
Abstract
Chronic intermittent hypobaric hypoxia (CIHH) has protective effects on heart and brain against ischemia injury through mobilizing endogenous adaptive mechanisms. However, whether CIHH prevents against cognitive impairment was not elucidated. The present study aimed to investigate the effect and mechanism of CIHH treatment on ischemia/reperfusion (IR)-induced cognitive dysfunction. Mice were randomly divided into 8 groups: Control, Sham, CIHH (simulating 5000 m high-altitude for 28 days, 6 h per day), IR (three 16-min occlusions of bilateral common carotid arteries interrupted by two 10-min intervals), CIHH + IR, PD98059 (inhibitor of MEK1/2) + CIHH + IR, PD98059 + Sham and PD98059 + IR group. Morris water maze and step-down passive avoidance tests were performed to evaluate the capability of learning and memory 1 month after ischemia. Thionine dyeing was to examine histological manifestations of pyramidal neurons in hippocampus CA1 region. Western blotting assay was for measurement of the protein expressions in ERK1/2-CREB-BDNF signaling pathway. There were a shorter escape latency and a longer percentage of time retaining in the target quadrant in Morris water maze test, fewer times of errors in the step-down avoidance test and a higher neuronal density of the hippocampal CA1 subfield in CIHH + IR group than in IR group. CIHH upregulated the expressions of BDNF, phosphorylated CREB, ERK1/2 and TrkB with or without ischemia. The protective effects of CIHH were abolished by PD98059 administration 15 min before ischemia. CIHH ameliorated ischemia-induced cognitive dysfunction through activation of ERK1/2-CREB-BDNF signaling pathway.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Emerging findings have indicated the critical role of cerebral ischemia in the pathogenesis of learning and memory dysfunction because it induces neuronal cell death [1, 2]. Many interventions have been explored to preserve the ability of learning and memory via increasing the neuron survival during ischemia/reperfusion (I/R), such as clearing excessive glutamate, maintaining redox balance or modulating neurotrophic factors.
The key role of Brain-derived neurotrophic factor (BDNF) has been widely discussed in the process of synaptic plasticity and learning by enhancing long-term potentiation [3], the cellular basis of learning and memory in the hippocampus [4, 5]. It is involved in the strengthening of synaptic transmission by binding of BDNF to the tropomyosin-related kinase B (TrkB) receptor [6]. Interestingly, cognitive impairment resulting from BDNF deficiency was rescued by reintroduction of BDNF. BDNF levels have previously been proved associated with cognitive impairment of schizophrenia patients [7, 8], suggesting that BDNF as a potential biomarker of learning and memory [6, 9]. TrkB activation by BDNF stimulates intracellular signaling cascades involved in plasticity, such as the extracellular signal-regulated kinases (ERK)/MAPK pathway [10]. The consolidation of recognition memory was associated with BDNF release in the dentate gyrus (DG), which was associated with significant increase in ERK activation and c-fos expression [11]. The transcription factor of BDNF is phosphorylated cyclic AMP response element binding protein (CREB). CREB interacts with the transcription co-activator, CREB-binding protein, to initiate transcription and translation of CREB target genes, which are required for synaptic plasticity, learning and memory [12–14]. Therefore, enhancing BDNF pathway serves as a potential target for cognitive improvement.
Accumulating evidence demonstrate that chronic intermittent hypobaric hypoxia (CIHH) has various beneficial actions on human being and has been widely used in athletes’ training to enhance the tolerance to anoxia [15]. Furthermore, CIHH exerts an anti-hypertension effect [15, 16] and protects heart and liver against I/R or hypoxia/reoxygenation injuries [17–19]. Recent study suggested the involvement of glial glutamate and glucocorticoid receptor in CIHH-mediated protection against brain IR injury [20, 21]. However, whether CIHH has protective effects on cognitive impairments induced by cerebral ischemia in mice remains unknown.
The purpose of this study is to investigate whether CIHH can ameliorate learning and memory dysfunction by ischemia/reperfusion via modulating ERK1/2-CREB-BDNF pathway.
