tat peptide (vehicle), CCT2, or Tat-CCT2 treatment. Low-magnified picture is demonstrated to show the His-tag immunoreactivity in brain regions including cerebral cortex, hippocampus, and thalamus. The densities of His-tag immunoreactive structures in the hippocampal CA1 region are quantified based on pixel numbers and pixel densities. The optical density is normalized to a percentile value compared to the vehicle group. (B) Spontaneous motor activity is recorded, and locomotor activity based on distance traveled is traced 1 d after ischemia in the sham-operated (control), vehicle-treated, 0.5 mg/kg CCT2-treated (CCT2), and 0.2 mg/kg Tat-CCT2-treated [Tat-CCT2 (0.2)] groups. (C) Mature neurons in the hippocampus are confirmed by NeuN immunohistochemical staining 4 d after ischemia to detect surviving neurons. Scale bar = 400 μm. The number of NeuN-immunostained neurons is calculated in the CA1 region, and the number is normalized to percentile value vs. control group. (A, B, and C) Data are analyzed using one-way analysis of variance followed by Tukey’s multiple comparison post hoc test [n = 5 in each group (A and C), n = 10 in each group (B); ap < 0.05, significantly different from the control group; bp < 0.05, significantly different from the vehicle group; cp < 0.05, significantly different from the CCT2 group; dp < 0.05, significantly different from the Tat-CCT2 (0.2) group]. Data are presented as mean ± standard deviation
Effects of Tat-CCT2 on Glial Activation Induced by Transient Forebrain Ischemia in Gerbils
The ischemia-induced activation of microglia and astrocytes, as well as the effects of Tat-CCT on glial activation by ischemia, were assessed by immunohistochemical staining for Iba-1 and GFAP, respectively, 4 d after ischemia. In the control group, Iba-1-immunoreactive microglia and GFAP-immunoreactive astrocytes exhibited thin and long processes with a small cytoplasm. In the vehicle-treated group, Iba-1-immunoreactive microglia exhibited retracted processes with round cytoplasm (phagocytic form), which was mainly observed in the stratum pyramidale of the CA1 region. In other regions (stratum oriens and radiatum), Iba-1-immunoreactive microglia exhibited a hypertrophied cytoplasm with thickened processes (activated form). In this group, the GFAP-immunoreactive astrocytes had a punctuated cytoplasm with thickened processes. Iba-1 and GFAP immunoreactivity in the CA1 region significantly increased to 419.6% ± 77.0% and 320.0% ± 40.4%, respectively, compared with those of the respective control group. In the group treated with 0.5 mg/kg CCT2, the distribution pattern and morphology of Iba-1-immunoreactive microglia and GFAP-immunoreactive astrocytes were similar to those in the vehicle-treated group. In addition, the Iba-1 and GFAP immunoreactivity levels were similar to those in the vehicle-treated group. In the 0.2 mg/kg Tat-CCT2-treated group, fewer Iba-1-immunoreactive microglia were found in the stratum pyramidale and some GFAP-immunoreactive astrocytes showed a resting form. In this group, Iba-1 and GFAP immunoreactivity significantly decreased to 300.0% ± 61.2% and 234.0% ± 30.5% of that of the control group, respectively, compared to those of the vehicle- or CCT2-treated groups. In the group treated with 0.5 mg/kg Tat-CCT2, a few Iba-1 immunoreactive microglia were observed in the stratum pyramidale, and only some GFAP-immunoreactive astrocytes had punctuated the cytoplasm with thickened processes. In this group, Iba-1 and GFAP immunoreactivity further decreased to 244.6% ± 41.9% and 169.5% ± 29.6% of the control group, respectively (Fig. 4A).
Effects of Tat-CCT2 on lipid peroxidation after transient forebrain ischemia in gerbils.
The most common byproducts of lipid peroxidation, 4-HNE and 8-iso-PGF2α, were measured in the hippocampus 6 h after ischemia. In the vehicle-treated group, 4-HNE and 8-iso-PGF2α levels were significantly increased to 283.8% ± 32.9% and 313.6% ± 41.4% of those in the control group, respectively. In the 0.5 mg/kg CCT2- and 0.2 mg/kg Tat-CCT2-treated groups, similar levels of 4-HNE and 8-iso-PGF2α were observed compared to those in the vehicle-treated group, respectively. In the group treated with 0.5 mg/kg Tat-CCT2, 4-HNE and 8-iso-PGF2α levels were significantly decreased to 185.7% ± 30.0% and 180.6% ± 30.1% of those of the control group, respectively, compared to vehicle- or 0.5 mg/kg CCT2-treated groups (Fig. 4B).
