
Abstract
Parkinson's disease (PD) is a common age-related neurodegenerative disorder whose pathogenesis is not completely understood. Mitochondrial dysfunction and increased oxidative stress have been considered as major causes and central events responsible for the progressive degeneration of dopaminergic (DA) neurons in PD. Therefore, investigating mitochondrial disorders plays a role in understanding the pathogenesis of PD and can be an important therapeutic target for this disease. This study discusses the effect of environmental, genetic and biological factors on mitochondrial dysfunction and also focuses on the mitochondrial molecular mechanisms underlying neurodegeneration, and its possible therapeutic targets in PD, including reactive oxygen species generation, calcium overload, inflammasome activation, apoptosis, mitophagy, mitochondrial biogenesis, and mitochondrial dynamics. Other potential therapeutic strategies such as mitochondrial transfer/transplantation, targeting microRNAs, using stem cells, photobiomodulation, diet, and exercise were also discussed in this review, which may provide valuable insights into clinical aspects. A better understanding of the roles of mitochondria in the pathophysiology of PD may provide a rationale for designing novel therapeutic interventions in our fight against PD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Parkinson’s disease (PD) is the second most prevalent, age-related neurodegenerative disease, affecting 1 to 3% of individuals older than 65 years [1]. PD is pathologically characterized by the extensive degeneration of dopaminergic (DA) neurons in the substantia nigra pars compacta (SNpc) and accumulation of alpha-synuclein (α-syn) amyloid, termed Lewy bodies (LBs), in surviving neurons [2]. The cardinal motor signs of PD include rest tremor, bradykinesia, rigidity, and postural instability. In addition, a lot of non-motor features are also well recognized in PD patients, including anosmia, depression, hyposmia, dementia, and pain [3].
Although many studies have been performed on the pathogenesis of PD, the underlying causes of the selective loss of DA neurons or abnormal aggregation of α-syn have not yet been clarified [4]. Furthermore, available treatments for PD have symptomatic effects; there is no cure or disease-modifying therapy available to slow the progression of PD [5]. Thus, the biggest current challenge is to achieve effective treatments based on an understanding of the underlying PD mechanisms [6].
More recent studies have shown that mitochondrial dysfunction has a prominent role in the development and devastating consequences of central nervous system (CNS) diseases, including ischemic stroke, Alzheimer’s disease (AD) and PD [7,8,9]. Mitochondria are dynamic organelles that are essential for various cellular functions, such as cell proliferation and differentiation [10, 11], apoptosis [12], calcium homeostasis [13], energy production [14], and redox signaling [15]. Mitochondria are the main site of reactive oxygen species (ROS) production in CNS injuries [16]. Defects in the function of enzymes involved in mitochondrial electron transport chain (ETC) lead to the excessive production of ROS, which can induce oxidative damage in the mitochondria DNA (mtDNA), proteins, and lipids [17], and finally results in the mitochondrial dysfunction that implicates PD pathogenesis. Therefore, prevention of mitochondrial dysfunction is an effective strategy for future PD therapies [18]. In this review, we focus on the current knowledge regarding the role of mitochondrial dysfunction in the pathogenesis of PD, followed by discussing novel therapeutic strategies targeting mitochondrial dysfunction.
Pathophysiology of Parkinson’s Disease
PD is a multifactorial disease caused by an interplay between various environmental and genetic factors, and manifested with a wide range of clinical symptoms [19]. However, aging has been identified as the biggest risk factor for initiation and progression of PD [20]. A study determined the combined effects of natural aging, genetic disorder, and environmental toxicity in pathogenesis of PD [21]. To date, a number of different genes and environmental factors have been recognized as risk factors for PD. Scientists have identified more than 20 genes that can cause PD when mutated or over-expressed, including Single-nucleotide polymorphism rs356219 in the α-syn (SNCA), Leucine-rich repeat kinase 2 (LRRK2), parkinsonism associated deglycase (PARK7), PTEN-induced kinase 1 (PINK1), parkin RBR E3 ubiquitin protein ligase (PRKN), deglycase DJ1, and glucocerebrosidase gene 1 (GBA1) [22]. Furthermore, numerous systematic reviews and meta-analyses have identified that environmental factors, such as pesticide exposure, traumatic brain injury, anxiety, and depression are associated with onset and severity of PD [1, 23, 24].
These risk factors are associated with a number of cellular and molecular mechanisms underlying PD initiation and progression, including oxidative stress, mitochondrial dysfunction, apoptosis, abnormal protein aggregation, excitotoxicity, and neuroinflammation, which ultimately leads to accumulation of misfolded α-syn and DA neuronal death; However, the main reason is still unknown [25, 26].
A better understanding of the molecular mechanisms implicated in PD pathogenesis may contribute to the development of prospective therapies for this disease [27, 28]. Among various cellular factors, mitochondria can be considered as a main factor in PD pathogenesis due to their prominent role in the main cellular functions [29, 30]. Therefore, mitochondrial dysfunction triggers a sequence of events through energy deficiency, oxidative stress, inflammatory and apoptotic cascades [9], which eventually leads to neuronal damage in PD [31, 32].
Mitochondrial Dysfunction in Parkinson’s Disease
Mitochondria are double-membrane–bound organelles located in the eukaryotic cells and have their own genomic DNA, which is known as mtDNA. These organelles are often known as the “powerhouses” or “energy factories” of the cell due to its pivotal role in the generation of adenosine triphosphate (ATP) through oxidative phosphorylation process coupled to the ETC [33]. Besides their prominent role in cellular energy production, mitochondria also play a critical role in various metabolic processes, including production of amino acids, lipids and second messengers, regulation of intracellular calcium, autophagy, and many other functions [34, 35].
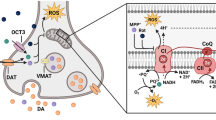
It therefore comes as no surprise that dysfunction of these vital organelles has been implicated in the pathophysiology of various diseases, including neurodegenerative disorders and ischemic stroke [9, 16, 36]. Overwhelming evidence indicates that disruption of mitochondrial function and integrity is a major causative factor in the progression of PD [37, 38]; because many lines of evidence suggest that risk factors involved in the pathogenesis of PD exert their effects by triggering mitochondrial dysfunction [39]. In a rotenone-induced PD mice model, a significant decrease in mitochondrial complex I activity was observed in the striatum [21]. Mitochondrial dysfunction can be caused by multiple PD-related risk factors including environmental, genetic, and biological factors [40]. Here, the effect of these factors on the pathogenesis and progression of PD through mitochondrial dysfunction are discussed (Fig. 1).

Schematic representation of various pathogenic mechanisms associated with mitochondrial dysfunction in Parkinson’s disease (PD). Mitochondrial dysfunction was increased after several environmental, genetic, and biological factors of PD. Following PD, α-syn interact with the ETC increased the production of mitochondrial ROS, promoting mtDNA damage. Dysfunction in ETC decreased complex I activity, decreased ATP production, and decreased respiratory capacity. It was also induced MPTP opening, calcium diffusion, cytochrome C release, and mitochondrial swelling, which increased inflammatory responses by releasing succinate and mitochondrial DAMPs, and induced apoptosis. In the PD, mitochondrial fragmentation was increased by increasing Drp1-mediated fission, and interaction with fusion proteins, such as MFN1/2 and OPA1. Mitochondrial dysfunction triggers NLRP3 inflammasome activation, leading to the caspase-1 activation, and maturation of IL-1β and IL-18. Mitochondrial dysfunction also causes abnormal mitophagy, which affects ROS formation. After mitochondrial dysfunction and absence of Parkin, PARIS was bound to the PGC1α gene promoter and suppresses its expression, which impaired mitochondrial biogenesis. ETC Electron transport chain; mtDNA mitochondrial DNA; DAMPs mitochondrial damage-associated molecular patterns; AP-1 activator protein-1; NLRP NOD (nucleotide-binding oligomerization domain)-like receptor (NLR) Pyrin domain containing 3; ASC apoptosis-associated speck-like protein containing a caspase recruitment domain; ASIC acid-sensing ion channel; ATP adenosine triphosphate; ROS reactive oxygen species; RNS reactive nitrogen species; Δψm mitochondrial membrane potential; MPTP mitochondrial permeability transition pore; Drp dynamin-related proteins; Mfn1/2 mitofusins; Opa1 optic atrophy 1; PINK1 PTEN-induced putative kinase 1; LC3 microtubule-associated protein 1A/1B light chain 3; IL interleukin; Pre precursor; PGC-1α PARIS Parkin-interacting substrate; peroxisome proliferator-activated receptor gamma-co-activator 1-alpha
Environmental Factors and Mitochondrial Dysfunction in PD
Numerous meta-analyses and epidemiological studies have examined the role of environmental factors in the pathogenesis of PD and demonstrated that long term exposure to environmental toxicants such as pesticides, air pollutants, and heavy metals represents potential risk factors for PD [41,42,43].
The impact of environmental toxins in PD has been demonstrated since the discovery of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) in the early 1980s [44], which induces selective degeneration of DA neurons by inhibiting mitochondrial complex I, leading to PD symptoms, such as rigidity, tremor, bradykinesia, and cognitive deficits [45]. MPTP is a protoxicant and lipid-soluble compound that easily crosses the Blood–Brain Barrier (BBB) [46]. Following systemic administration, MPTP crosses the BBB and is metabolized to its toxic metabolite 1-methyl-4-phenylpyridinium ion (MPP+), by astroglial monoamine oxidase B (MAO-B) [47]. MPP+ is specifically transported into the DA neurons through the plasma membrane dopamine transporter (DAT), and then accumulates in mitochondria [48]. Within the mitochondria, accumulated MPP+ inhibits complex I of the ETC, causing DA neuronal death by reducing ATP synthesis and increasing ROS generation [49].
Besides MPTP, several other environmental toxins like rotenone, fenazaquin, trichloroethylene, paraquat, tebunfenpyrad, and fenpyroximate were identified to induce lossing of the nigral dopaminergic neurons in vivo models, and implicates in mitochondrial dysfunction in PD pathogenesis [50,51,52]. Like MPTP, these neurotoxins also inhibit mitochondrial complex I [53], triggering mitochondrial dysfunction by inducing ROS production, impairing ATP synthesis, increasing membrane permeability, and decreasing mitochondrial motility, which ultimately leads to neuronal damage in SN [54, 55]. Inhibition of complex I to a small degree has been shown to significantly increase ROS production [56], which in turn inhibits complex I [57], creating a vicious cycle leading to mitochondrial damage and neuronal death [16]. Altogether, these data suggest that environmental exposures contribute to the pathogenesis of sporadic PD, and mitochondrial dysfunction seems to play a prominent role in these devastating effects of environmental toxicants.
Genetic Factors and Mitochondrial Dysfunction in PD
More than two decades ago, PD was considered to be a non-genetic sporadic origin disorder [58]. In 1997, the identification of mutations in SNCA gene (encoding αSyn) in families with autosomal dominant PD revealed the association between genetic factors and pathogenesis of PD [59]. Subsequent studies discovered mutations in several genes, such as PRKN [60], PINK1 [61], and LRRK2 [62], which were associated with autosomal dominant or autosomal recessive forms of PD [63]. Moreover, to investigate the influence of genetics on the occurrence of sporadic PD, Tanner et al. in a twin study indicated that monozygotic (identical) twins had higher concordance rates than dizygotic (non-identical) twins in those with early-onset disease (onset before age 50), suggesting a genetic basis for early-onset of PD [64]. About 5–10% of patients with PD follow a classical Mendelian type of inheritance and a family history of PD is reported in 15 to 20% of patients [65, 66]. To date, more than 15 genes and more than 40 independent loci have been identified as risk factors for monogenic forms and sporadic forms of PD, respectively [67]. It has been demonstrated that both mtDNA and nuclear DNA (nuDNA) genomes are involved in pathogenesis of PD [68, 69]. Since accurate function of different mitochondrial complexes is achieved by mtDNA and nuDNA encoded peptides, its mutations trigger mitochondrial dysfunction and oxidative stress [70].
mtDNA Defects and PD
Mitochondria are the only cellular organelles possessing their own genomes, which encode and produce protein components of respiratory chain complexes [71]. Human mtDNA is a double-stranded circular DNA molecule with 16,569 DNA base pairs that encodes 37 genes [72]. In addition, mtDNA has a higher mutation rate (about 77 times higher) than nuDNA [73], possibly due to less efficient repair of DNA damage, a high replication rate, and mitochondrial ROS [74]. Consequently, deleterious mutations in mtDNA have been shown to disrupt mitochondrial protein synthesis, leading to inadequate ATP production and increased free radicals [68]. A great deal of evidence supports the role of some mtDNA variations in the mitochondrial dysfunction and pathogenesis of PD [75]. Although no specific genetic mutation in mtDNA has been identified as a hallmark of PD, a significant increase in mtDNA deletions has been observed in substantia nigra DA neurons of PD patients [76], which is probably related to dysfunction of the mitochondrial respiratory chain (MRC) and increased oxidative stress [77]. It has been shown that in mice with deletion of mitochondrial transcription factor A (Tfam) gene, mitochondrial function in DA neurons of the substantia nigra was impaired, which induced PD-like symptoms [78]. Furthermore, an increased number and variety of mtDNA deletions/rearrangements in the substantia nigra of PD patients have been reported in comparison to aged controls and patients with other motor dysfunctions. Although these increased mtDNA deletions/rearrangements was not limited to the substantia nigra and were also demonstrated in other brain regions of PD patients. These findings suggest a relatively specific association between total mtDNA deletions/rearrangements and PD pathogenesis [77]. Studies have also shown that mutations in nuclear-encoded mitochondrial proteins involved in mtDNA synthesis, such as polymerase gamma (POLG), lead to the defects or deletion of mtDNA, which is ultimately associated with mitochondrial dysfunction and the pathogenesis of PD [79]. These results indicate the important role of primary and secondary mtDNA variations in PD [75].
Nuclear DNA Mutations and PD
A number of nuclear-encoded factors are known to be required for mtDNA replication, transcription, and translation. Variation in at least one of these factors induces mitochondrial dysfunction by impairment of mitochondrial biogenesis, dynamics, trafficking, and mitophagy [80]. Many nuclear genes related to mitochondrial function have been identified, and their potential role in PD has been investigated in previous studies [81].
For instance, PRKN (PARK2) gene, which makes the Parkin protein, is known to be the most frequent cause of autosomal recessive early-onset parkinsonism [82]. Parkin is a multifunctional E3 ubiquitin ligase, and plays an essential role in mitochondrial quality control (QC), mitochondrial homeostasis, and mitophagy [83]. Mitochondrial QC is a key process to maintain cellular homeostasis by coordinating various functions including mitochondrial dynamics, biogenesis, and mitophagy [84]. As a result, pathogenic mutations in PARK2 gene inactivate Parkin, resulting in mitochondrial QC dysregulation, increased ROS, and neuronal damage [85]. Until now, over 200 different mutations have been reported throughout the PRKN gene, including insertions, deletions, and point mutations [86, 87].
PINK1 is the second most frequent gene associated with autosomal recessive early-onset PD, after PARK2 [88]. PINK1 encodes a mitochondrial-localized serine/threonine kinase and, together with Parkin, is involved in mitochondrial QC, as well as in mitochondrial dynamic and size [83, 89]. Genetic studies indicate that mutations in the PINK1 disrupt complex I of the mitochondrial ETC and increased abnormalities in mitochondrial morphology, leading to accumulation of damaged mitochondria and increased sensitivity to apoptotic stress [90].
PARK7 is another genetic cause of autosomal recessive early-onset forms of PD that encodes the protein DJ-1 [91]. DJ-1 is a multifunctional protein with protective effects on the nervous system that prevents oxidative stress-induced mitochondrial impairment and neuronal death [92]. The antioxidant property of DJ-1 is due to its binding with p47phox and inactivation of NADPH oxidase (Nox), which defects in its encoding gene increase NOX4 expression and ROS generation [93]. DJ-1 mutations account for about 1–2% of autosomal recessive PD, but represent the highest prevalence of non-motor symptoms (∼57%) in PD patients [94].
LRRK2 gene (or PARK8), which encodes the protein Lrrk2, is the most frequent genetic cause of both familial and sporadic PD [95]. Although the precise mechanism of the LRRK2 in mitochondrial function is not fully understood, studies have shown that overexpression and mutations in this gene affect the mitochondrial dynamics and morphology, leading to a decline in mitochondrial membrane potential and ATP synthesis [96]. To date, more than 100 different missense/nonsense mutations within LRRK2 gene have been reported [97], only a small percentage of them are pathogenic and associated with PD [98].