Materials and Methods
Drug and Chemicals
Protease inhibitor and phosphoric acid protease inhibitors were purchased from Roche Mannheim (#: 17669600, Germany). PVDF filters and the enhanced chemiluminescence detecting reagents were from Millipore Corporation (Billerica, MA, USA). The antibodies to CREB (#: 9197) and p-CREB (#: 9198) were from Cell Signaling (Danvers, MA, USA). BDNF (#: ab108319) and PD98059 (#: ab146592) were supplied by Abcam (Cambridge, MA, USA). p-TrkB (for detecting phosphorylated at Tyr705, #: 11328-1, Signalway Antibody) and TrkB (#: sc-12) from Santa Cruz Biotechnology (Santa Cruz, CA, USA), ERK1/2 (#: 05-1152) and p-ERK1/2 (for detecting ERK1/2 MAPKs phosphorylated at Thr202/Tyr204 and Thr185/Tyr187, Millipore, #: 05-797R) from Millipore Corporation and a rabbit antibody to GAPDH was from Sigma-Aldrich (#: G8795, St. Louis, MO, USA). Biotin labeled anti-rabbit IgG was from KPL Scaffold Inc (#: 074-1506, La Mirada, CA, USA) and anti-mouse IgG was from Earthox life sciences (#: E030110-01, CA, USA). Sodium nitroprusside was purchased from Beijing Double-Crane Pharmaceutical Co., Ltd (Beijing, China). Other drugs were from Sigma-Aldrich.
Animals and Protocols
Male ICR mice (weighing 20–30 g) were purchased from Beijing Vital River Laboratory Animal Center. The mice were allowed access to water and food ad libitum. The environment was maintained at a constant temperature and humidity with a natural light:dark cycle (12:12 h). Animal experiments were carried out in accordance to the National Institutes of Health guide for care and use of laboratory animals (NIH Publications No. 8023, revised 1978).
One hundred and forty-five mice were randomly assigned into 8 groups: (1) Control group: mice without surgery and treatment. (2) Sham group: mice subjected only to surgery but no occlusion of the bilateral common carotid arteries. (3) CIHH group: mice exposed to hypobaric hypoxia simulating 5000 m altitude (PB = 53.87 kPa, PO2 = 11.2 kPa) in a hypobaric chamber for 28 days, 6 h/day from 10:00 AM to 4:00 PM. The following three protocols were applied to this group. In 35 mice, the dynamic expressions of BDNF, p-TrkB, TrkB, CREB and p-CREB were examined at 0, 12 h, 1, 2, 3, 7 and 14 day post 28-day CIHH by western bolt analysis. In 5 mice, the hippocampus was harvested 4 d after CIHH to compare the expression of related proteins among groups. In the remaining 10 mice, the mice were subjected to behavioral test and subsequent neuropathology evaluation. (4) Ischemia/reperfusion (IR) group: mice were challenged by bilateral common carotid arteries occlusion for three 16 min with two intervals of 10 min. Five samples of hippocampus were harvested 3 day after I/R for proteins assay among groups. Ten mice were used for behavioral test 30 day after I/R and then the hippocampus was kept to evaluate the histological changes. (5) CIHH + IR group: mice were given I/R 1 day after CIHH treatment. The following procedure is similar with IR group. (6) PD98059 + CIHH + IR group: 5 µl PD98059 (inhibitor of MEK1/2, 2 mmol/L) dissolved in 2% DMSO was microinjected into the right lateral cerebral ventricle at coordinate −0.9 mm anteroposterior, ±1.5 mm mediolateral, and −3.5 mm dorsoventral from the bregma 15 min before I/R [22]. The subsequent procedure is similar with CIHH + IR group. (7) PD98059 + Sham group, the procedure applied to Sham group except for PD98059. (8) PD98059 + IR group: Similar procedure with IR group was performed except for PD98059. The schematic of the experiment was illustrated in Fig. 1.

The schematic of the experiment. Con control group, Sham sham group, CIHH CIHH treatment group, IR ischemia and reperfusion group, MWM Morris water maze test, Step Step-down avoidance test, NE neuropathological evaluation, PD98059 inhibitor of MEK1/2. The red arrows represent the time point for operation in Sham or IR groups. The square head arrow represents the sample collecting for western bolt analysis. The letters of a, b, c, d, e, f, g and h with the square head arrow in CIHH group represent the different time points of sample collecting
Ischemia/Reperfusion
The modified model of acquired cognitive impairment is established as described previously [23]. Briefly, the ICR mice were anesthetized via intraperitoneal injection with 350 mg/kg 10% chloral hydrate. After the bilateral carotid arteries were separated, a thread was passed below each carotid artery for facilitate the closure. Sodium nitroprusside was intraperitoneally injected (3.5 mg/kg) to cause hypotension. Bilateral carotid arteries were occluded by arteriole clips for 16 min (ischemia), and subsequently loosened for 10 min (reperfusion). This process was repeated for three times. Core body temperature was maintained at 37.0 ± 0.5 °C during surgery with heated blanket. Brain temperature was monitored with a tympanic membrane probe into the ear and maintained at 36.5 ± 0.2 °C. The mice were then placed in cages for rearing.