Effects of Tat-CCT2 on autophagy after transient forebrain ischemia in gerbils.
The autophagic/lysosomal pathway in the hippocampus was assessed by western blotting performed 12 h after ischemia. LC3B and Beclin-1 protein levels in the vehicle-treated group significantly increased to 140.0% ± 10.9% and 187.6% ± 21.0%, respectively, compared with those of the control group. No significant differences were observed in LC3B and Beclin-1 protein levels between the vehicle- and CCT2-treated groups. In the groups treated with 0.2 and 0.5 mg/kg Tat-CCT2, LC3B levels were further increased in a concentration-dependent manner to 179.3% ± 15.1% and 192.0% ± 24.1% of the control group, respectively, and were significantly higher in both groups. In contrast, Beclin-1 levels were significantly decreased compared with those of the vehicle-treated group and were 148.9% ± 9.3% and 124.0% ± 15.7% of the control group, respectively. Levels of p62 protein were significantly lower (46.1% ± 8.8% and 48.7% ± 8.8% of the respective control groups) in the vehicle- and CCT2-treated groups than those in the control group. However, in the 0.2 and 0.5 mg/kg Tat-CCT2-treated groups, the reduction in p62 was ameliorated to 66.8% ± 7.0% and 81.1% ± 11.8% of the control group, respectively (Fig. 4C).

Mechanisms of Tat-CCT2 and its control protein against ischemic damage in gerbils. (A) Astrocytes and microglia are visualized by GFAP and Iba-1 immunohistochemical staining in the hippocampal CA1 region 4 d after ischemia, respectively. Scale bar = 50 μm. The densities of GFAP and Iba-1 immunoreactive structures are quantified based on pixel numbers and pixel densities. The optical density is normalized to a percentile value compared with that of the control group. (B) The protein expression related to lipid peroxidation, including 4-HNE and 8-iso-PGF2α, are measured using ELISA in the hippocampus 6 h after ischemia, and the protein levels are assessed using the standard curve. (C) The protein expression related to autophagy, such as LC3B, Beclin-1, and p62 are assessed using western blotting in the hippocampus 1 d after ischemia. The band densities of LC3B, Beclin-1, p62, and β-actin are quantified using Gel Doc. The optical density is normalized to percentile value compared with that of control group. (A, B, and C) Data are analyzed using one-way analysis of variance followed by Tukey’s multiple comparison post hoc test [n = 5 in each group; ap < 0.05, significantly different from the control group; bp < 0.05, significantly different from the vehicle group; cp < 0.05, significantly different from the CCT2 group; dp < 0.05, significantly different from the Tat-CCT2 (0.2) group]. Data are presented as mean ± standard deviation
Discussion
CCTs are among the most abundant chaperones that deal with aggregation-prone proteins [16, 17] and facilitate autophagic flux by promoting the incorporation of autophagosomes and clearance of aggregated proteins after conformational changes [19]. In addition, CCT2 levels are negatively correlated with aging and neurodegenerative diseases [29]. CCT2 is upregulated in the rat brain 3 or 7 d after exposure to normobaric hyperoxia for 3 h [30]. In the present study, we investigated the effects of CCT2 on oxidative stress in HT22 cells and ischemic damage in the gerbil hippocampus to elucidate its role of CCT2 in oxidative or ischemic damage. To overcome the limitations of protein delivery in the intracellular space and in crossing the blood-brain barrier, we synthesized a Tat-CCT2 fusion protein, which has cell-penetrating signals derived from the human immunodeficiency virus. We confirmed the delivery of Tat-CCT2 protein in a concentration- and time-dependent manner into HT22 cells. In addition, the transduced Tat-CCT2 proteins exhibited a time-dependent manner degradation within the cell, and the delivered protein was visualized by immunohistochemical staining for His-tag because we inserted the His-tag pET15b vector. These results are consistent with those of our previous studies demonstrating that proteins fused with the Tat peptide can be delivered into the intracellular space of HT22 cells [22, 23]. We examined the neuroprotective effects of Tat-CCT2 against oxidative stress induced by 200 µM H2O2 in HT22 cells [23, 31, 32]. We observed that treatment with 8 µM Tat-CCT2 ameliorated H2O2-induced neuronal death, DNA fragmentation, and ROS formation in HT22 cells, based on the WST-1 assay, TUNEL assay, and DCF-DA staining. These results suggested that Tat-CCT2 efficiently alleviated neuronal damage by reducing ROS production or facilitating ROS removal in HT22 cells. This result is supported by previous studies showing that the presence of TRiC or CCT2 reduces protein aggregation related to various neurodegenerative diseases [19, 33]. In addition, the modulation of CCT2 γ subunit demonstrates protective effects against miltefosine-induced oxidative damage in Leishmania donovani [34].