The SNCA gene, encoding αSyn, is the first identified gene associated with late-onset autosomal dominant PD [99]. α-syn is a presynaptic neuronal protein and the principal component of Lewy bodies (LBs) and Lewy neurites (LNs) inclusions, which are pathological hallmarks of PD [100]. Overexpression of the SNCA gene causes the accumulation of α-syn aggregates in the mitochondria, which leads to various impairments in mitochondrial function, including complex I deficiency, reduction of membrane potential, movement deficiency, and disturbances in Ca2+ homeostasis [101, 102].
Epidemiological Factors and Mitochondrial Dysfunction in PD
The role of biological factors such as aging, sex, alcohol consumption, and smoking status in the initiation and progression of PD has been widely investigated in recent decades [103,104,105]. Aging is the greatest risk factor for the development and progression of neurodegenerative diseases including PD [106]. The effect of the aging process on the pathogenesis of PD appears to be partially mediated by mitochondrial dysfunction [107]. With aging, mitochondria become more vulnerable to environmental risk factors and the possibility of mitochondrial dysfunction is increased [108]. Studies have also indicated that the aging process is associated with a variety of mtDNA mutations, particularly deletions and point mutations [109, 110]. The results of these mutations are reduction of ATP production, increasing free radicals and oxidative stress, and ultimately disruption of mitochondrial structure and function [111].
Clinical and epidemiological studies have shown that the prevalence of PD is different between men and women. On average, the incidence of PD is significantly higher in men than in women, but the mortality rate is higher in women [112, 113]. The association between body weight and the risk of developing PD has also been investigated in meta-analysis and epidemiological studies, which show that obesity (defined as body mass index ≥ 23 kg/m2) is associated with a higher risk of PD [114, 115]. However, the effects of biological factors on mitochondrial function related to PD have not been investigated in detail and require further studies.
Consequences of Mitochondrial Dysfunction Following Parkinson’s Disease
Mitochondrial dysfunction was demonstrated to contribute to the pathogenesis of PD, which is caused by calcium accumulation, aberrant production of ROS, ATP depletion, increasing mitophagy, increasing mtDNA damage, defective mitochondrial biogenesis/ dynamics, triggering apoptosis, and finally cell death (Fig. 1).
ATP Depletion and ROS Production
Complex I (NADH-quinone oxidoreductase) acts as an input point for the electrons of the mitochondrial matrix in the ETC by catalyzing NADH electron transfer in the ETC subunits [116]. In PD patients and animal PD models, activities of complex I of the mitochondrial respiratory chain were significantly decreased [117]. Inhibition of mitochondrial complex I reduced ATP synthesis and increased ROS production, leading to respiratory failure. Partial inhibition of complex I in the nerve terminals is adequate for in situ mitochondria to produce more ROS [29, 57]. Indeed, inhibition of complex I increased ROS production, which in turn inhibits complex I, and this vicious cycle in DA neurons results in overproduction of oxidative stress over time and ATP depletion, ultimately leading to neuronal loss in the nigrostriatal pathway [57].
Further evidence of mitochondrial dysfunction associated with oxidative stress and damage to dopamine cells comes from findings that mutations in genes of proteins like αSyn, parkin, PINK, or DJ-1are related to familial forms of PD [118,119,120].
Accumulation of α-syn in dopamine neurons was demonstrated to decreased complex I activity and enhanced ROS production, and induction of neuronal loss [121]. α-syn inclusions were reported to increase mitochondrial oxidative stress in dendrites of DA neurons [122]. Oxidative stress increased accumulation, uptake, and oligomerization of extracellular α-syn in oligodendrocytes [123], as well as posttranslational modifications in α-syn that increment dopamine toxicity [124]. The activity of mitochondrial complex I is impaired in humans with parkin mutations [125]. In addition, PINK1 knockout (KO) in human and rodent dopamine neurons leads to ROS production and lessening of membrane potential [126].
With increased oxidative stress, DJ-1 expression is enhanced in reactive astrocytes [127], and excessive expression of DJ-1 is detected in reactive astrocytes in sporadic PD [128]. DJ-1 knockdown or KO in astrocytes impairs astrocyte-mediated neuroprotection against oxidative stress by deregulation of inflammatory responses and mitochondrial complex I damage (Fig. 2) [53, 129].
Calcium (Ca2+) Overload
Mitochondrial dysfunction can induce excitotoxicity by decreasing both cellular ATP and Ca2+ overload [130]. Inhibition of complex I can interfere with energy/ATP generation, which causes partial neuronal depolarization, and is attributed to a decrease in Na+/K+-ATPase activity [130]. Mitochondria can rapidly take up Ca2+ from the cytosol through a Ca2+ uniporter that depends on mitochondrial membrane potential (ΔΨm). The formation of ROS due to mitochondrial ETC failure impairs mitochondrial membranes and disrupts the mechanism of absorption and storage of Ca2+, thus increasing intracellular Ca2+ levels and intensifying the excitotoxicity [131]. Disturbed ΔΨm results in enhanced sensitivity to Ca2+ overload. This indicates that mitochondrial-driven excitotoxicity is an important factor in PD [131].
Mitophagy Impairment
Recently, many studies have revealed complex molecular signaling governing the identification and selective elimination of damaged mitochondria from the cell via autophagy, which is a mitochondrial quality control process, and called mitophagy [132].
Mitochondrial dysfunction and mitophagy reduction have been suggested as major components in determining pathological heterogeneity and selective susceptibility of certain brain regions in PD [133, 134]. Mutations of Parkin and PINK1, two important mitophagy proteins, lead to autosomal recessive early-onset PD (EOPD) [135]. F-box only protein 7 (FBXO7), as an adapter protein of the E3-ubiquitin ligase complex responsible for degradative and non-degradative protein ubiquitination, has multifunctional mitochondria actions and may affect mitophagy by interactions with PINK1 and Parkin [136,137,138]. Lastly, the sorting of the 13C (VPS13C) vacuolar protein, whose mutations have also been linked to autosomal recessive EOPD, is partially at the level of the outer mitochondrial membrane (OMM). Lack of VPS13C is related to decreased mitochondrial ΔΨm, metabolic effects, and changes in mitochondrial morphology. Furthermore, VPS13C has been suggested to function with Parkin and PINK1 to regulate mitophagy [139, 140]
Other evidence for mitochondrial dysfunction related to quality control is mutations in αSyn, GBA1, and LRRK2 which contribute to enhanced or impaired autophagy [141, 142].
Cells with the mutation in LRRK2 show enhanced numbers of fragmented mitochondria [143] and decreased ΔΨm [144]. Additionally, fibroblasts in patients with LRRK2 mutations have enhanced uptake of mitochondrial calcium [145]. Reduced ΔΨm and calcium dysregulation initiated clearance of injured mitochondria from the dendritic compartment in primary neurons, [145, 146]. Dynamin-related protein-1 (Drp1) phosphorylation by mutant LRRK2 also encourages fission and excessive autophagy. [147].
Mutations in DJ-1 (Park7) are a rare recessive form of familial PD [148]. Alteration of DJ-1 results in increased mitophagy levels through ROS and PINK1/Parkin [149, 150]. It is difficult to know if the accumulation of DJ-1 of mitochondria depends on PINK1/Parkin signaling [149] or functions in parallel [151] to regulate mitochondrial function during oxidative stress.
Sterol regulatory element-binding protein 1 (SREBP-1), a regulator of lipogenesis, is a risk locus for PD [152, 153]. A screen to detect genes that promote mitophagy in Drosophila S2 cells showed SREBP-1 to regulate mitophagy [154]. Cardiolipin is a mitochondrial-specific lipid residing in the inner mitochondrial membrane [155, 156] that is enhanced in pink1 mutant flies [153, 157]. Neurons expressing mutant α-syn show fragmented mitochondria as well as OMM-exposed cardiolipin, which initiates mitophagy [158]. α-syn is a major component of Lewy bodies that accumulate in mitochondrial membranes [119, 159]. Intriguingly, cardiolipin and dopamine facilitate the formation of Lewy body-like complexes containing α-syn and cytochrome c, which may serve to prevent the transmission of apoptotic signals from damaged neuritic mitochondria [160].
Dynamic Impairment
The unique energetic requirements of neurons need well-orchestrated maintenance and distribution of mitochondria. Therefore, mitochondrial dynamical properties, including fusion (fusing with one another), fission (actively divide), biogenesis, trafficking (actively transported through axons and dendrites), and degradation, are vital for neurons [161]. The mitochondrial dynamics malfunction is associated with PD [162]. The unique features of neurons that degenerate in PD can predispose those neural populations to be sensitive to changes in mitochondrial dynamics. Accumulating evidence of PD-associated toxins confirms that mitochondrial transport, fusion, and fission can be implicated in pathogenesis. [163]. Moreover, PD-related mutations affect genes that encode proteins that have specific functions in mitochondrial dynamics. Two proteins associated with familial forms of the disease, PINK1, and parkin, interact in a common route to regulate mitochondrial fusion/fission [164, 165]. Additionally, parkin can play an important role in maintenance of mitochondrial homeostasis by targeting mitophagy [166]. Furthermore, mutations in LRRK2, DJ-1, and α-syn vacuolar protein sorting-related protein 35 (VPS35) demonstrate the relevance of mitochondrial malfunction as the main cause of neural death in PD [167,168,169,170].
Biogenesis Impairment
Mitochondrial biogenesis is an essential event involving the coordination of transcription, translation, import of nuclear-encoded components, as well as the expression of mitochondrial genes by which new mitochondria are produced from existing mitochondria [171, 172]. Parkin induces the proteasomal degradation of Parkin-interacting substrate (PARIS), which acts as a transcriptional repressor of peroxisome proliferator-activated receptor gamma-co-activator 1-alpha (PGC-1α) [173]. PGC-1α stimulates mitochondrial biogenesis and it has been strongly involved in the pathogenesis of idiopathic PD [174]. In the absence of Parkin, PARIS is bound to the PGC1α gene promoter and suppresses its expression [175]. PARIS is highly expressed in the SNpc [175], and provisional Parkin KO in rodents results in progressive degeneration of DA neurons that are conditional on the expression of PARIS [176, 177]. Furthermore, PARIS overexpression leads to reduction in expression of PGC1α and selective death of DA neurons in the SNpc [175, 178]. Overall, the loss of Parkin function suppresses mitochondrial biogenesis through an accumulation of PARIS [179].
NLRP3 Inflammasome Activation
Neuroinflammation, which appeared in the early stages of PD, gradually causes disease progression. Mitochondrial dysfunction and the increased activity of NLRP3 inflammasome complex plays an important role in induction and maintaining neuroinflammation [180]. Additionally, inflammatory factors associated with mitochondria can induce formation of inflammasome complexes, which is responsible for activating, maturation, and releasing proinflammatory cytokines, including interleukin-1β/18 (IL-1β/18) [180, 181]. Following mitochondrial damage, NLRP3 binds to a protein associated with apoptosis that contains a CARD (PYCARD/ASC) and forms inflammasomes in the brain. Inflammasomes serve as a substrate for caspase 1 to induce the maturation of IL-1β and IL-18 and induce neuronal pyroptosis, a type of cell death that possesses the potential for inflammation, to rupture microglia to further release IL-1β/18 [181]. Furthermore, chronic inflammation may be associated with the formation of α-syn oligomers, eventually resulting in DA neuronal death in PD. Overexpressing or decreasing the clearance of α-syn in neurons can lead to the formation of α-syn tetramers, which are secreted by exosomes. Exosomes containing α-syn are then identified by microglia and endocytosis, which activates the NLRP3 inflammasome [182, 183]. Evidence has confirmed an association between Parkin and NLRP3 inflammasomes [184]. When mitochondrial damage occurs, PINK1 on the mitochondrial membrane will recruit and phosphorylate Parkin in an active state, which triggers mitochondrial ubiquitination [185]. Parkin dysfunction leads to the accumulation of damaged mitochondria and overexpression of ROS, which are an important factors in the activation of the NLRP3 inflammation [186]. It was also shown that loss of parkin activity results in spontaneous neuronal NLRP3 inflammasome assembly in mouse and human DA neurons, which cause DA neuronal death [187]. Normally, parkin inhibits priming of the inflammasome by ubiquitinating and targeting NLRP3 for proteasomal degradation, and losing of its activity participates to the assembly of an active NLRP3 inflammasome complex via mitochondrial-derived reactive oxygen species (mitoROS) generation [187].
The NLRP3 inflammasome pathway was also evidenced to be triggered by certain models of neurotoxin-induced PD, including 6-hydroxydopamine (6-OHDA), MPTP, lipopolysaccharides (LPS), and rotenone [184]. Because, these neurotoxins inducing mitochondrial dysfunction, which may indirectly activates NLRP3 inflammasomes by increasing ROS production [188,189,190]. However, the underlying mechanism is not clear. The LPS is a classic NLRP3 trigger that can bind to the Toll-like receptor 4 (TLR4) and facilitate NLRP3 movement via the NF-kB pathway [184]. Overall, aggregation of αSyn, neurotoxins, disrupted mitophagy, and mitochondrial ROS are the main regulators of the activation of microglial NLRP3 inflammasome in the SN.
Triggering Apoptosis
Activation of the mitochondrial-dependent apoptosis pathway may aid in SNpc DA neurodegeneration in PD. It consists of a sequence of procedures including enhanced ROS generation, permeabilization of the outer mitochondrial membrane, releasing cytochrome c into the cytoplasm, and ATP depletion, as well as activation of caspase-9 and caspase 3 [191]. Mitochondrial outer membrane permeabilization (MOMP) shows the point-of-no-return in mitochondria-mediated apoptosis and is strongly regulated by B-cell lymphoma 2 (Bcl-2)family proteins, which are characterized by one or more BCL-2 homology (BH) domains (i.e., BH1–4) and can be divided into three classes: anti-apoptotic (e.g., Bcl-2 and Bcl-xL), pro-apoptotic (e.g., Bcl-2-associated X protein (BAX) and Bcl-2 homologues antagonist/killer (Bak)), and BH3-only proteins [191]. Bax and Bak proteins form a channel within the OMM and cause the permeabilization of OMM [192]. When this happens, mitochondrial-dependent apoptotic death is identified. Bcl 2 proteins are able to bind with Bax/Bak, and decrease the OMM permeability caused by Bax/Bak [192]. Although the precise mechanism through which pro-apoptotic proteins, like Bax, induce MOMP is still a matter of debate, it requires that these proteins be translocated and inserted into the mitochondrial membranes. Hence, they may elicit the release of mitochondrial apoptogenic factors, like cytochrome c, through at least two different mechanisms described: The first relates to the opening of the mitochondrial permeability transition pore complex, and the second is dependent on the formation of channels directly through these proteins in the mitochondrial membranes [192, 193]. Cytochrome c is an intermembrane space protein that is particularly important in caspase activation. When cytochrome c is released in the cytosol, it induces mitochondria-mediated apoptosis [191,192,193]. Cytochrome c binds to the caspase adaptor molecule Apaf-1, resulting in the formation of a multimeric Apaf-1/cytochrome c complex. Apaf1 recruits caspase-9, forming together a major collective complex, called apoptosome. This recruitment happens through the exposure of the caspase activation and recruitment domains in Apaf-1 [191, 194]. As a result, procaspase-9 is activated by proteolysis and then dissociated from this complex. Once activated, caspase-9 activates executioner caspases-3, -6, and/or -7, which mediate proteolytic events that cause severe cell death [191, 192, 194].α-syn has been shown to have a strong affinity for mitochondria. This may be due to its affinity for cardiolipin, which is found throughout the IMM [141, 195]. It has been shown that oligomerization and aggregation of α-syn cause deficits in complex I activities, leading to reduced ATP production, IMM depolarization, and release of mitochondrial apoptogenic factors like cytochrome c into the cytosol. After its release into the cytoplasm, cytochrome c interacts with anti-apoptotic and pro-survival proteins and induces mitochondrial-mediated apoptosis [141].
Therapeutic Strategies for Mitochondrial Dysfunction
The role of mitochondrial dysfunction and mitochondrial oxidative stress in many neurological diseases is well known. Therefore, trying to find a way to improve the lost function of mitochondria could be an attractive therapeutic approach for such diseases. Many of these strategies are used to improve the function of mitochondria by increasing ATP production, reducing oxidative stress, modulating mitochondrial biogenesis and dynamics, decreasing mitochondrial-related signaling pathways that induce inflammation and apoptotic cell death. Other potential therapeutic strategies such as mitochondrial transfer/transplantation, targeting microRNAs, using stem cells and photobiomodulation were also discussed here.
Recovering ATP Production
ATP level is one of the most important parameters indicating overall health in the cells, and ATP depletion appears to be the most common dying cellular phenotype [196]. Mitochondrial bioenergetics dysfunction may be reduced due to impaired energy substrate importation, energy substrate processing (oxidative phosphorylation; OXPHOS), ADP (adenosine diphosphate) export/ATP (adenosine triphosphate) export, and mitochondrial fission/fusion alters [197]. Factors that increase mitochondrial mass and stimulates mitobiogenesis (such as recombinant human TFAM, rhTFAM) or stimulate LRPPRC expression may improve bioenergy and reduce nerve damage. One promising treatment strategy for PD is to maintain ATP levels by inhibiting ATP consumption or increasing ATP production, or both [197].