Morris Water Maze Test
The Morris water maze (Jiliang Software Technology Co., Ltd Shanghai, China) consisted of a large circular pool (120 cm in diameter, 60 cm in height, water depth of 45 cm, and water temperature 21 ± 1 °C). A 6-cm-diameter platform was submerged 1.5 cm below the water surface in the center of one quadrant. The mice received 6 trials a day for 5 consecutive days and performance was assessed by measuring the time (escape latency) to find the hidden platform. Each mouse was placed in one of three quadrants (not containing the platform) in a random manner, with the head facing the wall. If the mouse could not find the escape platform within 60 s, the experimenter gently guided it onto the platform and allowed it to stay there for 10 s. The mouse was then dried and placed in a cage for a rest of at least 1 h. Spatial memory was then analyzed by the probe trial on the 6th day, during which the platform was removed from the pool, and the animal was tracked during a 60-s exploration of the pool. Memory was then inferred based on the percentages of time spent in the target quadrant searching for the platform.
Step-Down Avoidance Test
A passive avoidance reaction tank, 11 × 11 × 35 cm in size (Jiliang Software Technology Co., Ltd, Shanghai, China) was used as the jumping test device. An energized copper grid was placed on the bottom. An insulated rubber mat with diameter and height of 5 × 5 cm was placed on the right of the device, which was used as the safe platform for the mouse to avoid an electric shock. A voltage regulator was used to regulate the electric current. The mice were placed in the jumping device to adapt for 3 min and then a 0.25 mA alternating current was turned on to shock the mice. The mice jumped onto the safe platform to avoid electric shock. The frequency of electric shock (number of errors) within 5 min was recorded, which was considered as their learning performance. After 24 h, the test was repeated. The mice were placed on the safe platform and an electric shock was induced when mice jumped to energized copper grid. For this time the number of electric shocks was taken as the number of errors, which was considered as the memory performance.
Neuropathological Evaluation
One day after the finish of behavioral test, mice were anesthetized with chloral hydrate and perfused with normal saline followed by 4% paraformaldehyde through ascending aorta. A 2-mm-thick brain slice containing the bilateral dorsal hippocampus was excised coronally and fixed in 4% paraformaldehyde. Two days later, the slice was embedded in paraffin and sectioned (5 µm). The sections were stained with thionine. The DND (delayed neuronal death) of pyramidal neurons in the CA1 hippocampus was evaluated under light microscope and scored by histological grade (HG) and neuronal density (ND) [24]. HG was divided into four grades and the standard is as following: grade 0, no neuron death; grade I, scattered single neuron death; grade II, mass neuron death; grade III, almost complete neuron death. ND of the hippocampal CA1 subfield (i.e., the number of intact pyramidal cells per 1-mm linear length of CA1 stratum pyramidale) was determined according to the method of Kirino et al [25]. The average numbers of pyramidal neurons in three areas of CA1 hippocampus were calculated to establish the ND value.
Western Blotting
The hippocampus was quickly separated and homogenized in lysis buffer containing protease inhibitor and phosphoric acid protease inhibitors. The supernatant was kept at −80 °C and protein concentration was determined by Coomassie assay. The samples were electrophoresed on SDS–PAGE and transferred to PVDF membranes. After blocking in 5% defatted milk for 1 h, the membrane were incubated overnight at 4 °C with the rabbit anti-BDNF, CREB, p-CREB, TrkB, p-TrkB, ERK1/2 and p-ERK1/2 primary antibodies and a rabbit antibody to GAPDH. After washing with Tris Buffered Saline with Tween 20 (TBST), the membranes were incubated with biotin labeled anti-rabbit IgG for 1 h at 37 °C. After washing with TBST again, the signals developed with enhanced chemiluminescence system. Band images were quantified by the ratio of the band’s IOD of target band to that of GAPDH or the phosphorylated band to the total one.
Statistical Analyses
A Kruskal–Wallis analysis of variance on ranks with Dunn’s multiple comparisons test were performed to test for differences in HG between groups. Other data were presented as means ± SD, and were tested by one-way ANOVA, combined with a LSD test as a multiple comparison method to test differences between groups. P < 0.05 was considered significant.