Furthermore, we examined the effects of CCT2 on ischemic damage in the gerbil hippocampi. Transient forebrain ischemia significantly increases hyperlocomotor activity 1 d after ischemia, characterized by animals spending more time navigating their path due to functional impairment of memory [27, 28]. Neuronal damage was confirmed by immunohistochemical staining for NeuN 4 d after ischemia, and selective neuronal death was detected in the hippocampal CA1 region [23, 32, 35, 36]. In addition, astrocytes and microglia were visualized by immunohistochemical staining for GFAP and Iba-1, respectively, because ischemia-induced neuronal death causes morphological changes in the hippocampal CA1 region [37,38,39]. Treatment with Tat-CCT2 significantly ameliorated ischemia-induced hyperlocomotor activity 1 d after ischemia, as well as neuronal death and glial activation in the hippocampal CA1 region 4 d after ischemia, suggesting the neuroprotective potential of Tat-CCT2 against ischemic damage in gerbils. This result was supported by a previous study showing that UC-MSC‐EVs treatment mitigates liver damage 6 h after ischemia by reducing proinflammatory cytokines, but no positive effects have been observed with UC‐MSC‐EVsshCCT2 treatment [20]. To elucidate the neuroprotective mechanisms of Tat-CCT2 against ischemic damage in gerbils, we examined markers for lipid peroxidation, including 4-HNE and 8-iso-PGF2α, as well as for autophagy, including LC3B, p62, and Beclin-1, because autophagy could be a major contributor to cell death and recovery after ischemia in gerbils [40, 41]. In addition, ROS production and transient forebrain ischemia activates hypoxia-inducible factor-1α, nuclear factor erythroid 2–related factor 2, p53 and forkhead box O-3 (FoxO3) [42]. Consistent with previous studies [23, 32], 4-HNE and 8-iso-PGF2α levels were significantly increased in the hippocampus after ischemia. In the present study, treatment with Tat-CCT2 significantly ameliorated ischemia-induced increases in lipid peroxidation in the hippocampus. This result is consistent with our in vitro study, which demonstrated that Tat-CCT2 has ability to reduce ROS generation in HT22 cells.
In addition, LC3B and Beclin-1 levels were significantly increased in the hippocampus 1 d after ischemia, and treatment with Tat-CCT2 further increased LC3B levels, suggesting the enhancement of autophagic activity [43]. In contrast, Beclin-1 protein levels were ameliorated after Tat-CCT2 treatment, indicating the inhibition of autophagic initiation by Tat-CCT2 treatment. Moreover, we observed that p62 was significantly decreased in the hippocampus 1 d after ischemia, and this reduction was significantly alleviated by Tat-CCT2 treatment. This study suggested that Tat-CCT2 treatment increases the autophagic substrate and activity in the hippocampus but inhibits autophagic initiation, thereby, stimulating the removal of damaged organelles and protein aggregates induced by ischemia. This result is consistent with the findings of a previous study showing that CCT prevents and clears toxic protein aggregates to reduce neuronal damage [19]. In addition, the enhancement of autophagy by α7nAChR activation protects neurons from oxygen and glucose deprivation, and treatment with 3-methyladenine (3-MA), an autophagy inhibitor, attenuates the neuroprotective effects of α7nAChR in primary cortical neurons [44]. In addition, 3-MA treatment facilitates cell death, ROS formation, and anti-apoptotic protein expression in HT22 cells after exposure to H2O2 [31]. However, several lines of evidence suggested that autophagy serves as a double-edged sword in ischemic brain disease [12, 45]. The inhibition of autophagy by intracerebroventricular injections of 3-MA following occlusion of the middle cerebral artery reduces brain damage [46]. More recently, intraperitoneal injection of 3-MA was shown to significantly decreases hippocampal injury after hypoxic-ischemic damage in neonatal rats [47]. However, it remains to be determined which cell types (neurons, astrocytes, or microglia) are mainly involved in autophagy during ischemic damage.