ATP maintenance via ATP regulators was demonstrated to mitigate pathological phenotypes in the mouse model of PD [196]. Resveratrol as a polyphenol antioxidant demonstrated a protective effect in PD by decreasing oxidative stress and increase mitochondrial biogenesis, which was associated with increased complex I and citrate synthase activities, basal oxygen consumption, and mitochondrial ATP production [198, 199]. Enhancing NAD+ was reported to salvage metabolism and exert neuroprotection in a PINK1 model of PD. As we know, disruption of PINK1 affects mitochondrial bioenergy and affects cellular stores of metabolites required for mitochondrial function, including NAD+ and ATP [200]. Mitochondrial transfer from Wharton's jelly-derived mesenchymal stem cells to mitochondria-defective cells was reported to recapture impaired mitochondrial function by affecting oxidative phosphorylation and bioenergy [201].
Decreasing ROS Production
Increasing ROS production was well evidenced that induced autosomal PD [202]. The enhancement in ROS production is mainly due to an imbalance between mitophagy and mitochondrial biogenesis, and damaged mtDNA, and leads to ETC dysfunction [203]. Deletion of the NOD2 (Nucleotide-binding oligomerization domain-containing protein 2) gene was demonstrated to reduce the Bax/Bcl-2 ratio, cytochrome C, caspase-3 activation, and ROS in 6-OHDA induced PD model [204]. Inhibition of Nox1 was demonstrated to reduce ROS production and oxidative stress in DA neurons [205, 206]. Furthermore, inhibiting Nox2 decreased ROS production, and demonstrated protective effects on SN in MPTP-receiving mice the model [207, 208]. In addition, using Mito-Q (a mitochondria-targeted antioxidant) was reported to stabilize mitochondria in the presence of 6-OHDA by suppressing ROS formation, and reducing mitochondrial fragmentation [209]. Using monoamine oxidase type B (MAO-B) inhibitor reduced the pathogenesis of PD by decreasing the ROS production [210].
Increasing Mitophagy
Destruction of damaged mitochondria is begun through a selective autophagy pathway called mitophagy. Pharmacological enhancement of mitophagy and acceleration of damaged mitochondria removal were considered for the development of PD treatment. Mutation in the two key mitophagy genes, which code the PINK1 and Parkin proteins, were reported as the most common causes of PD in people under 45 years of age [202, 211]. However, evidence for their role in mitophagy in vivo is still scarce, and findings in Drosophila provide evidence that Pink1 and parkin are not essential for bulk basal mitophagy, and mitolysosomes in flight muscles were not detected in Pink1 or parkin null flies. While, these flies exhibit locomotor defects and dopaminergic neuron loss [212]. Hence, in Drosophila, reports reveal that mitophagy increases with aging, and this age-dependent rise is abrogated by PINK1 or parkin deficiency in Drosophila, and mitophagy occurs in muscle cells and dopaminergic neurons, even in the absence of exogenous mitochondrial toxins [213].
Mitophagy augmentation can be done by two pathways: PINK1-Parkin-related pathways and PINK1-Parkin-independent pathways [202]. Modulating mitophagy pathways may be an avenue to treat a subset of early-onset PD that may additionally provide therapeutic opportunities in sporadic disease. The PINK1/Parkin mitophagy pathway, as well as alternative mitophagy pathways controlled by BNIP3L/Nix and FUNDC1, are emerging targets to enhance mitophagy to treat PD [214]. It was demonstrated that celastrol (as a plant-derived triterpene) inhibits dopaminergic neuronal death of PD through activating mitophagy. Celastrol decreased nerve damage in the SN and striatum and increased mitophagy by enhancing PINK1 and DJ-1 in the striatum [215]. PINK1 and DJ-1 can induce mitophagy and play a neuroprotective role in neurodegenerative disorders [215].
It has been made clear that PINK1 induces mitophagy even in the absence of Parkin, which shows the potential and significant effect of PINK1 on mitophagy and can be considered as a therapeutic pathway. To develop treatment, many studies have been performed today to find molecules that are effective in regulating mitophagy, and many of them have been identified [202].
Suppression or removal of OMA1 (IMM-embedded metalloprotease) was reported to restore PINK1 stabilization after mitochondrial membrane depolarization and increased mitophagy. OMA1 is activated by a wide range of stress stimuli, causing fragmentation and bioenergetic loss of mitochondria. This, in turn, prepares the cell to increase the stress response through mechanisms such as apoptosis and autophagy [202, 216]. Hence, deletion of the USP33 gene increased the formation of K63-related ubiquitin chains in Parkin and then accelerates mitophagy by stabilizing the Parkin, and increasing its transfer rate to depolarized mitochondria [217]. Inhibition or silencing of PPEF2 (protein phosphatase with EF-hand domain 2) and PTEN-L phosphatase were demonstrated to increase mitophagy. PPEF2 is a PINK1 phosphatase antagonist that dephosphorylates ubiquitin and inhibits PINK1-mediated mitophagy [202, 218].
Improving Mitochondrial Dynamic
Mitochondria are highly dynamic organelles that are controlled by fission and fusion proteins. Breaking off and rejoining the mitochondrial network as well as their trafficking is critical to maintaining the function of neurons, primarily DA neurons, which have long axons with numerous synapses [219]. Spatially, relocating mitochondria provides metabolic energy for different regions of neurons from the dendrites to soma, axons, and synapses [220]. Most of the genetic, mitochondria-related PD proteins are involved in mitochondrial dynamics and the trafficking process [221]. The importance of the balance of mitochondrial dynamics and trafficking in DA neurons makes it a crucial therapeutic target for PD. For instance, Rappold and coworkers, by inhibiting a mitochondrial fission protein named Drp1 were able to prevent neurotoxicity and cell death, and rescue the synaptic dysfunction in the MPTP-injured and Pink1-knockout mouse PD models [222]. Moreover, this research team applied this therapeutic strategy in the hA53T-α-syn rat model of PD. They showed that mitochondrial division inhibitor (mdivi-1) which blocks Drp1 and subsequently mitochondrial fragmentation could effectively reduce α-syn-induced neurodegeneration, α-syn aggregation, mitochondrial impairment, oxidative stress, and motor abnormalities in PD rats [223]. α-syn aggregation, one of the main pathological hallmarks of PD, may occur due to mitochondrial dysfunction. Indeed, damaged mitochondria and reduced energy leads to microtubule disassembly and subsequent accumulation of α-syn protein. On the other hand, α-syn deposition impairs mitochondrial dynamics, and trafficking results in disordered distribution of organelles and accumulation of autophagosomes in synapses [224]. As regards damaged mitochondria induce microtubule disassembly, and stabilization of microtubules may prevent the accumulation of α-syn protein. Esteves et al. showed that NAP (davunetide) repairs microtubule network assembly and so improves microtubule-mediated mitochondrial trafficking [225]. More research is needed to determine the exact role of mislocation of mitochondria and intercellular mitochondrial transfer in PD etiology.
Improving Mitochondrial Biogenesis
Mitochondrial biogenesis is affected by environmental stresses such as exercise, calorie restriction, low temperature, oxidative stress, cell division, and renewal and differentiation. Mitochondrial biogenesis is associated not only with changes in number but also in size and mass [226]. Decreased biogenesis and mitochondrial function are seen in many pathological conditions, including neurodegenerative disorders. Therefore, increasing mitochondrial biogenesis in neurons can be pursued as a therapeutic goal.
Several regulatory factors are involved in modulating mitochondrial function, including PGC-1α, PGC-1β, and PRC. PGC1α is known as the major regulator in the mitochondrial translation and transcription machine. PGC1α interacts strongly with NRFs [83, 227]. NRF-1 and NRF-2 are important contributors to the sequence of events that lead to increased transcription of key mitochondrial enzymes and have been shown to interact with Tfam, which directs mtDNA transcription and replication. In addition to NRFs, PGC-1α also interacts with other transcription factors such as PPARs, thyroid hormone, glucocorticoids, estrogen, and ERRs [226].
A mutation in the parkin-encoding gene, PARK2, reduces the removal of damaged mitochondria in human DA neurons, which are involved in both familial and sporadic forms of Parkinson's disease (PD), by losing the ability to interact with PINK1 [228, 229]. Parkin mutation increases PARIS expression and affects mitochondrial quality control by regulating PGC1α-TFEB signaling. Rapamycin regenerates PGC1α-TFEB signaling independently of parkin activity and reduces mitochondrial dysfunction [230]. The combination of resveratrol and equol in the cell culture medium also increases NRF1 as well as TFAM by activating PGC1α /sirt1 and finally increases biogenesis [199, 231]
RNS60 is an electrokinetic modified saline that contains charge-stabilized nanobubbles but has no active drug substance. Administration of RNS60 both in culture and in vivo increases mitochondrial biogenesis by increasing PGC1α in neurons and other brain cells. PGC1α activates transcription factors responsible for mitochondrial biogenesis, including NRF-1, NRF-2, and PPARγ. RNS60 stimulates PGC1α via CREB activation by PI3K class IA [232]. Drug activation of dopamine D1 receptors also significantly improves mitochondrial biogenesis, ATP levels, and mitochondrial membrane potential, and defends DAergic nigral neurons against neurotoxicity induced by 6-OHDA in adult mice [233].
Oral ferulic acid supplementation reduces mitochondrial Drp1 expression and increases PGC1α gene and protein expression. We found that oral FA supplementation reduced 6-OHDA-induced oxidative stress, DNA fragmentation, morphological changes, and the cascade of apoptosis [227]. It has been shown that one to four weeks of physical exercise increases mitochondrial function and biogenesis in rats with PD by increasing PGC-1α protein levels and stimulating PGC-1α and TFAM gene expression, and phosphorylation rates in skeletal muscle, and improving complex I activity Induced by 6-OHDA [234]. Physical activity is also linked to lower α-Syn expression and upregulation of mitochondrial COX-I, COX-IV, and mtHSP70 proteins in MPTP-induced Parkinson's disease mouse models [235]. Calorie restriction also affects mitochondrial function. Calorie restriction in humans increases the expression of genes involved in the regulation of mitochondrial biogenesis, including PGC-1α, Tfam, eNOS, and SIRT1 [226]. Another environmental stress is frostbite. Cold increases the expression of PGC-1α and the expression of key enzymes in mitochondria such as ATP synthetase subunits (β subunit) and COX (cytochrome c oxidase) (COX-II and COX- IV) [236].
Some components of signaling pathways that activate the transcription cascade and act as regulators of mitochondrial biogenesis by increasing PGC-1α expression, such as AMPK, CaMKIV, nitric oxide, SIRT1, TORC, Calcineurin, Sin3A, p38 MAPK, and RIP140 [226].
Mitochondrial Transfer
It has been explored that cells for decreasing stress and increasing energy production, use an intercellular mitochondrial transferring mechanism [237]. The defective intercellular mitochondrial transferring system may be the culprit for the propagation of α-syn deposition seen in DA neurons. Because PD has a complex and multifaceted nature, access to a worthwhile therapeutic approach depends on the development of a combined-disease modifying approach. Targeting and restoration of the intracellular and intercellular mitochondrial dynamics and trafficking defects may be an important hotspot for achieving an efficient therapy for PD [162].
It was found that healthy microglia donate mitochondria to α-syn overloaded cells, as well as α-syn are transferred between microglia through tunneling nanotubes [238]. This α-syn cargo was effectively degraded in the neighboring naive microglia, which attenuated the inflammatory microglia profile. This degradation strategy was represented in the cells loud the LRRK2 mutation [238].
Mitochondrial Transplantation
Transcellular transfer of mitochondria dynamically happens in response to CNS damage and is involved in maintaining and homeostasis of the CNS. Likewise, mitochondrial transplantation therapy is a novel approach to overcoming and improvement of mitochondria-related disorders including PD. Evidence shows that supplementing exogenous mitochondria to the injured site rather than focusing on a particular perspective of mitochondrial function may be a better strategy for targeting the neurodegeneration process [237]. Chang et al. transferred the mitochondrial with or without cell-penetrating peptide-based (Pep-1) conjugation into the 6-OHDA-treated PC12 cells and showed that Pep-1 mitochondrial delivery was able to rescue mitochondrial function, enhanced cell viability and improves neurite outgrowth. They also injected xenogenic/allogenic peptide-labeled mitochondria into the medial forebrain bundle of 6-OHDA-induced PD rat models and reported that mitochondrial graft rescued DA neuron loss, enhanced tyrosine hydroxylase–positive DA neurons in the SNc and striatum, improved motor function of PD rats, and restored mitochondrial function by restoring normal levels of mitochondrial complex I protein and ameliorating oxidative stress [239]. Moreover, Shi et al. in another study demonstrated that intravenous administration of mitochondria to MPTP-induced PD rats reduced ROS levels, improved the electron transport chain function, restricted necrosis and apoptosis, and eventually prevented PD progression [240]. Some important issues that determine the success of mitochondrial transplantation are quality and source of the isolated mitochondria, delivery system, and adequate uptake of supplemental mitochondria by neurons [241]. A more recent study investigated the efficacy of intranasal delivery of allogeneic mitochondria conjugated with Pep-1 or unconjugated to the ipsilateral sides of 6-OHDA-lesioned rats once a week for three months to bypass the blood–brain barrier. Its results unveiled that mitochondrial could penetrate the accessory olfactory bulb and migrate through the rostral migratory stream neurons and express in striatal, but not by DA neurons of SN. Intranasal mitochondria (with or without Pep-1) infusion leads to improvement of movement disorders of PD rats, and increased the survival and recovery of DA neuron in lesion site. This recovery was related to improvement of mitochondrial function and lessening of oxidative stress in SN. Remarkably, mitochondria conjugated with Pep-1 inhibited plasma levels of inflammatory cytokines [242].
Using Stem Cells
Stem cells are a population of unspecialized cells that originate from adult body tissues and embryos. They are able to differentiate into different types of cells and tissue [243]. Stem cell-based therapies are emerging as one of the most promising approaches for the treatment of PD [244]. Due to the considerable technical difficulties in earning normal purified mitochondria for transplantation, and the limited half-life of obtained mitochondria, finding a permanent source of healthy mitochondria is important for therapeutic purposes. In favour of this concept, a study showed that astrocytic conditioned media or iPSCs-derived astrocytes can rescue degeneration of DA neurons through intercellular mitochondrial transfer in a rotenone-induced PD model. Therefore, iPSCs-derived astrocytes can consider mitochondria donors to the injured DA neurons to provide healthy mitochondria and attenuate PD symptoms. iPSCs-derived astrocytes may provide a novel insight toward cellular therapy for PD in the future [245].
Micro RNAs as Therapeutic Targets
Micro RNAs (miRNAs) are a large family of non-coding RNAs with 20–24 nucleotides in length that have a critical role in modification of gene expression [246]. More than 30% of genes are regulated by miRNAs, and one specific miRNA can mediate the expression of different genes [247]. A large body of studies exhibits the association of miRNA dysregulation with PD pathogenesis [248]. Interestingly, most of these miRNAs are implicated in mitochondrial homeostasis destruction in PD (Table 1). Altered miRNA levels drive ROS production, which leads to mitochondrial impairment. Electron transport chain leakage that occurs during mitochondrial dysfunction leads to ROS formation. Indeed, mitochondrial dysfunction and ROS formation cause oxidative stress and a cascade of events results in neuronal network alteration and degeneration of DA neurons in PD [249]. Based on a study, miRNAs are regulators of the gene expression in the cortex of PD patients and have distinct expression patterns in PD brains compared with healthy ones [250]. In addition, decreased activity of complex I mitochondria was identified in the frontal cortex and SNpc of PD patients [251]. Peroxisome proliferator-activated receptor-gamma coactivator 1-alpha (PGC1α) is a mitochondrial gene-activator with a crucial role in mitochondrial homeostasis which diminishes in PD patients [252].