Results
The Effect of CIHH on Cognitive Dysfunction Induced by Cerebral Repetitive I/R
There were no significant differences in physiologic parameters, such as body weight, blood pressure and heart rate during experiment among groups.
In the Morris water maze, the time of escape latency to find the submerged platform was analyzed from the first to the fifth day, and became shorter in all groups. No significant difference of latency time was detected between sham and CIHH group (22.21 ± 8.20 vs. 23.64 ± 10.84 s). Ischemia resulted in longer escape latency than sham group (40.06 ± 9.50 vs. 22.21 ± 8.20 s, p < 0.05) and CIHH pretreatment reversed the impairment partially (31.31 ± 8.65 vs. 40.06 ± 9.50 s, p < 0.05, Fig. 2a). In the spatial probe test, we plotted the performance of different groups by analyzing the percentage of time in the target quadrant where the hidden platform had previously been in. The mice in IR group showed a shorter percentage of time in the target quadrant than Sham group (24.11 ± 5.84 vs. 38.66 ± 7.87%, P < 0.05). The mice in CIHH + IR group was favorable to cognitive improvement in searching for the target quadrant compared with the IR group (33.80 ± 6.47 vs. 24.11 ± 5.84%, P < 0.05, Fig. 2b), yet no changes were observed between Sham and CIHH group.

Effects of CIHH on the escape latency (a), percentage time in target quadrant (b) and times of errors (c) in mice. Sham sham group, CIHH CIHH treatment group, IR ischemia and reperfusion group. Data were expressed as mean ± SD, n = 10 for each group, *P < 0.05 vs. sham group, # P < 0.05 vs. IR group, + P < 0.05 vs. CIHH + IR group
In the step-down avoidance test, Sham and CIHH group remained no change in learning and memory ability. IR group were coupled with increased number of errors than Sham group (5.3 ± 1.64 vs. 2.6 ± 0.84, P < 0.05), Which was reversed by CIHH pretreatment (3.7 ± 1.42 vs. 5.3 ± 1.64, P < 0.05, Fig. 2c).
These results suggested that cerebral ischemia could cause the learning and memory disability, while CIHH could protect the brain tissue against the cognitive disorder.
In order to test whether activation of ERK pathway was pivotal for the neuroprotective effect of CIHH, MAPK/ERK Kinase (MEK), the upstream kinase of ERK, was inhibited pharmacologically with PD098059 (2 mmol/L in 5 µl), a MEK inhibitor. Compared PD98059 + CIHH + IR group with CIHH + IR group, the escape latency is 38.062 ± 10.49733 vs. 31.31 ± 8.65 (p < 0.05), the percentage time retaining in the target quadrant is 26.11 ± 6.11 vs. 33.80 ± 6.47% (p < 0.05) and the number of errors is 4.9 ± 1.20 vs. 3.7 ± 1.42 (p < 0.05) (Fig. 2). The result showed that in the presence of the MEK inhibitors, the neuroprotective effects of CIHH against cognition impairment were obviously abrogated, suggesting that ERK pathway mediated the neuroprotective effect of CIHH against cognitive impairment produced by I/R.
The Effect of CIHH on Pyramidal Neurons in Hippocampal CA1 Region After Cerebral I/R
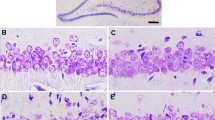
One day after the behavioral tests, the neuropathological evaluation on hippocampus CA1 was performed and the neuroprotective effect of CIHH against cerebral I/R was confirmed. The pyramidal neurons in the CA1 hippocampus were arranged in order with 2–3 cell layers, intact outlines, full nuclei, and clear nucleoli in sham rats. The HG values were 0, and ND was 261.21 ± 34.33 mm−1. The histological characteristics of the CA1 hippocampi in the CIHH group were similar to Sham group. However, almost all neurons died 30 days after ischemia in IR group, which were represented by increase in HG and significant decrease in ND (37.46 ± 18.21 vs. 261.21 ± 34.33 mm−1, P < 0.05) compared with Sham groups. The above noted lesions were prevented by CIHH pretreatment (ND: 210.33 ± 49.52 in CIHH + IR group vs. 37.46 ± 18.21 mm−1 in IR group).