Many preclinical studies have been conducted to identify neuroprotective agents against ischemic damage, but clinical trials have found weak or no evidence for neuroprotection, with the exception of tissue plasminogen activator or hypothermia. This discrepancy was due to the failure to adequately consider age, vascular condition, diet, blood-brain barrier integrity, and other factors [48]. In the present study, we synthesized Tat-CCT2 protein to deliver it to the intracellular space and reduce H2O2-induced oxidative damage in HT22 cells, as well as ischemic damage in gerbils.
In conclusion, Tat-CCT2 reduced the neuronal damage caused by oxidative stress in HT22 cells and ischemic death in gerbils by reducing oxidative stress and facilitating autophagy during brain ischemia.
Data Availability
The datasets and supporting materials generated and/or analyzed during the current study are available from the corresponding author upon reasonable request.
References
Waller A, Fakes K, Carey M et al (2023) Quality of life and mood disorders of mild to moderate stroke survivors in the early post-hospital discharge phase: a cross-sectional survey study. BMC Psychol 11:32
Wongsa D, Soivong P, Chaiard J, Davidson PM (2023) Patterns of health-related quality of life among stroke survivors: a longitudinal study. West J Nurs Res 45:511–519
Tsao CW, Aday AW, Almarzooq ZI et al (2023) Heart disease and stroke statistics-2023 update: a report from the american heart association. Circulation 147:e93–e621
Kuchinka J, Nowak E, Szczurkowski A, Kuder T (2008) Arteries supplying the base of the brain in the mongolian gerbil (Meriones unguiculatus). Pol J Vet Sci 11:295–299
Kim YY, Chao JR, Kim C et al (2019) Comparing the superficial vasculature of the central nervous system in six laboratory animals: a hypothesis about the role of the “circle of Willis. Anat Rec (Hoboken) 302:2049–2061
Lin CS, Polsky K, Nadler JV, Crain BJ (1990) Selective neocortical and thalamic cell death in the gerbil after transient ischemia. Neuroscience 35:289–299
Ahn JH, Song M, Kim H et al (2019) Differential regional infarction, neuronal loss and gliosis in the gerbil cerebral hemisphere following 30 min of unilateral common carotid artery occlusion. Metab Brain Dis 34:223–233
Krupska O, Kowalczyk T, Beręsewicz-Haller M et al (2021) Hippocampal sector-specific metabolic profiles reflect endogenous strategy for ischemia-reperfusion insult resistance. Mol Neurobiol 58:1621–1633
Li M, Tang H, Li Z, Tang W (2022) Emerging treatment strategies for cerebral ischemia-reperfusion injury. Neuroscience 507:112–124
Puleo MG, Miceli S, Di Chiara T et al (2022) Molecular mechanisms of inflammasome in ischemic stroke pathogenesis. Pharmaceuticals (Basel) 15:1168
Zhang Q, Jia M, Wang Y, Wang Q, Wu J (2022) Cell death mechanisms in cerebral ischemia-reperfusion injury. Neurochem Res 47:3525–3542
Lu X, Zhang J, Ding Y, Wu J, Chen G (2022) Novel therapeutic strategies for ischemic stroke: Recent insights into autophagy. Oxid Med Cell Longev 2022:3450207
Zeng X, Zhang YD, Ma RY et al (2022) Activated Drp1 regulates p62-mediated autophagic flux and aggravates inflammation in cerebral ischemia-reperfusion via the ROS-RIP1/RIP3-exosome axis. Mil Med Res 9:25
Hipp MS, Kasturi P, Hartl FU (2019) The proteostasis network and its decline in ageing. Nat Rev Mol Cell Biol 20:421–435
Kim YE, Hipp MS, Bracher A, Hayer-Hartl M, Hartl FU (2013) Molecular chaperone functions in protein folding and proteostasis. Annu Rev Biochem 82:323–355