It has been reported that the overexpression of miR-124 by regulating the impaired autophagy and apoptosis process could reduce DA neuron loss and reverse the striatal levels of dopamine in the MPTP-treated mice model of PD. Likewise, upregulated miR-124 by targeting protein bcl-2-like protein 11 (Bim) decreases the translocation of protein bcl-2-like protein 4 (Bax) to lysosome and mitochondria. This cascade of events eventually inhibits mitochondria apoptotic signaling pathways and improves autophagy activity [253]. Moreover, investigations unveiled the association between the DJ-1 levels in the SNpc and mitochondrial function. MiR-494, by diminishing the DJ-1 expression, induces oxidative stress and exacerbates neuronal damage [254]. Another study revealed that the decreased level of miR34b/c may be through DJ-1 downregulation result in mitochondrial impairment in PD brains [255]. Furthermore, miR-205 regulates the expression of LRRK2 which is a PD-related protein. Notably, LRRK2 protein can change the mitochondrial dynamics and integrity via targeting a dynamin-like protein (DLP1). Interestingly, reduced expression levels of miR-205 besides enhanced expression levels of LRRK2 were detected in the brains of patients with sporadic PD [256]. Therefore, upregulation of miR-205 may be a therapeutic approach for the suppression of abnormal LRRK2 overexpression in PD. Results of another study suggested that tumor necrosis factor-α by improving the miR-27a and miR-103 levels may inhibit the expression of the mitochondrial complex-I subunit. High miR-155 and miR-27a expression may promote oxidative stress, mitochondrial dysfunction and decrease the transcription of ATP synthase membrane subunit c locus 3 (ATP5G3), a mitochondrial complex-V subunit [257]. Another example is miR-7, which has the capacity to stabilize the potential of mitochondrial membrane by suppression of the voltage-dependent anion channel 1 (VDAC1) expression. Thus, miR-7 could be another target for improving the mitochondrial impairment in PD [258]. Additionally, miRNA181a/b downregulation has a neuroprotective effect on mitochondrial dysfunction in comorbid neurodegenerative conditions such as PD. This miRNA regulates respiratory chain assembly and mitochondrial biogenesis and also influences mitochondrial antioxidants suggesting a potential target for the treatment of diseases with mitochondrial dysfunction, including PD [259]. Small drugs like L-DOPA are capable of adjusting miRNA profiles and thereby improving neurodegeneration status in PD [260]. Ebert et al. developed cell expressed-miRNA inhibitors called miRNA sponges with the capability to inhibit various miRNA concomitantly [261]. Also, there is also the prospect of developing the use of miRNAs in diagnostic assays. For instance, salivary miR-223 and miR-153 levels have been introduced as potential diagnostic markers of idiopathic PD [262]. In short, crosstalk between mitochondrial impairment, oxidative stress, and miRNAs dysfunction plays a critical role in the onset and progression of PD and may provide potential targeted molecules for novel, personalized therapeutic approaches. Controlling miRNA expression also provides the therapeutic potential to modify various mitochondrial pathways including biogenesis, bioenergetics, oxidative stress, apoptosis, and mitophagy [263].
Other Therapeutic Strategies
Photobiomodulation
There is an agreement that apoptotic mechanisms are the main leading cause of DA neuron death in PD. The apoptotic process, a slow breakdown of cellular components, is mediated by two major mechanisms including Lewy body accumulation and mitochondrial dysfunction [264, 265]. Photobiomodulation, which refers to the application of red to infrared wavelength (λ = 600-1070 nm) on body tissues through influences of mitochondrial activity, exerts beneficial effects in PD studies. For that reason, photobiomodulation has been reported as a potential therapeutic option for neurodegenerative diseases such as PD [266,267,268]. While the exact mechanism by which photobiomodulation displays neuroprotective effects in distressed neurons is presently not fully understood, the two proposed mechanisms are direct and indirect stimulation of these neurons [267, 269]. In the direct stimulation procedure, the direct application of photons on the distressed neurons leads to chemical changes, and then the conversion of light energy to metabolic energy impacts the survival and function of neurons. Mechanistically, absorption of light photons by cytochrome c oxidase, unit IV, the best-known photoacceptor in the mitochondrial electron transport chain, leads to the release of nitric oxide which in turn triggers a cascade of events leading to electron transportation along the respiratory chain and proton translocation across the mitochondrial membrane. This proton gradient across the membrane eventually results in ATP production. Furthermore, released nitric oxide promotes the vasodilation of nearby vessels, and enhances blood (and lymphatic) flow. Interestingly, during this process small, and normal levels of ROS are released within neurons which activate nuclear transcription factors [270]. There is also evidence that photobiomodulation increases ATP levels in the lack of cytochrome c oxidase in mouse and human cell lines. This finding sheds light on the presence of other photoacceptor(s) within the neurons [271]. Water is another photoacceptor within the mitochondria. Increasing water viscosity within the folded membranes of the mitochondria impedes the production of ATP, causing distress in the neuron. Photobiomodulation reduces the viscosity of the water and ultimately enhances ATP synthase, and decreases ROS levels [272, 273]. Further evidence showed that chlorophyll metabolites within the mitochondria may be other photoacceptors, because when treated with photobiomodulation, they catalysed the coenzyme Q reduction, activated cytochrome c oxidase, and enhanced mitochondrial activity and ATP production [274]. Photobiomodulation can improve the activity of mitochondria in distressed neurons, and in this way stimulate the expression of some protective genes, such as neurotrophic factor genes [275]. Taken together, photobiomodulation by stimulation of intrinsic mechanisms protects distressed neurons and helps them to repair their damage. These intrinsic self-protective mechanisms surge energy production for the neuron and stimulate the expression of protective genes. Furthermore, photobiomodulation by increasing the local blood flow and perfusion of the region helps to maintain neurons’ survival and homeostasis [270, 276]. Two studies using in vitro models of PD showed the neuroprotective effects of photobiomodulation. Their findings reveal that photobiomodulation diminished oxidative stress, improved ATP levels, and reduced neural death [277, 278]. The culture study of human neuroblasts engineered to overexpress α-syn demonstrated that photobiomodulation improves mitochondrial function and reduces oxidative stress after exposure to parkinsonian toxins. Moreover, applying photobiomodulation to hybrid neural cells bearing mitochondrial DNA from PD patients leads to considerable improvement in mitochondrial movement along axons [279]. In addition, photobiomodulation helped to rescue defects in the mitochondria of mouse DA neurons and drosophila pink1 mutants [280]. Although direct stimulation shows remarkable value in animal models, some problems such as restricted penetration of light through the skull and brain tissue inhibit its translation to the clinic [267, 270]. Whereas intracranial photobiomodulation tries to bypass this inherent obstacle, achieving sufficient penetration of light energy to direct stimulation of the first affected parts of the PD brain is exceedingly improbable [267, 269, 281]. Other studies disclosed that non-invasive remote photobiomodulation (extracranial) has a neuroprotective effect [282, 283]. Human intranasal and transcranial photobiomodulation have demonstrated PD symptom improvement in patients [284, 285]. However, this therapeutic procedure may just be beneficial in the early stages of PD [286]. Also, a study detected circulating mitochondria in blood samples and showed that photobiomodulation can improve the function of these secreted intact mitochondria and may affect neural cell activities [287].
Exercise
Physical activity and exercise are widely used in rehabilitation protocols for the treatment of PD and their effects have been assessed in several animal and human studies. The result of these studies revealed that exercise can promote mitochondrial function and biogenesis in PD neurons [288]. For instance, a recent systematic review showed that treadmill training could improve the neural mitochondrial respiratory deficiency and neural mitochondrial quality-control dysregulation in PD, and suggested that treadmill exercise may slow down PD progression [289]. In addition, previous studies exhibited that treadmill training through modulation of the neural mitochondrial dynamics and improvement of mitochondrial respiration could attenuate PD symptoms and delay its progression in rodent models [290, 291]. exercise and fasting, by interacting with the adaptive cellular response to oxidative stress, protect neurons from oxidative damage in PD. Likewise, Curtis et al. introduce mitochondrial quality control as an appropriate target in exercise therapy for PD [292]. Long-term voluntary exercise markedly improved motor function, and nigrostriatal DA input in the mouse model of PD. Moreover, exercise enhanced oxygen consumption, oxidative phosphorylation, and conclusively ATP production. Thus, exercise increased mitochondrial aerobic metabolism in the nigrostriatal system [293]. In short, a large body of evidence demonstrates that regular exercise has health benefits with a great impact on brain health and may protect the brain against neurodegenerative diseases. Mitochondria seem to be the main players of these exercise-induced neuroprotective effects. Besides the direct effects of regular exercise on brain mitochondria, it can induce signaling from skeletal muscle to the brain via the muscle–brain axis. Theoretically, the release of various signaling molecules from muscles and direct mitochondrial transfer in response to exercise may mediate this signal transduction. Intensity, modality, duration, and frequency of exercise are the determinant factors for enhancing mitochondrial functions in the brain [294].
Diet
Another promising approach that has been proposed for treating PD is improving lifestyle, having a healthy diet, and protecting mitochondria via nutraceutical targeting of oxidative stress and inflammation. In addition, animal studies demonstrate that dietary management along with exercise can improve mitochondrial biogenesis and slow down PD progression [295]. Glutathione peroxidase and superoxide dismutase are two main players in the antioxidant system and their maintenance is critical for normal mitochondria function and the prevention of oxidative stress. Nowadays, targeting antioxidant systems is a novel therapeutic strategy for PD therapy. For example, the therapeutic effects of several antioxidants such as Vitamin E, vitamin C, and CoQ10 are commonly studied for the treatment of neurodegenerative diseases. Whereas in vitro and animal studies showed the effectiveness of antioxidant administration in some neurodegenerative disorders, it was not adequately efficient in patients owing to the chemical instability of antioxidants. Also, it has been clear that dietary secondary metabolites are potential antioxidants with neuroprotective effects [296]. Creatine (α-methyl-guanidinoacetic acid) is a current interesting agent that as a mitochondrial-enhancing antioxidant substance promotes energy transduction, stabilizes reactants, and inhibits mitochondrial permeability transition [297]. CoQ10 is another effective antioxidant that has up to 33% lower levels in blood and platelet mitochondria of PD patients compared with healthy controls. Studies showed that CoQ10 by modifying mitochondrial function can reverse striatal DA neuron loss, and PD progression [298]. These compounds may be disease-modifying, but large studies are needed to confirm their therapeutic potential before prescribing them to PD patients. Bajracharya and coworkers reviewed that dietary management and maintaining a low protein-to-carbohydrate (P: C) ratio in food may change mitochondrial energy metabolism, dynamics, and morphology and subsequently surge mitochondrial activity, motor function, and lifespan of PD [299]. Therefore, dietary macronutrient management may be another strategy for controlling of PD progression.
Conclusion and Perspectives
Mitochondria are powerhouses of the cells by the production of ATP, and also contribute to critical processes such as regulation of calcium accumulation, scavenging of free radicals, and mediation of apoptosis [300]. It was well evidence that mitochondria not only exist intracellularly but can also be feasible extracellularly in both physiological and pathophysiological conditions [301, 302]. Interestingly, dysfunction of mitochondria seems to have an important role in pathogenesis associated with PD [166], which can inhibit the normal functions of dopaminergic neurons by affecting normal function of mitochondrial complex I, and ROS production [303]. Mitochondrial dysfunctions contribute to PD severity by increasing oxidative stress and ROS production, which leads to calcium overload, NLRP3 inflammasome activation, mitophagy, and apoptotic cell death [166, 304]. Hence, defected bioenergetics, aberrant mitochondrial morphology and structure, and abnormal mitochondrial dynamics play an essential role in cell death induction [305], which may contribute to PD pathogenesis. Preclinical studies reported neuroprotective effects of targeting mitochondrial quality control and mitochondrial dynamics through genetic interventions or pharmacological agents [306].
Mitochondria represents an ideal target for candidate drug therapies, and its modulation will certainly have an impact on progression of PD [307]. However, targeting mitochondria by pharmacological agents confronts challenges in the clinic (Table 2), which may be due to a failure in addressing the multiple intertwined systems and pathways in the vastly delicate homeostatic cellular system of neural cells. Additionally, it may be difficult to warrant adequate delivery of therapeutic agents to their site of action. Besides, disease progression in these trials may be too advanced to recover appropriate function in mitochondria and target cell [307]. The future of neuroprotective agents in PD will possibly depend on a combination of treatments acting directly and indirectly on affected pathways using personalized precision medicine [308].
Massive evidence approving several therapeutic strategies against PD, but in the clinic, they failed to show beneficial and hopeful effects. To overcome this difficulty, more precise details of mitochondrial dysfunction and its underlying mechanisms are necessary in the pathogenesis of PD, to achieve accurate biomarkers for targeting mitochondria. Furthermore, treatments containing several approaches may possibly better respond to the complexity of mitochondrial dysfunction in PD. Besides, mitochondrial transplantation and mitochondrial transfer using stem cells may participate a pivotal role in our perspectives in treatment of PD. Therefore, targeting mitochondrial dysfunction by appropriate and accurate agents with limited side effects, may offer the best chance for development of an effective novel therapeutic agent in our fight against PD.
Data Availability
The authors provide data transparency.