With the presence of PD98059, the neuroprotective effect of CIHH against DND in hippocampus CA1 region was abolished. ND was much lower in PD98059 + CIHH + IR group than in CIHH + IR group (61.33 ± 25.27 vs. 210.33 ± 49.52 mm−1, P < 0.05, Fig. 3). The histological changes of neurons in hippocampus CA1 area were also examined upon PD98059 injection into Sham and IR animals, and there were no obvious effects (Fig. 3). These results indicated that CIHH could protect pyramidal neurons in the CA1 hippocampus against DND induced by ischemia via promoting ERK1/2 cascade.

Effects of CIHH on the histological grade and neuronal density in CA1 hippocampus. a Representative thionine-stained photomicrographs (Scale bar 400 µm in the first inset and 25 µm in others). b Group data of histological grade. Each dot represents one animal assigned that grade. c Group data of neuronal density. Sham sham group, CIHH CIHH treatment group, IR ischemia and reperfusion group, CIHH + IR CIHH + IR group, PD98059 + CIHH + IR PD98059 + CIHH + IR group, PD98059 + Sham PD98059 + Sham group, PD98059 + IR PD98059 + IR group. Data were expressed as mean ± SD, n = 8 for each group. *P < 0.05 vs. sham group, # P < 0.05 vs. IR group, + P < 0.05 vs. CIHH + IR group
Effects of CIHH Pretreatment on the Expressions of BDNF, p-TrkB and TrkB
The expression of BDNF was increased obviously at different time points at 0, 12 h, 1, 2, 3 and 7 day after 28-day CIHH treatment compared with control group. The maximal expression of BDNF was detected between 0 h and 2 day and then it declined and reached to control level at 14 day (Fig. 4a). The expressions of BDNF among groups were compared at 3 day after I/R or at corresponding time point in other groups. The expression of BDNF was significantly upregulated in IR and CIHH groups compared with Sham group,but the expression in CIHH + IR group was higher than that in IR group. The increased expression of BDNF was blocked in PD98059 + CIHH + IR group compared with CIHH + IR group even though PD98059 had no effect on BDNF in Sham and IR animals (Fig. 4b, c).

Effects of CIHH on the expressions of BDNF, p-TrkB and TrkB proteins. a The dynamic expressions of BDNF in time points of post-CIHH. b Effects of PD98059 on BDNF in Sham and IR animals. c The expressions of BDNF among groups. d The dynamic expressions of p-TrkB and TrkB in time points of post-CIHH. e The expressions of p-TrkB among groups. Samples used to compared the proteins expressions among groups were obtained 3 day after ischemia or at corresponding time points in groups. Sham sham group, CIHH CIHH treatment group, IR ischemia and reperfusion group, CIHH + IR CIHH + IR group, PD98059 + CIHH + IR PD98059 + CIHH + IR group. Data were expressed as mean ± SD, n = 5 for each group, *P < 0.05 vs. control or sham group, # P < 0.05 vs. IR group, + P < 0.05 vs. CIHH + IR group
The dynamic expression of TrkB, receptor of BDNF, was observed. The expression of p-TrkB was increased at time points at 0, 12 h, 1, 2 and 3 day after 28 day-CIHH treatment compared with control group, whereas TrkB expression unchanged. The increase was maintained from 0 h to 3 day, and decreased to control level at the 7 day (Fig. 4d). The expressions of TrkB among groups were also observed. The expression of TrkB was similar among groups, but the p-TrkB expression was increased in CIHH group and decreased in IR group compared with Sham group. The down-regulated expression of p-TrkB in IR group was restored in CIHH + IR group (Fig. 4e). The results suggested that the protective effect of CIHH on cognitive dysfunction induced by I/R might be carried out through upregulating the expression of BDNF and p-TrkB might be involved.
Effects of CIHH Pretreatment on the Expressions of p-ERK1/2, ERK1/2, p-CREB and CREB
No significant difference of ERK1/2 expression was detected among groups, but the expression of p-ERK was changed. The expression of p-ERK1/2 was upregulated by CIHH while downregulated by I/R compared with Sham group. Furthermore, the expression of p-ERK1/2 is higher in CIHH + IR group than in IR group. The increased expression of p-ERK 1/2 in CIHH + IR group was abolished by PD98059 (Fig. 5a).