Gestaut D, Limatola A, Joachimiak L, Frydman J (2019) The ATP-powered gymnastics of TRiC/CCT: an asymmetric protein folding machine with a symmetric origin story. Curr Opin Struct Biol 55:50–58
Jin M, Liu C, Han W, Cong Y (2019) TRiC/CCT chaperonin: structure and function. Subcell Biochem 93:625–654
Kampinga HH, Hageman J, Vos MJ et al (2009) Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones 14:105–111
Ma X, Lu C, Chen Y et al (2022) CCT2 is an aggrephagy receptor for clearance of solid protein aggregates. Cell 185:1325–1345e22
Zheng J, Lu T, Zhou C et al (2020) Extracellular vesicles derived from human umbilical cord mesenchymal stem cells protect liver ischemia/reperfusion injury by reducing CD154 expression on CD4 + T cells via CCT2. Adv Sci (Weinh) 7:1903746
Liu H, Rose ME, Ma X, Culver S, Dixon CE, Graham SH (2017) In vivo transduction of neurons with TAT-UCH-L1 protects brain against controlled cortical impact injury. PLoS ONE 12:e0178049
Feng B, Jia S, Li L et al (2023) TAT-LBD-Ngn2-improved cognitive functions after global cerebral ischemia by enhancing neurogenesis. Brain Behav 13:e2847
Kwon HJ, Hahn KR, Kang MS et al (2023) Tat-malate dehydrogenase fusion protein protects neurons from oxidative and ischemic damage by reduction of reactive oxygen species and modulation of glutathione redox system. Sci Rep 13:5653
Kwon HJ, Kim DS, Kim W et al (2020) Tat-cannabinoid receptor interacting protein reduces ischemia-induced neuronal damage and its possible relationship with 14-3-3η. Cells 9:1827
Percie du Sert N, Hurst V, Ahluwalia A et al (2020) The ARRIVE guidelines 2.0: updated guidelines for reporting animal research. PLoS Biol 18:e3000410
Radtke-Schuller S, Schuller G, Angenstein F, Grosser OS, Goldschmidt J, Budinger E (2016) Brain atlas of the mongolian gerbil (Meriones unguiculatus) in CT/MRI-aided stereotaxic coordinates. Brain Struct Funct 221(Suppl 1):1–272
Mileson BE, Schwartz RD (1991) The use of locomotor activity as a behavioral screen for neuronal damage following transient forebrain ischemia in gerbils. Neurosci Lett 128:71–76
Katsuta K, Umemura K, Ueyama N, Matsuoka N (2003) Pharmacological evidence for a correlation between hippocampal CA1 cell damage and hyperlocomotion following global cerebral ischemia in gerbils. Eur J Pharmacol 467:103–109
Brehme M, Voisine C, Rolland T et al (2014) A chaperome subnetwork safeguards proteostasis in aging and neurodegenerative disease. Cell Rep 9:1135–1150
Hinkelbein J, Feldmann RE Jr, Kalenka A (2010) Time-dependent alterations of cerebral proteins following short-term normobaric hyperoxia. Mol Cell Biochem 339:9–21
Deng A, Ma L, Zhou X, Wang X, Wang S, Chen X (2021) FoxO3 transcription factor promotes autophagy after oxidative stress injury in HT22 cells. Can J Physiol Pharmacol 99:627–634
Jung HY, Kwon HJ, Kim W et al (2022) The neuroprotective effects of phosphoglycerate mutase 5 are mediated by decreasing oxidative stress in HT22 hippocampal cells and gerbil hippocampus. Neurochem Int 157:105346
Shahmoradian SH, Galaz-Montoya JG, Schmid MF et al (2013) TRiC’s tricks inhibit huntingtin aggregation. Elife 2:e00710
Yadav S, Ali V, Singh Y, Kanojia S, Goyal N (2020) Leishmania donovani chaperonin TCP1γ subunit protects miltefosine induced oxidative damage. Int J Biol Macromol 165:2607–2620
Asai A, Tanahashi N, Qiu JH et al (2002) Selective proteasomal dysfunction in the hippocampal CA1 region after transient forebrain ischemia. J Cereb Blood Flow Metab 22:705–710