References
Ball N, Teo W-P, Chandra S, Chapman J (2019) Parkinson’s disease and the environment. Front Neurol 10:218
Lotankar S, Prabhavalkar KS, Bhatt LK (2017) Biomarkers for Parkinson’s disease: Recent advancement. Neurosci Bull 33:585–597
Choudhury GR, Daadi MM (2018) Charting the onset of Parkinson-like motor and non-motor symptoms in nonhuman primate model of Parkinson’s disease. PLoS ONE 13:e0202770
Pacelli C, Giguère N, Bourque M-J, Lévesque M, Slack RS, Trudeau L-É (2015) Elevated mitochondrial bioenergetics and axonal arborization size are key contributors to the vulnerability of dopamine neurons. Curr Biol 25:2349–2360
Paolini Paoletti F, Gaetani L, Parnetti L (2020) The challenge of disease-modifying therapies in Parkinson’s disease: Role of CSF biomarkers. Biomolecules 10:335
Prasuhn J, Davis RL, Kumar KR (2021) Targeting mitochondrial impairment in Parkinson's disease: challenges and opportunities. Front Cell Biol:1704
Tapias V (2019) Mitochondrial dysfunction and neurodegeneration. Front Neurosci:1372
Haas RH (2019) Mitochondrial dysfunction in aging and diseases of aging. Biology 8(2):48
Chen W, Huang J, Hu Y, Khoshnam SE, Sarkaki A (2020) Mitochondrial transfer as a therapeutic strategy against ischemic stroke. Transl Stroke Res 11:1214–1228
Cardoso AC, Lam NT, Savla JJ, Nakada Y, Pereira AHM, Elnwasany A, Menendez-Montes I, Ensley EL, Bezan Petric U, Sharma G (2020) Mitochondrial substrate utilization regulates cardiomyocyte cell-cycle progression. Nat Metab 2:167–178
Fang D, Maldonado EN (2018) VDAC regulation: A mitochondrial target to stop cell proliferation. Adv Cancer Res 138:41–69
Lei C, Liao J, Li Q, Shi J, Zhang H, Guo J, Han Q, Hu L, Li Y, Pan J (2021) Copper induces mitochondria-mediated apoptosis via AMPK-mTOR pathway in hypothalamus of Pigs. Ecotoxicol Environ Saf 220:112395
Calì T, Ottolini D, Brini M (2012) Mitochondrial Ca 2+ as a key regulator of mitochondrial activities. Adv Mitochon Med:53–73
Auger C, Vinaik R, Appanna VD, Jeschke MG (2021) Beyond mitochondria: Alternative energy-producing pathways from all strata of life. Metabolism 118:154733
Daiber A (2010) Redox signaling (cross-talk) from and to mitochondria involves mitochondrial pores and reactive oxygen species. Biochim Biophys Acta 1797:897–906
He Z, Ning N, Zhou Q, Khoshnam SE, Farzaneh M (2020) Mitochondria as a therapeutic target for ischemic stroke. Free Radical Biol Med 146:45–58
Bhatti JS, Bhatti GK, Reddy PH (2017) Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim Biophys Acta 1863:1066–1077
Prasuhn J, Brüggemann N (2021) Gene therapeutic approaches for the treatment of mitochondrial dysfunction in Parkinson’s Disease. Genes 12:1840
Pang SY-Y, Ho PW-L, Liu H-F, Leung C-T, Li L, Chang EES, Ramsden DB, Ho S-L (2019) The interplay of aging, genetics and environmental factors in the pathogenesis of Parkinson’s disease. Translat Neurodegene 8:1–11
Pringsheim T, Jette N, Frolkis A, Steeves TD (2014) The prevalence of Parkinson’s disease: a systematic review and meta-analysis. Mov Disord 29:1583–1590
Liu H-F, Ho PW-L, Leung GC-T, Lam CS-C, Pang SY-Y, Li L, Kung MH-W, Ramsden DB, Ho S-L (2017) Combined LRRK2 mutation, aging and chronic low dose oral rotenone as a model of Parkinson’s disease. Sci Rep 7:1–15
Guadagnolo D, Piane M, Torrisi MR, Pizzuti A, Petrucci S (2021) Genotype-phenotype correlations in monogenic Parkinson disease: A review on clinical and molecular findings. Front Neurol 12:648588
Georgiou A, Demetriou CA, Christou YP, Heraclides A, Leonidou E, Loukaides P, Yiasoumi E, Pantziaris M, Kleopa KA, Papacostas SS, Loizidou MA, Hadjisavvas A, Zamba-Papanicolaou E (2019) Genetic and environmental factors contributing to Parkinson’s disease: A case-control study in the cypriot population. Front Neurol 10:1047–1047
Bellou V, Belbasis L, Tzoulaki I, Evangelou E, Ioannidis JP (2016) Environmental risk factors and Parkinson’s disease: An umbrella review of meta-analyses. Parkinsonism Relat Disord 23:1–9
Pang SY-Y, Ho PW-L, Liu H-F, Leung C-T, Li L, Chang EES, Ramsden DB, Ho S-L (2019) The interplay of aging, genetics and environmental factors in the pathogenesis of Parkinson’s disease. Translat Neurodegen 8:23
Maiti P, Manna J, Dunbar GL (2017) Current understanding of the molecular mechanisms in Parkinson’s disease: Targets for potential treatments. Translat Neurodegen 6:1–35
Angelopoulou E, Pyrgelis E-S, Piperi C (2020) Neuroprotective potential of chrysin in Parkinson’s disease: Molecular mechanisms and clinical implications. Neurochem Int 132:104612
Esposito E, Cuzzocrea S (2010) New therapeutic strategy for Parkinson’s and Alzheimer’s disease. Curr Med Chem 17:2764–2774
Winklhofer KF, Haass C (2010) Mitochondrial dysfunction in Parkinson's disease. Biochim Biophys Acta 1802:29–44
Nicoletti V, Palermo G, Del Prete E, Mancuso M, Ceravolo R (2021) Understanding the multiple role of mitochondria in Parkinson’s disease and related disorders: Lesson from genetics and protein–Interaction Network. Front Cell Dev Biol 9:636506
Hwang O (2013) Role of oxidative stress in Parkinson’s disease. Exp Neurobiol 22:11
Guo JD, Zhao X, Li Y, Li GR, Liu XL (2018) Damage to dopaminergic neurons by oxidative stress in Parkinson’s disease. Int J Mol Med 41:1817–1825
Rodríguez-Varela C, Labarta E (2020) Clinical application of antioxidants to improve human oocyte mitochondrial function: A review. Antioxidants 9:1197
Inoue T, Maekawa H, Inagi R (2019) Organelle crosstalk in the kidney. Kidney Int 95:1318–1325
Krauss S (2001) Mitochondria: Structure and role in respiration. eLS
Ross JM, Coppotelli G, Olson L (2016) Mitochondrial dysfunction in ageing and Diseases. Int J Mol Sci 7(5):711
Park GH, Park JH, Chung KC (2021) Precise control of mitophagy through ubiquitin proteasome system and deubiquitin proteases and their dysfunction in Parkinson’s disease. BMB Rep 54:592
Huang M, Lou D, Charli A, Kong D, Jin H, Zenitsky G, Anantharam V, Kanthasamy A, Wang Z, Kanthasamy AG (2021) Mitochondrial dysfunction–induced H3K27 hyperacetylation perturbs enhancers in Parkinson’s disease. JCI insight 6
Moon HE, Paek SH (2015) Mitochondrial Dysfunction in Parkinson’s Disease. Exp Neurobiol 24:103–116
Zanon A, Pramstaller PP, Hicks AA, Pichler I (2018) Environmental and genetic variables influencing mitochondrial health and Parkinson’s disease penetrance. Parkinson’s Disease 2018:8684906
Coon S, Stark A, Peterson E, Gloi A, Kortsha G, Pounds J, Chettle D, Gorell J (2006) Whole-body lifetime occupational lead exposure and risk of Parkinson’s disease. Environ Health Perspect 114:1872–1876
Kamel F (2013) Paths from pesticides to Parkinson’s. Science 341:722–723
Tanner CM, Kamel F, Ross GW, Hoppin JA, Goldman SM, Korell M, Marras C, Bhudhikanok GS, Kasten M, Chade AR (2011) Rotenone, paraquat, and Parkinson’s disease. Environ Health Perspect 119:866–872
Langston JW, Ballard P, Tetrud JW, Irwin I (1983) Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 219:979–980
Casanova Y, Negro S, Barcia E (2022) Application of neurotoxin-and pesticide-induced animal models of Parkinson’s disease in the evaluation of new drug delivery systems. Acta Pharm 72:35–58
Chiueh C, Markey S, Burns R, Johannessen J, Pert A, Kopin I (1984) Neurochemical and behavioral effects of systematic and intranigral administration of N-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine in the rat. Eur J Pharmacol 100:189–194
Choi SJ, Panhelainen A, Schmitz Y, Larsen KE, Kanter E, Wu M, Sulzer D, Mosharov EV (2015) Changes in neuronal dopamine homeostasis following 1-methyl-4-phenylpyridinium (MPP+) exposure. J Biol Chem 290:6799–6809
Javitch JA (1984) Uptake of MPP^+ by dopaminergic neurons explains selectivity of parkinsonism inducing neurotoxin MPTP. Eur J Pharmacol 106:455–456
Javitch JA, D’Amato RJ, Strittmatter SM, Snyder SH (1985) Parkinsonism-inducing neurotoxin, N-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine: uptake of the metabolite N-methyl-4-phenylpyridine by dopamine neurons explains selective toxicity. Proc Natl Acad Sci 82:2173–2177
Chaturvedi RK, Beal MF (2008) Mitochondrial approaches for neuroprotection. Ann N Y Acad Sci 1147:395–412
Sherer TB, Richardson JR, Testa CM, Seo BB, Panov AV, Yagi T, Matsuno-Yagi A, Miller GW, Greenamyre JT (2007) Mechanism of toxicity of pesticides acting at complex I: Relevance to environmental etiologies of Parkinson’s disease. J Neurochem 100:1469–1479
Gash DM, Rutland K, Hudson NL, Sullivan PG, Bing G, Cass WA, Pandya JD, Liu M, Choi DY, Hunter RL, Gerhardt GA, Smith CD, Slevin JT, Prince TS (2008) Trichloroethylene: Parkinsonism and complex 1 mitochondrial neurotoxicity. Ann Neurol 63:184–192
Mullett SJ, Hinkle DA (2011) DJ-1 deficiency in astrocytes selectively enhances mitochondrial Complex I inhibitor-induced neurotoxicity. J Neurochem 117:375–387
Borland MK, Trimmer PA, Rubinstein JD, Keeney PM, Mohanakumar K, Liu L, Bennett JP (2008) Chronic, low-dose rotenone reproduces Lewy neurites found in early stages of Parkinson’s disease, reduces mitochondrial movement and slowly kills differentiated SH-SY5Y neural cells. Mol Neurodegener 3:1–12
Sherer TB, Richardson JR, Testa CM, Seo BB, Panov AV, Yagi T, Matsuno-Yagi A, Miller GW, Greenamyre JT (2007) Mechanism of toxicity of pesticides acting at complex I: relevance to environmental etiologies of Parkinson’s disease. J Neurochem 100:1469–1479
Adam-Vizi V (2005) Production of reactive oxygen species in brain mitochondria: contribution by electron transport chain and non-electron transport chain sources. Antioxid Redox Signal 7:1140–1149
Tretter L, Sipos I, Adam-Vizi V (2004) Initiation of neuronal damage by complex I deficiency and oxidative stress in Parkinson’s disease. Neurochem Res 29:569–577
Billingsley KJ, Bandres-Ciga S, Saez-Atienzar S, Singleton AB (2018) Genetic risk factors in Parkinson’s disease. Cell Tissue Res 373:9–20
Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R (1997) Mutation in the α-synuclein gene identified in families with Parkinson's disease. Science 276:2045–2047
Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392:605–608
Valente EM, Bentivoglio AR, Dixon PH, Ferraris A, Ialongo T, Frontali M, Albanese A, Wood NW (2001) Localization of a novel locus for autosomal recessive early-onset parkinsonism, PARK6, on human chromosome 1p35-p36. The American Journal of Human Genetics 68:895–900
Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB (2004) Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44:601–607
van den Munckhof P, Luk KC, Ste-Marie L, Montgomery J, Blanchet PJ, Sadikot AF, Drouin J (2003) Pitx3 is required for motor activity and for survival of a subset of midbrain dopaminergic neurons Development 130(11)2535-2542
Tanner CM, Ottman R, Goldman SM, Ellenberg J, Chan P, Mayeux R, Langston JW (1999) Parkinson disease in twins: An etiologic study. JAMA 281:341–346
Sellbach AN, Boyle RS, Silburn PA, Mellick GD (2006) Parkinson’s disease and family history. Parkinsonism Relat Disord 12:399–409
Bandres-Ciga S, Diez-Fairen M, Kim JJ, Singleton AB (2020) Genetics of Parkinson’s disease: An introspection of its journey towards precision medicine. Neurobiol Dis 137:104782
Reed X, Bandrés-Ciga S, Blauwendraat C, Cookson MR (2019) The role of monogenic genes in idiopathic Parkinson’s disease. Neurobiol Dis 124:230–239
Müller-Nedebock AC, van Der Westhuizen FH, Kõks S, Bardien S (2021) Nuclear genes associated with mitochondrial DNA processes as contributors to Parkinson’s disease risk. Mov Disord 36:815–831
Buneeva O, Fedchenko V, Kopylov A, Medvedev A (2020) Mitochondrial dysfunction in Parkinson's disease: Focus on mitochondrial DNA. Biomedicines 8(12):591
Pichaud N, Bérubé R, Côté G, Belzile C, Dufresne F, Morrow G, Tanguay RM, Rand DM, Blier PU (2019) Age dependent dysfunction of mitochondrial and ROS metabolism induced by mitonuclear mismatch. Front Genet 10:130
Watanabe K (2010) Unique features of animal mitochondrial translation systems–The non-universal genetic code, unusual features of the translational apparatus and their relevance to human mitochondrial diseases. Proceedings of the Japan Academy, Series B 86:11–39
Taanman J-W (1999) The mitochondrial genome: structure, transcription, translation and replication. Biochim Biophys Acta 1410:103–123
Torriani SFF, Penselin D, Knogge W, Felder M, Taudien S, Platzer M, McDonald BA, Brunner PC (2014) Comparative analysis of mitochondrial genomes from closely related Rhynchosporium species reveals extensive intron invasion. Fungal Genet Biol 62:34–42
Xu S, Schaack S, Seyfert A, Choi E, Lynch M, Cristescu ME (2012) High mutation rates in the mitochondrial genomes of Daphnia pulex. Mol Biol Evol 29:763–769
Andalib S, Vafaee MS, Gjedde A (2014) Parkinson’s disease and mitochondrial gene variations: A review. J Neurol Sci 346:11–19
Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH, Jaros E, Hersheson JS, Betts J, Klopstock T (2006) High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet 38:515–517
Gu G, Reyes PE, Golden GT, Woltjer RL, Hulette C, Montine TJ, Zhang J (2002) Mitochondrial DNA deletions/rearrangements in parkinson disease and related neurodegenerative disorders. J Neuropathol Exp Neurol 61:634–639
Ekstrand MI, Terzioglu M, Galter D, Zhu S, Hofstetter C, Lindqvist E, Thams S, Bergstrand A, Hansson FS, Trifunovic A (2007) Progressive parkinsonism in mice with respiratory-chain-deficient dopamine neurons. Proc Natl Acad Sci 104:1325–1330
Luoma P, Melberg A, Rinne JO, Kaukonen JA, Nupponen NN, Chalmers RM, Oldfors A, Rautakorpi I, Peltonen L, Majamaa K, Somer H, Suomalainen A (2004) Parkinsonism, premature menopause, and mitochondrial DNA polymerase gamma mutations: clinical and molecular genetic study. Lancet 364:875–882
Quinn PMJ, Moreira PI, Ambrósio AF, Alves CH (2020) PINK1/PARKIN signalling in neurodegeneration and neuroinflammation. Acta Neuropathol Commun 8:189
Selvaraj S, Piramanayagam S (2019) Impact of gene mutation in the development of Parkinson’s disease. Genes Diseases 6:120–128
Alafifi T, Bakhsh ARA, Elbashari M, Abouelnaga MEH, Eldimllawi AM (2020) A novel mutation of PARK-2 gene in a patient with early-onset Parkinson’s disease. Oman Med J 35:e140
Ge P, Dawson VL, Dawson TM (2020) PINK1 and Parkin mitochondrial quality control: A source of regional vulnerability in Parkinson’s disease. Mol Neurodegener 15:20
Picca A, Mankowski RT, Burman JL, Donisi L, Kim J-S, Marzetti E, Leeuwenburgh C (2018) Mitochondrial quality control mechanisms as molecular targets in cardiac ageing. Nat Rev Cardiol 15:543–554
Johnson BN, Berger AK, Cortese GP, Lavoie MJ (2012) The ubiquitin E3 ligase parkin regulates the proapoptotic function of Bax. Proc Natl Acad Sci U S A 109:6283–6288
Oczkowska A, Kozubski W, Lianeri M, Dorszewska J (2013) Mutations in PRKN and SNCA genes important for the progress of Parkinson’s disease. Curr Genomics 14:502–517
Li T, Kou D, Cui Y, Le W (2020) Whole exome sequencing identified a new compound heterozygous PRKN mutation in a Chinese family with early-onset Parkinson’s disease. Bioscience reports 40
Ibáñez P, Lesage S, Lohmann E, Thobois S, De Michele G, Borg M, Agid Y, Dürr A, Brice A (2006) Mutational analysis of the PINK1 gene in early-onset parkinsonism in Europe and North Africa. Brain 129:686–694
Yu W, Sun Y, Guo S, Lu B (2011) The PINK1/Parkin pathway regulates mitochondrial dynamics and function in mammalian hippocampal and dopaminergic neurons. Hum Mol Genet 20:3227–3240