Effects of CIHH on the expressions of p-ERK1/2, ERK1/2, p-CREB and CREB proteins. a The expressions of p-ERK1/2 and ERK1/2 among groups. b The dynamic expressions of p-CREB and CREB in time points of post-CIHH. c The expressions of p-CREB and CREB among groups. Samples used to compare the proteins expressions among groups were obtained 3 day after ischemia or at corresponding time points in groups. Sham Sham group, CIHH CIHH treatment group, IR ischemia and reperfusion group, CIHH + IR: CIHH + IR group, PD98059 + CIHH + IR: PD98059 + CIHH + IR group. Data were expressed as mean ± SD, n = 5 for each group. *P < 0.05 vs. sham group, # P < 0.05 vs. IR group, + P < 0.05 vs. CIHH + IR group
The expression of CREB was similar in every group, but the expression of p-CREB on ser-133 was increased by CIHH. The dynamic expression of p-CREB promoted obviously at time points of 0, 12 h, 1, 2, 3 and 7 day after 28-day CIHH treatment compared to control group. The maximal expression of p-CREB was between 0 h and 1 day time points, and then the expression of p-CREB decreased to control levels at 14 day (Fig. 5b).
Total CREB expressions were not significantly different among groups, but the expression of p-CREB on Ser 133 was increased by CIHH treatment. In addition, the downregulation of p-CREB induced by I/R were recovered when pretreatment with CIHH. Pretreated with PD98059, the up-regulated expression of p-CREB was reversed in PD98059 + CIHH + IR group, although there was no change in CREB expression (Fig. 5c). These results suggested that CIHH could enhance the expression of the related proteins in ERK1/2-CREB-BDNF-TrkB pathway and antagonize the downregulation of these proteins expression during I/R.
Discussion
This is the first study to investigate the protective effect of CIHH pretreatment on cognitive dysfunction induced by I/R and underlying mechanism in mice. The results showed that CIHH could ameliorate learning and memory impairment induced by repetitive cerebral I/R and antagonize DND in the hippocampus CA1. Additionly, CIHH promoted expressions of BDNF, p-TrkB, p-ERK1/2 and p-CREB in the hippocampus. Furthermore, CIHH could restore the changes of the proteins induced by I/R. Notablely, these effects were abrogated by blocking the activation of ERK1/2. So it demonstrated that CIHH has protective effect against learning and memory defects through activation of ERK1/2-CREB-BDNF pathway.
As a kind of stress stimulation, hypoxia is a double-edged sword. In this regard, severe hypoxia is involved in the genesis and prognosis of acute and chronic diseases such as diabetes, cardiovascular and cerebrovascular diseases. In contrast, the intensity and duration programed hypoxia produces a beneficial effect through an adaptive mechanism. CIHH has been reported to enhance athletic performance frequently through increasing tolerance to hypoxia [26]. CIHH can mobolize protective effect on heart against ischemia or hypoxia injury through triggering powerful endogenous adaptive mechanisms [27, 28]. Besides its beneficial effect on heart, limited research has been focused on its protective effect on brain [20], especially on cognition [29]. In this study, we demonstrated that learning and memory function of mice was severely impaired by repeated cerebral I/R combined with lower blood pressure, and CIHH protected against the learning and memory dysfunction by ischemia effectively for the first time. The promising results suggest a potential role of CIHH on prevention of cognitive dysfunction.
Previous studies have elucidated that the pathogenesis of vascular cognitive impairment is due to delayed neuronal death in the hippocampus CA1 area, and repetitive cerebral I/R is an important cause of vascular cognitive impairment [30]. In the hippocampus, the CA1, CA2/3 and DG areas are basic area involved in long-term memory formation [31]. Hippocampal neuronal death is a major factor in the progress of memory impairment in many brain disorders and prevention of hippocampal neuronal deaths provides a potential therapeutic strategy to ameliorate memory and cognitive impairment in many neurological disorders [32, 33]. In this study, Prevention of DND in hippocampus CA1 by CIHH pretreatment was consistent with the better behavioral performance. The result suggested that the benefits of CIHH on neuronal loss in hippocampus CA1 region caused by ischemia might be the basis of the restoration of learning and memory ability.
BDNF plays a crucial role in memory formation and storage. It is implicated in protein synthesis-dependent memory consolidation [34]. BDNF in the hippocampus is essential for the effects of various memory enhancing agents that promotes dendritic recovery and increases neuronal connectivity and memory facilitation [10, 35–37]. Infusion of BDNF and activation of BDNF signaling are promising targets for learning and memory enhancement and improvement of memory impairment observed in various brain disorders [38, 39]. In our study, CIHH elevated the expression of BDNF in hippocampus and reversed the downregulation of BDNF produced by ischemia. The results suggested that maintaining the expression of BDNF in neurons might be a reason of CIHH protective effect on cognitive dysfunction.