Lee TK, Kim H, Song M et al (2019) Time-course pattern of neuronal loss and gliosis in gerbil hippocampi following mild, severe, or lethal transient global cerebral ischemia. Neural Regen Res 14:1394–1403
Petito CK, Chung M, Halaby IA, Cooper AJ (1992) Influence of the neuronal environment on the pattern of reactive astrocytosis following cerebral ischemia. Prog Brain Res 94:381–387
Yan BC, Park JH, Ahn JH et al (2012) Comparison of glial activation in the hippocampal CA1 region between the young and adult gerbils after transient cerebral ischemia. Cell Mol Neurobiol 32:1127–1138
Hwang IK, Park JH, Lee TK et al (2017) CD74-immunoreactive activated M1 microglia are shown late in the gerbil hippocampal CA1 region following transient cerebral ischemia. Mol Med Rep 15:4148–4154
Kim W, Kwon HJ, Jung HY et al (2020) P27 protects neurons from ischemic damage by suppressing oxidative stress and increasing autophagy in the hippocampus. Int J Mol Sci 21:9496
Kawalec M, Wojtyniak P, Bielska E et al (2022) Mitochondrial dynamics, elimination and biogenesis during post-ischemic recovery in ischemia-resistant and ischemia-vulnerable gerbil hippocampal regions. Biochim Biophys Acta Mol Basis Dis 1869:166633
Redza-Dutordoir M, Averill-Bates DA (2021) Interactions between reactive oxygen species and autophagy: special issue: death mechanisms in cellular homeostasis. Biochim Biophys Acta Mol Cell Res 1868:119041
Cavaliere F, Fornarelli A, Bertan F et al (2019) The tricyclic antidepressant clomipramine inhibits neuronal autophagic flux. Sci Rep 9:4881
Xu ZQ, Zhang JJ, Kong N et al (2021) Autophagy is involved in neuroprotective effect of alpha7 nicotinic acetylcholine receptor on ischemic stroke. Front Pharmacol 12:676589
Chen W, Sun Y, Liu K, Sun X (2014) Autophagy: a double-edged sword for neuronal survival after cerebral ischemia. Neural Regen Res 9:1210–1216
Puyal J, Vaslin A, Mottier V, Clarke PG (2009) Postischemic treatment of neonatal cerebral ischemia should target autophagy. Ann Neurol 66:378–389
Xu LX, Tang XJ, Yang YY et al (2017) Neuroprotective effects of autophagy inhibition on hippocampal glutamate receptor subunits after hypoxia-ischemia-induced brain damage in newborn rats. Neural Regen Res 12:417–424
Lourbopoulos A, Mourouzis I, Xinaris C et al (2021) Translational block in stroke: a constructive and “out-of-the-box” reappraisal. Front Neurosci 15:652403
Acknowledgements
The authors are grateful to Ms. Hyun Sook Kim for providing technical support for this study. This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (NRF-2021R1F1A1049744 to Seung Myung Moon). This work was also supported by the Research Institute for Veterinary Science at Seoul National University.
Author information
Authors and Affiliations
Contributions
H. J. K., H. J. J., G. M. C., I. K. H., D. W. K., and S. M. M. conceived the study. H. J. K., D. W. K., and S. M. M. designed the study and prepared the manuscript. H. J. J., G. M. C., and I. K. H. conducted the animal experiments. H.J.K. and D.W.K. conducted biochemical experiments. H.J.K. and H.J.J. analyzed the data, and G.M.C. participated in discussions of the study. All authors read and approved the manuscript, all data were generated in-house, and no paper mill was used.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Kwon, H.J., Jeon, H.J., Choi, GM. et al. Tat-CCT2 Protects the Neurons from Ischemic Damage by Reducing Oxidative Stress and Activating Autophagic Removal of Damaged Protein in the Gerbil Hippocampus. Neurochem Res 48, 3585–3596 (2023). https://doi.org/10.1007/s11064-023-03995-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-023-03995-9