Morais VA, Verstreken P, Roethig A, Smet J, Snellinx A, Vanbrabant M, Haddad D, Frezza C, Mandemakers W, Vogt-Weisenhorn D (2009) Parkinson’s disease mutations in PINK1 result in decreased Complex I activity and deficient synaptic function. EMBO Mol Med 1:99–111
Antipova D, Bandopadhyay R (2017) Expression of DJ-1 in Neurodegenerative Disorders. Adv Exp Med Biol 1037:25–43
Pantcheva P, Elias M, Duncan K, Borlongan CV, Tajiri N, Kaneko Y (2014) The role of DJ-1 in the oxidative stress cell death cascade after stroke. Neural Regen Res 9:1430–1433
Ma MW, Wang J, Zhang Q, Wang R, Dhandapani KM, Vadlamudi RK, Brann DW (2017) NADPH oxidase in brain injury and neurodegenerative disorders. Mol Neurodegener 12:7
Kasten M, Hartmann C, Hampf J, Schaake S, Westenberger A, Vollstedt EJ, Balck A, Domingo A, Vulinovic F, Dulovic M, Zorn I, Madoev H, Zehnle H, Lembeck CM, Schawe L, Reginold J, Huang J, König IR, Bertram L, Marras C, Lohmann K, Lill CM, Klein C (2018) Genotype-Phenotype Relations for the Parkinson’s Disease Genes Parkin, PINK1, DJ1: MDSGene Systematic Review. Mov Disord 33:730–741
Funayama M, Hasegawa K, Kowa H, Saito M, Tsuji S, Obata F (2002) A new locus for Parkinson’s disease (PARK8) maps to chromosome 12p11. 2–q13. 1. Ann Neurol 51:296–301
Hewitt VL, Whitworth AJ (2017) Chapter 3: Mitochondrial fission and fusion. In: Verstreken P (ed) Parkinson’s Disease. Academic Press, San Diego, pp 77–111
Rubio JP, Topp S, Warren L, St.JeanWegmannKessnerNovembreShenFraserAponte PLDDJJDJ (2012) Deep sequencing of the LRRK2 gene in 14,002 individuals reveals evidence of purifying selection and independent origin of the p. Arg1628Pro mutation in Europe. Hum Mutat 33:1087–1098
Hernandez DG, Reed X, Singleton AB (2016) Genetics in Parkinson disease: Mendelian versus non-Mendelian inheritance. J Neurochem 139:59–74
Gasser T (2004) Genetics of Parkinson’s disease. Dialogues Clin Neurosci 6:295–301
Taguchi K, Watanabe Y, Tsujimura A, Tanaka M (2019) Expression of α-synuclein is regulated in a neuronal cell type-dependent manner. Anat Sci Int 94:11–22
Luth ES, Stavrovskaya IG, Bartels T, Kristal BS, Selkoe DJ (2014) Soluble, prefibrillar α-synuclein oligomers promote complex I-dependent, Ca2+-induced mitochondrial dysfunction. J Biol Chem 289:21490–21507
Parihar MS, Parihar A, Fujita M, Hashimoto M, Ghafourifar P (2009) Alpha-synuclein overexpression and aggregation exacerbates impairment of mitochondrial functions by augmenting oxidative stress in human neuroblastoma cells. Int J Biochem Cell Biol 41:2015–2024
Ragonese P, Salemi G, Morgante L, Aridon P, Epifanio A, Buffa D, Scoppa F, Savettieri G (2003) A case-control study on cigarette, alcohol, and coffee consumption preceding Parkinson’s disease. Neuroepidemiology 22:297–304
Ascherio A, Weisskopf MG, O’Reilly EJ, McCullough ML, Calle EE, Rodriguez C, Thun MJ (2004) Coffee consumption, gender, and Parkinson’s disease mortality in the cancer prevention study II cohort: The modifying effects of estrogen. Am J Epidemiol 160:977–984
Palacios N, Gao X, O’Reilly E, Schwarzschild M, McCullough ML, Mayo T, Gapstur SM, Ascherio AA (2012) Alcohol and risk of Parkinson’s disease in a large, prospective cohort of men and women. Mov Disord 27:980–987
Collier TJ, Kanaan NM, Kordower JH (2011) Ageing as a primary risk factor for Parkinson’s disease: evidence from studies of non-human primates. Nat Rev Neurosci 12:359–366
Park JS, Davis RL, Sue CM (2018) Mitochondrial dysfunction in Parkinson’s disease: New mechanistic insights and therapeutic perspectives. Curr Neurol Neurosci Rep 18:21
VanItallie TB (2008) Parkinson disease: primacy of age as a risk factor for mitochondrial dysfunction. Metabolism 57:S50–S55
Michikawa Y, Mazzucchelli F, Bresolin N, Scarlato G, Attardi G (1999) Aging-dependent large accumulation of point mutations in the human mtDNA control region for replication. Science 286:774–779
Youngmi Kim P, Jeong JH (2010) Mitochondria: The secret chamber of therapeutic targets for age-associated degenerative diseases. Biomolecules & Therapeutics 18:235–245
Rango M, Bresolin N (2018) Brain mitochondria, aging, and Parkinson's disease. Genes (Basel) 9(5):250
Zhang Z-X, Román GC (1993) Worldwide occurrence of Parkinson’s disease: An updated review. Neuroepidemiology 12:195–208
Cerri S, Mus L, Blandini F (2019) Parkinson’s disease in women and men: What’s the difference? J Parkinsons Dis 9:501–515
Wang Y-L, Wang Y-T, Li J-F, Zhang Y-Z, Yin H-L, Han B (2015) Body mass index and risk of Parkinson’s disease: A dose-response meta-analysis of prospective studies. PLoS ONE 10:e0131778
Kyrozis A, Ghika A, Stathopoulos P, Vassilopoulos D, Trichopoulos D, Trichopoulou A (2013) Dietary and lifestyle variables in relation to incidence of Parkinson’s disease in Greece. Eur J Epidemiol 28:67–77
Wright JJ, Biner O, Chung I, Burger N, Bridges HR, Hirst J (2022) Reverse electron transfer by respiratory complex I catalyzed in a modular proteoliposome system. J Am Chem Soc 144:6791–6801
Correia SC, Santos RX, Perry G, Zhu X, Moreira PI, Smith MA (2012) Mitochondrial importance in Alzheimer’s, Huntington’s and Parkinson’s diseases. Neurodegenerative diseases:205–221
Imaizumi Y, Okada Y, Akamatsu W, Koike M, Kuzumaki N, Hayakawa H, Nihira T, Kobayashi T, Ohyama M, Sato S (2012) Mitochondrial dysfunction associated with increased oxidative stress and α-synuclein accumulation in PARK2 iPSC-derived neurons and postmortem brain tissue. Mol Brain 5:1–13
Haque ME, Akther M, Azam S, Kim IS, Lin Y, Lee YH, Choi DK (2022) Targeting α-synuclein aggregation and its role in mitochondrial dysfunction in Parkinson’s disease. Br J Pharmacol 179:23–45
Hauser DN, Hastings TG (2013) Mitochondrial dysfunction and oxidative stress in Parkinson’s disease and monogenic parkinsonism. Neurobiol Dis 51:35–42
Martin LJ, Pan Y, Price AC, Sterling W, Copeland NG, Jenkins NA, Price DL, Lee MK (2006) Parkinson’s disease α-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J Neurosci 26:41–50
Dryanovski DI, Guzman JN, Xie Z, Galteri DJ, Volpicelli-Daley LA, Lee VM-Y, Miller RJ, Schumacker PT, Surmeier DJ (2013) Calcium entry and α-synuclein inclusions elevate dendritic mitochondrial oxidant stress in dopaminergic neurons. J Neurosci 33:10154–10164
Pukaß K, Richter-Landsberg C (2014) Oxidative stress promotes uptake, accumulation, and oligomerization of extracellular α-synuclein in oligodendrocytes. J Mol Neurosci 52:339–352
Xiang W, Schlachetzki JC, Helling S, Bussmann JC, Berlinghof M, Schäffer TE, Marcus K, Winkler J, Klucken J, Becker C-M (2013) Oxidative stress-induced posttranslational modifications of alpha-synuclein: Specific modification of alpha-synuclein by 4-hydroxy-2-nonenal increases dopaminergic toxicity. Mol Cell Neurosci 54:71–83
Müftüoglu M, Elibol B, Dalmızrak Ö, Ercan A, Kulaksız G, Ögüs H, Dalkara T, Özer N (2004) Mitochondrial complex I and IV activities in leukocytes from patients with parkin mutations. Movement disorders: official journal of the Movement Disorder Society 19:544–548
Wood-Kaczmar A, Gandhi S, Yao Z, Abramov AS, Miljan EA, Keen G, Stanyer L, Hargreaves I, Klupsch K, Deas E (2008) PINK1 is necessary for long term survival and mitochondrial function in human dopaminergic neurons. PLoS ONE 3:e2455
Rizor A, Pajarillo E, Johnson J, Aschner M, Lee E (2019) Astrocytic oxidative/nitrosative stress contributes to Parkinson’s disease pathogenesis: the dual role of reactive astrocytes. Antioxidants 8:265
Frøyset AK, Edson AJ, Gharbi N, Khan EA, Dondorp D, Bai Q, Tiraboschi E, Suster ML, Connolly JB, Burton EA (2018) Astroglial DJ-1 over-expression up-regulates proteins involved in redox regulation and is neuroprotective in vivo. Redox Biol 16:237–247
Di Nottia M, Masciullo M, Verrigni D, Petrillo S, Modoni A, Rizzo V, Di Giuda D, Rizza T, Niceta M, Torraco A (2017) DJ-1 modulates mitochondrial response to oxidative stress: clues from a novel diagnosis of PARK7. Clin Genet 92:18–25
Ludtmann MH, Abramov AY (2018) Mitochondrial calcium imbalance in Parkinson’s disease. Neurosci Lett 663:86–90
Carbone C, Costa A, Provensi G, Mannaioni G, Masi A (2017) The hyperpolarization-activated current determines synaptic excitability, calcium activity and specific viability of substantia nigra dopaminergic neurons. Front Cell Neurosci 11:187
Clark EH, de la Torre AV, Hoshikawa T, Briston T (2021) Targeting mitophagy in Parkinson's disease. J Biol Chem 296:100209
Bose A, Beal MF (2016) Mitochondrial dysfunction in Parkinson’s disease. J Neurochem 139:216–231
Pickrell AM, Youle RJ (2015) The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 85:257–273
Guo JF, Xiao B, Liao B, Zhang XW, Nie LL, Zhang YH, Shen L, Jiang H, Xia K, Pan Q (2008) Mutation analysis of Parkin, PINK1, DJ-1 and ATP13A2 genes in Chinese patients with autosomal recessive early-onset Parkinsonism. Mov Disord 23:2074–2079
Burchell VS, Nelson DE, Sanchez-Martinez A, Delgado-Camprubi M, Ivatt RM, Pogson JH, Randle SJ, Wray S, Lewis PA, Houlden H (2013) The Parkinson’s disease–linked proteins Fbxo7 and Parkin interact to mediate mitophagy. Nat Neurosci 16:1257–1265
Liu Y, Lear TB, Verma M, Wang KZ, Otero PA, McKelvey AC, Dunn SR, Steer E, Bateman NW, Wu C (2020) Chemical inhibition of FBXO7 reduces inflammation and confers neuroprotection by stabilizing the mitochondrial kinase PINK1. JCI insight 5
Zhou ZD, Lee JCT, Tan EK (2018) Pathophysiological mechanisms linking F-box only protein 7 (FBXO7) and Parkinson’s disease (PD). Mutat Res/Rev Mutat Res 778:72–78
Lesage S, Drouet V, Majounie E, Deramecourt V, Jacoupy M, Nicolas A, Cormier-Dequaire F, Hassoun SM, Pujol C, Ciura S (2016) Loss of VPS13C function in autosomal-recessive parkinsonism causes mitochondrial dysfunction and increases PINK1/Parkin-dependent mitophagy. The Am J Human Gen 98:500–513
Kumar N, Leonzino M, Hancock-Cerutti W, Horenkamp FA, Li P, Lees JA, Wheeler H, Reinisch KM, De Camilli P (2018) VPS13A and VPS13C are lipid transport proteins differentially localized at ER contact sites. J Cell Biol 217:3625–3639
Ludtmann MH, Angelova PR, Horrocks MH, Choi ML, Rodrigues M, Baev AY, Berezhnov AV, Yao Z, Little D, Banushi B (2018) α-synuclein oligomers interact with ATP synthase and open the permeability transition pore in Parkinson’s disease. Nat Commun 9:1–16
Kuo S-H, Tasset I, Cuervo AM, Sulzer D (2022) Misfolded GBA/β-glucocerebrosidase impairs ER-quality control by chaperone-mediated autophagy in Parkinson disease. Autophagy 18(12):3050-3052
Walter J, Bolognin S, Antony PM, Nickels SL, Poovathingal SK, Salamanca L, Magni S, Perfeito R, Hoel F, Qing X (2019) Neural stem cells of Parkinson’s disease patients exhibit aberrant mitochondrial morphology and functionality. Stem cell reports 12:878–889
Mortiboys H, Johansen KK, Aasly JO, Bandmann O (2010) Mitochondrial impairment in patients with Parkinson disease with the G2019S mutation in LRRK2. Neurology 75:2017–2020
Verma M, Callio J, Otero PA, Sekler I, Wills ZP, Chu CT (2017) Mitochondrial calcium dysregulation contributes to dendrite degeneration mediated by PD/LBD-associated LRRK2 mutants. J Neurosci 37:11151–11165
Cherra SJ III, Steer E, Gusdon AM, Kiselyov K, Chu CT (2013) Mutant LRRK2 elicits calcium imbalance and depletion of dendritic mitochondria in neurons. Am J Pathol 182:474–484
Su Y-C, Qi X (2013) Inhibition of excessive mitochondrial fission reduced aberrant autophagy and neuronal damage caused by LRRK2 G2019S mutation. Hum Mol Genet 22:4545–4561
Taipa R, Pereira C, Reis I, Alonso I, Bastos-Lima A, Melo-Pires M, Magalhães M (2016) DJ-1 linked parkinsonism (PARK7) is associated with Lewy body pathology. Brain 139:1680–1687
Joselin AP, Hewitt SJ, Callaghan SM, Kim RH, Chung Y-H, Mak TW, Shen J, Slack RS, Park DS (2012) ROS-dependent regulation of Parkin and DJ-1 localization during oxidative stress in neurons. Hum Mol Genet 21:4888–4903
Salazar C, Ruiz-Hincapie P, Ruiz LM (2018) The interplay among PINK1/PARKIN/Dj-1 network during mitochondrial quality control in cancer biology: Protein interaction analysis. Cells 7:154
Thomas KJ, McCoy MK, Blackinton J, Beilina A, van der Brug M, Sandebring A, Miller D, Maric D, Cedazo-Minguez A, Cookson MR (2011) DJ-1 acts in parallel to the PINK1/parkin pathway to control mitochondrial function and autophagy. Hum Mol Genet 20:40–50
Do CB, Tung JY, Dorfman E, Kiefer AK, Drabant EM, Francke U, Mountain JL, Goldman SM, Tanner CM, Langston JW (2011) Web-based genome-wide association study identifies two novel loci and a substantial genetic component for Parkinson’s disease. PLoS Genet 7:e1002141
Vos M, Klein C (2021) The importance of Drosophila melanogaster research to uncover cellular pathways underlying Parkinson’s disease. Cells 10:579
Ivatt RM, Sanchez-Martinez A, Godena VK, Brown S, Ziviani E, Whitworth AJ (2014) Genome-wide RNAi screen identifies the Parkinson disease GWAS risk locus SREBF1 as a regulator of mitophagy. Proc Natl Acad Sci 111:8494–8499
Schlame M, Brody S, Hostetler KY (1993) Mitochondrial cardiolipin in diverse eukaryotes: Comparison of biosynthetic reactions and molecular acyl species. Eur J Biochem 212:727–733
Li X-X, Tsoi B, Li Y-F, Kurihara H, He R-R (2015) Cardiolipin and its different properties in mitophagy and apoptosis. J Histochem Cytochem 63:301–311
Vos M, Geens A, Böhm C, Deaulmerie L, Swerts J, Rossi M, Craessaerts K, Leites EP, Seibler P, Rakovic A (2017) Cardiolipin promotes electron transport between ubiquinone and complex I to rescue PINK1 deficiency. J Cell Biol 216:695–708
Ryan T, Bamm VV, Stykel MG, Coackley CL, Humphries KM, Jamieson-Williams R, Ambasudhan R, Mosser DD, Lipton SA, Harauz G (2018) Cardiolipin exposure on the outer mitochondrial membrane modulates α-synuclein. Nat Commun 9:1–17
Rocha EM, De Miranda B, Sanders LH (2018) Alpha-synuclein: Pathology, mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurobiol Dis 109:249–257
Bayır H, Kapralov AA, Jiang J, Huang Z, Tyurina YY, Tyurin VA, Zhao Q, Belikova NA, Vlasova II, Maeda A (2009) Peroxidase mechanism of lipid-dependent cross-linking of synuclein with cytochrome c. J Biol Chem 284:15951–15969
Baum T, Gama V (2021) Dynamic properties of mitochondria during human corticogenesis. Development 148:dev194183
Valdinocci D, Simões RF, Kovarova J, Cunha-Oliveira T, Neuzil J, Pountney DL (2019) Intracellular and intercellular mitochondrial dynamics in Parkinson’s disease. Front Neurosci 13:930
Yang D, Ying J, Wang X, Zhao T, Yoon S, Fang Y, Zheng Q, Liu X, Yu W, Hua F (2021) Mitochondrial dynamics: a key role in neurodegeneration and a potential target for neurodegenerative disease. Front Neurosci 15:654785
Wasner K, Smajic S, Ghelfi J, Delcambre S, Prada‐Medina CA, Knappe E, Arena G, Mulica P, Agyeah G, Rakovic A (2022) Parkin Deficiency Impairs Mitochondrial DNA Dynamics and Propagates Inflammation. Mov Disord 37(7):1405-1415
Gonçalves FB, Morais VA (2021) PINK1: a bridge between mitochondria and Parkinson’s disease. Life 11:371
Malpartida AB, Williamson M, Narendra DP, Wade-Martins R, Ryan BJ (2021) Mitochondrial dysfunction and mitophagy in Parkinson’s disease: from mechanism to therapy. Trends Biochem Sci 46:329–343
Williams ET, Chen X, Moore DJ (2017) VPS35, the retromer complex and Parkinson’s disease. J Parkinsons Dis 7:219–233
Chan SL, Tan E-K (2017) Targeting LRRK2 in Parkinson’s disease: an update on recent developments. Expert Opin Ther Targets 21:601–610
Wang X, Petrie TG, Liu Y, Liu J, Fujioka H, Zhu X (2012) Parkinson’s disease-associated DJ-1 mutations impair mitochondrial dynamics and cause mitochondrial dysfunction. J Neurochem 121:830–839
Pozo Devoto VM, Falzone TL (2017) Mitochondrial dynamics in Parkinson’s disease: a role for α-synuclein? Dis Model Mech 10:1075–1087
Chandra G, Shenoi R, Anand R, Rajamma U, Mohanakumar K (2019) Reinforcing mitochondrial functions in aging brain: An insight into Parkinson’s disease therapeutics. J Chem Neuroanat 95:29–42
Gureev AP, Shaforostova EA, Popov VN (2019) Regulation of mitochondrial biogenesis as a way for active longevity: interaction between the Nrf2 and PGC-1α signaling pathways. Front Genet 10:435
Lin C-Y, Huang Y-N, Fu R-H, Liao Y-H, Kuo T-Y, Tsai C-W (2021) Promotion of mitochondrial biogenesis via the regulation of PARIS and PGC-1α by parkin as a mechanism of neuroprotection by carnosic acid. Phytomedicine 80:153369
Wang Y, Chen C, Huang W, Huang M, Wang J, Chen X, Ye Q (2019) Beneficial effects of PGC-1α in the substantia nigra of a mouse model of MPTP-induced dopaminergic neurotoxicity. Aging (Albany NY) 11:8937
Zheng L, Bernard-Marissal N, Moullan N, D’Amico D, Auwerx J, Moore DJ, Knott G, Aebischer P, Schneider BL (2017) Parkin functionally interacts with PGC-1α to preserve mitochondria and protect dopaminergic neurons. Hum Mol Genet 26:582–598
Jiang H, Kang S-U, Zhang S, Karuppagounder S, Xu J, Lee Y-K, Kang B-G, Lee Y, Zhang J, Pletnikova O (2016) Adult conditional knockout of PGC-1α leads to loss of dopamine neurons. eNeuro 3(4)
Lee Y, Stevens DA, Kang S-U, Jiang H, Lee Y-I, Ko HS, Scarffe LA, Umanah GE, Kang H, Ham S (2017) PINK1 primes Parkin-mediated ubiquitination of PARIS in dopaminergic neuronal survival. Cell Rep 18:918–932
Dickey AS, Spada ARL (2016) Transcription modulation of mitochondrial function and related pathways as a therapeutic opportunity in parkinson’s disease. In: Mitochondrial Mechanisms of Degeneration and Repair in Parkinson's Disease. Springer, pp 231–253
Pirooznia SK, Yuan C, Khan MR, Karuppagounder SS, Wang L, Xiong Y, Kang SU, Lee Y, Dawson VL, Dawson TM (2020) PARIS induced defects in mitochondrial biogenesis drive dopamine neuron loss under conditions of parkin or PINK1 deficiency. Mol Neurodegener 15:1–21
Litwiniuk A, Baranowska-Bik A, Domańska A, Kalisz M, Bik W (2021) Contribution of mitochondrial dysfunction combined with NLRP3 inflammasome activation in selected neurodegenerative diseases. Pharmaceuticals 14:1221
Haque ME, Akther M, Jakaria M, Kim IS, Azam S, Choi DK (2020) Targeting the microglial NLRP3 inflammasome and its role in Parkinson’s disease. Mov Disord 35:20–33
Bliederhaeuser C, Grozdanov V, Speidel A, Zondler L, Ruf WP, Bayer H, Kiechle M, Feiler MS, Freischmidt A, Brenner D (2016) Age-dependent defects of alpha-synuclein oligomer uptake in microglia and monocytes. Acta Neuropathol 131:379–391
Panicker N, Sarkar S, Harischandra DS, Neal M, Kam T-I, Jin H, Saminathan H, Langley M, Charli A, Samidurai M (2019) Fyn kinase regulates misfolded α-synuclein uptake and NLRP3 inflammasome activation in microglia. J Exp Med 216:1411–1430
Yan Y-Q, Fang Y, Zheng R, Pu J-L, Zhang B-R (2020) NLRP3 Inflammasomes in Parkinson’s disease and their Regulation by Parkin. Neuroscience 446:323–334
Gladkova C, Maslen SL, Skehel JM, Komander D (2018) Mechanism of parkin activation by PINK1. Nature 559:410–414
Zhou R, Yazdi AS, Menu P, Tschopp J (2011) A role for mitochondria in NLRP3 inflammasome activation. Nature 469:221–225
Panicker N, Kam T-I, Wang H, Neifert S, Chou S-C, Kumar M, Brahmachari S, Jhaldiyal A, Hinkle JT, Akkentli F (2022) Neuronal NLRP3 is a parkin substrate that drives neurodegeneration in Parkinson’s disease. Neuron 110(15):2422-2437
Qiu Z, He Y, Ming H, Lei S, Leng Y, Xia Z-y (2019) Lipopolysaccharide (LPS) aggravates high glucose-and hypoxia/reoxygenation-induced injury through activating ROS-dependent NLRP3 inflammasome-mediated pyroptosis in H9C2 cardiomyocytes. Journal of diabetes research 2019
Lee E, Hwang I, Park S, Hong S, Hwang B, Cho Y, Son J, Yu J-W (2019) MPTP-driven NLRP3 inflammasome activation in microglia plays a central role in dopaminergic neurodegeneration. Cell Death Differ 26:213–228
Han C, Shen H, Yang Y, Sheng Y, Wang J, Li W, Zhou X, Guo L, Zhai L, Guan Q (2020) Antrodia camphorata polysaccharide resists 6-OHDA-induced dopaminergic neuronal damage by inhibiting ROS-NLRP3 activation. Brain and Behavior 10:e01824
Erekat NS (2018) Apoptosis and its Role in Parkinson’s Disease. Exon Publications:65–82
Wolf P, Schoeniger A, Edlich F (2022) Pro-apoptotic complexes of BAX and BAK on the outer mitochondrial membrane. Biochim Biophys Acta:119317
Galluzzi L, Blomgren K, Kroemer G (2009) Mitochondrial membrane permeabilization in neuronal injury. Nat Rev Neurosci 10:481–494
He C, Bassik MC, Moresi V, Sun K, Wei Y, Zou Z, An Z, Loh J, Fisher J, Sun Q (2012) Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature 481:511–515
Rostovtseva TK, Gurnev PA, Protchenko O, Hoogerheide DP, Yap TL, Philpott CC, Lee JC, Bezrukov SM (2015) α-Synuclein shows high affinity interaction with voltage-dependent anion channel, suggesting mechanisms of mitochondrial regulation and toxicity in Parkinson disease. J Biol Chem 290:18467–18477
Nakano M, Imamura H, Sasaoka N, Yamamoto M, Uemura N, Shudo T, Fuchigami T, Takahashi R, Kakizuka A (2017) ATP maintenance via two types of ATP regulators mitigates pathological phenotypes in mouse models of Parkinson’s disease. EBioMedicine 22:225–241
Bennett JP Jr, Keeney PM (2020) Alzheimer’s and Parkinson’s brain tissues have reduced expression of genes for mtDNA OXPHOS Proteins, mitobiogenesis regulator PGC-1α protein and mtRNA stabilizing protein LRPPRC (LRP130). Mitochondrion 53:154–157
Wei H, Zhang Z, Saha A, Peng S, Chandra G, Quezado Z, Mukherjee AB (2011) Disruption of adaptive energy metabolism and elevated ribosomal p-S6K1 levels contribute to INCL pathogenesis: Partial rescue by resveratrol. Hum Mol Genet 20:1111–1121
Ferretta A, Gaballo A, Tanzarella P, Piccoli C, Capitanio N, Nico B, Annese T, Di Paola M, Dell'Aquila C, De Mari M, Ferranini E, Bonifati V, Pacelli C, Cocco T (2014) Effect of resveratrol on mitochondrial function: Implications in parkin-associated familiar Parkinson's disease. Biochim Biophys Acta 1842:902–915
Lehmann S, Loh SH, Martins LM (2017) Enhancing NAD(+) salvage metabolism is neuroprotective in a PINK1 model of Parkinson’s disease. Biology open 6:141–147
Lin HY, Liou CW, Chen SD, Hsu TY, Chuang JH, Wang PW, Huang ST, Tiao MM, Chen JB, Lin TK, Chuang YC (2015) Mitochondrial transfer from Wharton’s jelly-derived mesenchymal stem cells to mitochondria-defective cells recaptures impaired mitochondrial function. Mitochondrion 22:31–44
Clark EH, Vázquez de la Torre A, Hoshikawa T, Briston T (2021) Targeting mitophagy in Parkinson’s disease. J Biol Chem 296:100209
Gureev AP, Shaforostova EA, Popov VN (2019) Regulation of mitochondrial biogenesis as a way for active longevity: Interaction between the Nrf2 and PGC-1α signaling pathways. Fronti Gen 10
Cheng L, Chen L, Wei X, Wang Y, Ren Z, Zeng S, Zhang X, Wen H, Gao C, Liu H (2018) NOD2 promotes dopaminergic degeneration regulated by NADPH oxidase 2 in 6-hydroxydopamine model of Parkinson’s disease. J Neuroinflammation 15:243
Gao HM, Liu B, Hong JS (2003) Critical role for microglial NADPH oxidase in rotenone-induced degeneration of dopaminergic neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience 23:6181–6187
Cristóvão AC, Choi DH, Baltazar G, Beal MF, Kim YS (2009) The role of NADPH oxidase 1-derived reactive oxygen species in paraquat-mediated dopaminergic cell death. Antioxid Redox Signal 11:2105–2118
Langley M, Ghosh A, Charli A, Sarkar S, Ay M, Luo J, Zielonka J, Brenza T, Bennett B, Jin H, Ghaisas S, Schlichtmann B, Kim D, Anantharam V, Kanthasamy A, Narasimhan B, Kalyanaraman B, Kanthasamy AG (2017) Mito-apocynin prevents mitochondrial dysfunction, microglial activation, oxidative damage, and progressive neurodegeneration in MitoPark Transgenic Mice. Antioxid Redox Signal 27:1048–1066
Ghosh A, Langley MR, Harischandra DS, Neal ML, Jin H, Anantharam V, Joseph J, Brenza T, Narasimhan B, Kanthasamy A, Kalyanaraman B, Kanthasamy AG (2016) Mitoapocynin treatment protects against neuroinflammation and dopaminergic neurodegeneration in a preclinical animal model of Parkinson’s disease. J Neuroimmune Pharmacol 11:259–278
Xi Y, Feng D, Tao K, Wang R, Shi Y, Qin H, Murphy MP, Yang Q, Zhao G (2018) MitoQ protects dopaminergic neurons in a 6-OHDA induced PD model by enhancing Mfn2-dependent mitochondrial fusion via activation of PGC-1α. Biochim Biophys Acta 1864:2859–2870
Schapira AHV (2007) CHAPTER 71 - PARKINSON’S DISEASE. In: Schapira AHV, Byrne E, DiMauro S, Frackowiak RSJ, Johnson RT, Mizuno Y, Samuels MA, Silberstein SD, Wszolek ZK (eds) Neurology and Clinical Neuroscience. Mosby, Philadelphia, pp 927–960
Tolkovsky AM (2009) Mitophagy. Biochim Biophys Acta 1793:1508–1515
Lee JJ, Sanchez-Martinez A, Martinez Zarate A, Benincá C, Mayor U, Clague MJ, Whitworth AJ (2018) Basal mitophagy is widespread in Drosophila but minimally affected by loss of Pink1 or parkin. J Cell Biol 217:1613–1622
Cornelissen T, Vilain S, Vints K, Gounko N, Verstreken P, Vandenberghe W (2018) Deficiency of parkin and PINK1 impairs age-dependent mitophagy in Drosophila. eLife 7
Masaldan S, Callegari S, Dewson G (2022) Therapeutic targeting of mitophagy in Parkinson’s disease. Biochem Soc Trans 50:783–797
Lin MW, Lin CC, Chen YH, Yang HB, Hung SY (2019) Celastrol inhibits dopaminergic neuronal death of Parkinson's disease through activating mitophagy. Antioxidants (Basel, Switzerland) 9
Gilkerson R, De La Torre P, St. Vallier S (2021) Mitochondrial OMA1 and OPA1 as gatekeepers of organellar structure/function and cellular stress response. Fronti Cell Dev Biol 9
Niu K, Fang H, Chen Z, Zhu Y, Tan Q, Wei D, Li Y, Balajee AS, Zhao Y (2020) USP33 deubiquitinates PRKN/parkin and antagonizes its role in mitophagy. Autophagy 16:724–734
Wall CE, Rose CM, Adrian M, Zeng YJ, Kirkpatrick DS, Bingol B (2019) PPEF2 opposes PINK1-mediated mitochondrial quality control by dephosphorylating ubiquitin. Cell Rep 29:3280-3292.e3287
Bolam JP, Pissadaki EK (2012) Living on the edge with too many mouths to feed: why dopamine neurons die. Mov Disord 27:1478–1483
Parrado-Fernández C, Schreiner B, Ankarcrona M, Conti MM, Cookson MR, Kivipelto M, Cedazo-Mínguez Á, Sandebring-Matton A (2018) Reduction of PINK 1 or DJ-1 impair mitochondrial motility in neurites and alter ER-mitochondria contacts. J Cell Mol Med 22:5439–5449
Gao J, Wang L, Liu J, Xie F, Su B, Wang X (2017) Abnormalities of mitochondrial dynamics in neurodegenerative diseases. Antioxidants 6:25
Rappold PM, Cui M, Grima JC, Fan RZ, de Mesy-Bentley KL, Chen L, Zhuang X, Bowers WJ, Tieu K (2014) Drp1 inhibition attenuates neurotoxicity and dopamine release deficits in vivo. Nat Commun 5:1–13
Bido S, Soria FN, Fan RZ, Bezard E, Tieu K (2017) Mitochondrial division inhibitor-1 is neuroprotective in the A53T-α-synuclein rat model of Parkinson’s disease. Sci Rep 7:1–13
Hunn BH, Cragg SJ, Bolam JP, Spillantini M-G, Wade-Martins R (2015) Impaired intracellular trafficking defines early Parkinson’s disease. Trends Neurosci 38:178–188
Esteves A, Gozes I, Cardoso S (2014) The rescue of microtubule-dependent traffic recovers mitochondrial function in Parkinson's disease. Biochim Biophys Acta 1842:7–21
Jornayvaz FR, Shulman GI (2010) Regulation of mitochondrial biogenesis. Essays Biochem 47:69–84
Anis E, Zafeer MF, Firdaus F, Islam SN, Anees Khan A, Ali A, Hossain MM (2020) Ferulic acid reinstates mitochondrial dynamics through PGC1α expression modulation in 6-hydroxydopamine lesioned rats. Phytotherapy research : PTR 34:214–226
Kumar M, Acevedo-Cintrón J, Jhaldiyal A, Wang H, Andrabi SA, Eacker S, Karuppagounder SS, Brahmachari S, Chen R, Kim H, Ko HS, Dawson VL, Dawson TM (2020) Defects in mitochondrial biogenesis drive mitochondrial alterations in PARKIN-deficient human dopamine neurons. Stem Cell Reports 15:629–645
Ivankovic D, Chau KY, Schapira AH, Gegg ME (2016) Mitochondrial and lysosomal biogenesis are activated following PINK1/parkin-mediated mitophagy. J Neurochem 136:388–402
Siddiqui A, Bhaumik D, Chinta SJ, Rane A, Rajagopalan S, Lieu CA, Lithgow GJ, Andersen JK (2015) Mitochondrial quality control via the PGC1α-TFEB signaling pathway is compromised by Parkin Q311X mutation but independently restored by rapamycin. J Neurosci 35:12833–12844
Davinelli S, Sapere N, Visentin M, Zella D, Scapagnini G (2013) Enhancement of mitochondrial biogenesis with polyphenols: combined effects of resveratrol and equol in human endothelial cells. Immunity & Ageing 10:28
Chandra G, Kundu M, Rangasamy sb, Dasarathy S, Ghosh S, Watson R, Pahan K (2018) Increase in mitochondrial biogenesis in neuronal cells by RNS60, a physically-modified saline, via phosphatidylinositol 3-kinase-mediated upregulation of PGC1α. J Neuroimmun Pharmacol 13(2): 143-162
Mishra A, Singh S, Tiwari V, Bano S, Shukla S (2020) Dopamine D1 receptor agonism induces dynamin related protein-1 inhibition to improve mitochondrial biogenesis and dopaminergic neurogenesis in rat model of Parkinson’s disease. Behav Brain Res 378:112304
Ferreira AFF, Binda KH, Singulani MP, Pereira CPM, Ferrari GD, Alberici LC, Real CC, Britto LR (2020) Physical exercise protects against mitochondria alterations in the 6-hidroxydopamine rat model of Parkinson’s disease. Behav Brain Res 387:112607
Koo J-H, Cho J-Y, Lee U-B (2017) Treadmill exercise alleviates motor deficits and improves mitochondrial import machinery in an MPTP-induced mouse model of Parkinson’s disease. Exp Gerontol 89:20–29
Irrcher I, Adhihetty PJ, Sheehan T, Joseph AM, Hood DA (2003) PPARgamma coactivator-1alpha expression during thyroid hormone- and contractile activity-induced mitochondrial adaptations. Am J Physiol Cell Physiol 284:C1669-1677
Shanmughapriya S, Langford D, Natarajaseenivasan K (2020) Inter and intracellular mitochondrial trafficking in health and disease. Ageing Res Rev 62:101128
Scheiblich H, Dansokho C, Mercan D, Schmidt SV, Bousset L, Wischhof L, Eikens F, Odainic A, Spitzer J, Griep A (2021) Microglia jointly degrade fibrillar alpha-synuclein cargo by distribution through tunneling nanotubes. Cell 184(5089–5106):e5021
Chang J-C, Wu S-L, Liu K-H, Chen Y-H, Chuang C-S, Cheng F-C, Su H-L, Wei Y-H, Kuo S-J, Liu C-S (2016) Allogeneic/xenogeneic transplantation of peptide-labeled mitochondria in Parkinson’s disease: restoration of mitochondria functions and attenuation of 6-hydroxydopamine–induced neurotoxicity. Transl Res 170(40–56):e43
Shi X, Zhao M, Fu C, Fu A (2017) Intravenous administration of mitochondria for treating experimental Parkinson’s disease. Mitochondrion 34:91–100
Chang C-Y, Liang M-Z, Chen L (2019) Current progress of mitochondrial transplantation that promotes neuronal regeneration. Translational neurodegeneration 8:1–12
Chang J-C, Chao Y-C, Chang H-S, Wu Y-L, Chang H-J, Lin Y-S, Cheng W-L, Lin T-T, Liu C-S (2021) Intranasal delivery of mitochondria for treatment of Parkinson’s Disease model rats lesioned with 6-hydroxydopamine. Sci Rep 11:1–14
Zakrzewski W, Dobrzyński M, Szymonowicz M, Rybak Z (2019) Stem cells: past, present, and future. Stem Cell Res Ther 10:68
Stoker TB, Greenland JC (2018) Parkinson’s disease: pathogenesis and clinical aspects [internet].
Cheng X-Y, Biswas S, Li J, Mao C-J, Chechneva O, Chen J, Li K, Li J, Zhang J-R, Liu C-F (2020) Human iPSCs derived astrocytes rescue rotenone-induced mitochondrial dysfunction and dopaminergic neurodegeneration in vitro by donating functional mitochondria. Translat Neurodegen 9:1–14
Ying H, Ebrahimi M, Keivan M, Khoshnam SE, Salahi S, Farzaneh M (2021) miRNAs; a novel strategy for the treatment of COVID-19. Cell Biol Int 45:2045–2053
Nilsen TW (2007) Mechanisms of microRNA-mediated gene regulation in animal cells. Trends Genet 23:243–249
Nies YH, Mohamad Najib NH, Lim WL, Kamaruzzaman MA, Yahaya MF, Teoh SL (2021) MicroRNA dysregulation in Parkinson’s disease: a narrative review. Front Neurosci 15:660379
Sanders LH, Greenamyre JT (2013) Oxidative damage to macromolecules in human Parkinson disease and the rotenone model. Free Radical Biol Med 62:111–120
Hoss AG, Labadorf A, Beach TG, Latourelle JC, Myers RH (2016) microRNA profiles in Parkinson’s disease prefrontal cortex. Frontiers in aging neuroscience 8:36
Parker WD Jr, Parks JK, Swerdlow RH (2008) Complex I deficiency in Parkinson’s disease frontal cortex. Brain Res 1189:215–218
Zheng B, Liao Z, Locascio J, Lesniak K, Roderick S, Watt M (2010) A Potential Therapeutic Target for Early Intervention in Parkinson’s Disease. Sci Transl Med 2:52–73
Wang H, Ye Y, Zhu Z, Mo L, Lin C, Wang Q, Wang H, Gong X, He X, Lu G (2016) MiR-124 regulates apoptosis and autophagy process in MPTP model of Parkinson’s disease by targeting to Bim. Brain Pathol 26:167–176
Xiong R, Wang Z, Zhao Z, Li H, Chen W, Zhang B, Wang L, Wu L, Li W, Ding J (2014) MicroRNA-494 reduces DJ-1 expression and exacerbates neurodegeneration. Neurobiol Aging 35:705–714
Miñones-Moyano E, Porta S, Escaramís G, Rabionet R, Iraola S, Kagerbauer B, Espinosa-Parrilla Y, Ferrer I, Estivill X, Martí E (2011) MicroRNA profiling of Parkinson’s disease brains identifies early downregulation of miR-34b/c which modulate mitochondrial function. Hum Mol Genet 20:3067–3078
Cho HJ, Liu G, Jin SM, Parisiadou L, Xie C, Yu J, Sun L, Ma B, Ding J, Vancraenenbroeck R (2013) MicroRNA-205 regulates the expression of Parkinson’s disease-related leucine-rich repeat kinase 2 protein. Hum Mol Genet 22:608–620
Prajapati P, Sripada L, Singh K, Bhatelia K, Singh R, Singh R (2015) TNF-α regulates miRNA targeting mitochondrial complex-I and induces cell death in dopaminergic cells. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease 1852:451–461
Chaudhuri AD, Choi DC, Kabaria S, Tran A, Junn E (2016) MicroRNA-7 regulates the function of mitochondrial permeability transition pore by targeting VDAC1 expression. J Biol Chem 291:6483–6493
Indrieri A, Carrella S, Romano A, Spaziano A, Marrocco E, Fernandez-Vizarra E, Barbato S, Pizzo M, Ezhova Y, Golia FM (2019) miR-181a/b downregulation exerts a protective action on mitochondrial disease models. EMBO Mol Med 11:e8734
Serafin A, Foco L, Zanigni S, Blankenburg H, Picard A, Zanon A, Giannini G, Pichler I, Facheris MF, Cortelli P (2015) Overexpression of blood microRNAs 103a, 30b, and 29a in l-dopa–treated patients with PD. Neurology 84:645–653
Ebert MS, Neilson JR, Sharp PA (2007) MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nat Methods 4:721–726
Cressatti M, Juwara L, Galindez JM, Velly AM, Nkurunziza ES, Marier S, Canie O, Gornistky M, Schipper HM (2020) Salivary microR-153 and microR-223 levels as potential diagnostic biomarkers of idiopathic Parkinson’s disease. Mov Disord 35:468–477
Catanesi M, d’Angelo M, Tupone MG, Benedetti E, Giordano A, Castelli V, Cimini A (2020) MicroRNAs dysregulation and mitochondrial dysfunction in neurodegenerative diseases. Int J Mol Sci 21:5986
Surmeier DJ (2018) Determinants of dopaminergic neuron loss in Parkinson’s disease. FEBS J 285:3657–3668
Johnstone DM, Hamilton C, Gordon LC, Moro C, Torres N, Nicklason F, Stone J, Benabid A-L, Mitrofanis J (2021) Exploring the use of intracranial and extracranial (remote) photobiomodulation devices in Parkinson’s disease: A comparison of direct and indirect systemic stimulations. J Alzheimers Dis 83:1399–1413
Zhang J, Yuan L, Zhang X, Hamblin M, Zhu T, Meng F, Li Y, Chen Y, Yin K (2016) Altered long non-coding RNA transcriptomic profiles in brain microvascular endothelium after cerebral ischemia. Exp Neurol 277:162–170
Mitrofanis J (2019) Run in the Light: Exploring exercise and photobiomodulation in Parkinson's disease. Morgan & Claypool Publishers
Enengl J, Hamblin MR, Dungel P (2020) Photobiomodulation for Alzheimer’s disease: Translating basic research to clinical application. J Alzheimers Dis 75:1073–1082
Johnstone DM, Moro C, Stone J, Benabid A-L, Mitrofanis J (2016) Turning on lights to stop neurodegeneration: the potential of near infrared light therapy in Alzheimer’s and Parkinson’s disease. Front Neurosci 9:500
Hamblin MR (2016) Shining light on the head: photobiomodulation for brain disorders. BBA clinical 6:113–124
Lima PL, Pereira CV, Nissanka N, Arguello T, Gavini G, da Costa Maranduba CM, Diaz F, Moraes CT (2019) Photobiomodulation enhancement of cell proliferation at 660 nm does not require cytochrome c oxidase. J Photochem Photobiol, B 194:71–75
Sommer AP, Haddad MK, Fecht H-J (2015) Light effect on water viscosity: implication for ATP biosynthesis. Sci Rep 5:1–6
Wang Y, Huang Y-Y, Wang Y, Lyu P, Hamblin MR (2017) Photobiomodulation of human adipose-derived stem cells using 810 nm and 980 nm lasers operates via different mechanisms of action. Biochim Biophys Acta 1861:441–449
Xu C, Zhang J, Mihai DM, Washington I (2014) Light-harvesting chlorophyll pigments enable mammalian mitochondria to capture photonic energy and produce ATP. J Cell Sci 127:388–399
Xuan W, Agrawal T, Huang L, Gupta GK, Hamblin MR (2015) Low-level laser therapy for traumatic brain injury in mice increases brain derived neurotrophic factor (BDNF) and synaptogenesis. J Biophotonics 8:502–511
Stone J, Mitrofanis J, Johnstone DM, Falsini B, Bisti S, Adam P, Nuevo AB, George-Weinstein M, Mason R, Eells J (2018) Acquired resilience: an evolved system of tissue protection in mammals. Dose-Response 16:1559325818803428
Salehpour F, Hamblin MR (2020) Photobiomodulation for Parkinson’s disease in animal models: A systematic review. Biomolecules 10:610
Liang HL, Whelan HT, Eells JT, Wong-Riley MT (2008) Near-infrared light via light-emitting diode treatment is therapeutic against rotenone-and 1-methyl-4-phenylpyridinium ion-induced neurotoxicity. Neuroscience 153:963–974
Ying R, Liang HL, Whelan HT, Eells JT, Wong-Riley MT (2008) Pretreatment with near-infrared light via light-emitting diode provides added benefit against rotenone-and MPP+-induced neurotoxicity. Brain Res 1243:167–173
Trimmer PA, Schwartz KM, Borland MK, De Taboada L, Streeter J, Oron U (2009) Reduced axonal transport in Parkinson’s disease cybrid neurites is restored by light therapy. Mol Neurodegener 4:1–11
Oueslati A, Lovisa B, Perrin J, Wagnières G, van den Bergh H, Tardy Y, Lashuel HA (2015) Photobiomodulation suppresses alpha-synuclein-induced toxicity in an AAV-based rat genetic model of Parkinson’s disease. PLoS ONE 10:e0140880
Johnstone D, El Massri N, Moro C, Spana S, Wang X, Torres N, Chabrol C, De Jaeger X, Reinhart F, Purushothuman S (2014) Indirect application of near infrared light induces neuroprotection in a mouse model of parkinsonism–an abscopal neuroprotective effect. Neuroscience 274:93–101
Kim B, Mitrofanis J, Stone J, Johnstone DM (2018) Remote tissue conditioning is neuroprotective against MPTP insult in mice. IBRO reports 4:14–17
Hamilton CL, El Khoury H, Hamilton D, Nicklason F, Mitrofanis J (2019) “Buckets”: early observations on the use of red and infrared light helmets in Parkinson’s disease patients. Photobiomodul Photomed Laser Surg 37:615–622
Santos L, del Olmo-Aguado S, Valenzuela PL, Winge K, Iglesias-Soler E, Argüelles-Luis J, Álvarez-Valle S, Parcero-Iglesias GJ, Fernández-Martínez A, Lucia A (2019) Photobiomodulation in Parkinson’s disease: A randomized controlled trial. Brain Stimul 12:810–812
Foo ASC, Soong TW, Yeo TT, Lim K-L (2020) Mitochondrial dysfunction and Parkinson’s disease—Near-infrared photobiomodulation as a potential therapeutic strategy. Front Aging Neuroscie 12:89
Farfara D, Tuby H, Trudler D, Doron-Mandel E, Maltz L, Vassar RJ, Frenkel D, Oron U (2015) Low-level laser therapy ameliorates disease progression in a mouse model of Alzheimer’s disease. J Mol Neurosci 55:430–436
Ferreira AFF, Binda KH, Singulani MP, Pereira CPM, Ferrari GD, Alberici LC, Real CC, Britto LR (2020) Physical exercise protects against mitochondria alterations in the 6-hidroxydopamine rat model of Parkinson’s disease. Behav Brain Res 387:112607
Nhu NT, Cheng Y-J, Lee S-D (2021) Effects of treadmill exercise on neural mitochondrial functions in Parkinson’s disease: A ssystematic review of animal studies. Biomedicines 9:1011
Chuang C-S, Chang J-C, Cheng F-C, Liu K-H, Su H-L, Liu C-S (2017) Modulation of mitochondrial dynamics by treadmill training to improve gait and mitochondrial deficiency in a rat model of Parkinson’s disease. Life Sci 191:236–244
Tuon T, Souza PS, Santos MF, Pereira FT, Pedroso GS, Luciano TF, De Souza CT, Dutra RC, Silveira PC, Pinho RA (2015) Physical training regulates mitochondrial parameters and neuroinflammatory mechanisms in an experimental model of Parkinson’s disease. Oxid Med Cell Longev 2015
Curtis WM, Seeds WA, Mattson MP, Bradshaw PC (2022) NADPH and mitochondrial quality control as targets for a circadian-based fasting and exercise therapy for the treatment of Parkinson’s disease. Cells 11:2416
Lai J-H, Chen K-Y, Wu JC-C, Olson L, Brené S, Huang C-Z, Chen Y-H, Kang S-J, Ma K-H, Hoffer BJ (2019) Voluntary exercise delays progressive deterioration of markers of metabolism and behavior in a mouse model of Parkinson’s disease. Brain Res 1720:146301
Burtscher J, Millet GP, Place N, Kayser B, Zanou N (2021) The muscle-brain axis and neurodegenerative diseases: The key role of mitochondria in exercise-induced neuroprotection. Int J Mol Sci 22:6479
Bajracharya R, Ballard JWO (2018) Dietary management and physical exercise can improve climbing defects and mitochondrial activity in Drosophila melanogaster parkin null mutants. Fly 12:95–104
Agnihotri A, Aruoma OI (2020) Alzheimer’s disease and Parkinson’s disease: a nutritional toxicology perspective of the impact of oxidative stress, mitochondrial dysfunction, nutrigenomics and environmental chemicals. J Am Coll Nutr 39:16–27
Tarnopolsky MA, Beal MF (2001) Potential for creatine and other therapies targeting cellular energy dysfunction in neurological disorders. Ann Neurol 49:561–574
Kones R (2010) Parkinson’s disease: mitochondrial molecular pathology, inflammation, statins, and therapeutic neuroprotective nutrition. Nutr Clin Pract 25:371–389
Bajracharya R, Youngson NA, Ballard JWO (2019) Dietary macronutrient management to treat mitochondrial dysfunction in Parkinson’s disease. Int J Mol Sci 20:1850
Kann O, Kovács R (2007) Mitochondria and neuronal activity. Am J Physiol Cell Physiol 292:C641–C657
Dache ZAA, Otandault A, Tanos R, Pastor B, Meddeb R, Sanchez C, Arena G, Lasorsa L, Bennett E, Grange T (2020) Blood contains circulating cell free respiratory competent mitochondria. FASEB J 34:3616–3630
Caicedo A, Zambrano K, Sanon S, Gavilanes AW (2021) Extracellular mitochondria in the cerebrospinal fluid (CSF): Potential types and key roles in central nervous system (CNS) physiology and pathogenesis. Mitochondrion 58:255–269
Haddad D, Nakamura K (2015) Understanding the susceptibility of dopamine neurons to mitochondrial stressors in Parkinson’s disease. FEBS Lett 589:3702–3713
Dias V, Junn E, Mouradian MM (2013) The role of oxidative stress in Parkinson’s disease. J Parkinsons Dis 3:461–491
Suomalainen A, Battersby BJ (2018) Mitochondrial diseases: the contribution of organelle stress responses to pathology. Nat Rev Mol Cell Biol 19:77–92
Tucker D, Lu Y, Zhang Q (2018) From mitochondrial function to neuroprotection—an emerging role for methylene blue. Mol Neurobiol 55:5137–5153
Prasuhn J, Davis RL, Kumar KR (2021) Targeting mitochondrial impairment in Parkinson’s disease: challenges and opportunities. Frontiers in cell and developmental biology 8:615461
Titova N, Chaudhuri KR (2017) Personalized medicine in Parkinson’s disease: time to be precise. Mov Disord 32:1147
Kabaria S, Choi DC, Chaudhuri AD, Mouradian MM, Junn E (2015) Inhibition of miR-34b and miR-34c enhances α-synuclein expression in Parkinson’s disease. FEBS Lett 589:319–325
Funding
No funding.
Author information
Authors and Affiliations
Contributions
Conceptualization: S.M.V. and A.N.; writing-original draft preparation: All authors; writing-review and editing: S.E.K.; supervision: S.E.K. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Moradi Vastegani, S., Nasrolahi, A., Ghaderi, S. et al. Mitochondrial Dysfunction and Parkinson’s Disease: Pathogenesis and Therapeutic Strategies. Neurochem Res 48, 2285–2308 (2023). https://doi.org/10.1007/s11064-023-03904-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-023-03904-0