BDNF stimulates ERK-promoted activation of CREB, which is critical for long-term memory formation. At the same time, the activation of CREB could increase BDNF production [40, 41]. As an important transcription factor, CREB is implicated in control of adaptive neuronal responses and contributes to several critical functions of BDNF-mediated cell survival [42]. Specifically, phosphorylated CREB can activate the transcription of genes involved in cell survival and memory [36, 43]. It was reported that dephosphorylation of ERK1/2 and CREB could participate in neurocognitive impairments induced by chronic intermittent normoxic hypoxia simulating sleep apnea syndromes [44]. In this study, the expression of p-CREB and BDNF proteins were increased after CIHH. Furthermore, CIHH could reverse the downregulated expression of p-CREB and BDNF induced by repetitive ischemia. The result suggested the neuroprotective effect of CIHH might be mediated by activating the CREB signal pathway and promoting the expression of its downstream pro-survival gene, BDNF, in turn preventing neuronal death in hippocampus CA1 region and further preserving the learning and memory ability during I/R stress.
The upstream phosphorylating enzyme of phosphorylated CREB is ERK1/2 that is required for BDNF-induced increase in spine density on hippocampal pyramidal neurons and promotes memory [45]. ERK1/2, a key player in both upstream and downstream signaling of BDNF [12, 46], is activated by BDNF signaling through the Ras/Raf/MEK signaling cascade [47, 48]. Moreover, ERK1/2 activation may further generate BDNF or mediate the neurotropic actions of BDNF, including synaptic strength [49] and neurogenesis [50]. Previous work has shown that CREB can be a prosurvival target of ERK1/2 [51]. And BDNF, ERK1/2 and CREB compose a positive feedback to promote the production of each other [48, 49]. In our study, CIHH not only increased the expression of p-ERK1/2, but also reversed the downregulated expression induced by ischemia. And CIHH-induced upregulated expressions of BDNF and p-CREB were mostly blocked by PD98059 administration. Thus, it can be speculated that ERK1/2 may be an upstream signaling molecule involved in the CIHH-induced increase of BDNF and p-CREB expression.
It is reported that BDNF exerts its cognition-promoting effect mainly via combining the receptor of phosphorylated TrkB and activating MAPK/ERK or phosphatidylinositol-3-kinase and phosphorylated protein kinase B (PI3K/Akt) pathways [11]. Other studies have shown that activation of TrkB receptors elicits a complex program of changes in gene expression, which will cause multiple changes in the proteome [52]. In our study, CIHH augmented the expression of p-TrkB and it also reversed the decrease of the expression of p-TrkB induced by I/R. Therefore, the combined upregulation of BDNF and p-TrkB suggests that CIHH could modulate BDNF/TrkB pathway to exert cognition-promoting effect.
In summary, our results demonstrate for the first time that CIHH ameliorated I/R-induced cognitive dysfunction through activation of ERK1/2-CREB-BDNF signaling pathway in mice.
References
Lei Y, Guo Q, Li Y, Jiang H, Ni W, Gu Y (2014) Characteristics of cognitive impairment in adults with cerebral ischemia. Zhonghua Yi Xue Za Zhi 94:984–989
Briones TL, Woods J, Wadowska M (2014) Chronic neuroinflammation and cognitive impairment following transient global cerebral ischemia: role of fractalkine/CX3CR1 signaling. J Neuroinflammation 11:13. doi:10.1186/1742-2094-11-13
Lee YS, Silva AJ (2009) The molecular and cellular biology of enhanced cognition. Nat Rev Neurosci 10:126–140. doi:10.1038/nrn2572
Lipsky RH, Marini AM (2007) Brain-derived neurotrophic factor in neuronal survival and behavior-related plasticity. Ann N Y Acad Sci 1122:130–143. doi:10.1196/annals.1403.009
Okuyama S, Morita M, Sawamoto A, Terugo T, Nakajima M, Furukawa Y (2015) Edaravone enhances brain-derived neurotrophic factor production in the ischemic mouse brain. Pharmaceuticals (Basel) 8:176–185. doi:10.3390/ph8020176
Buckley PF, Pillai A, Howell KR (2011) Brain-derived neurotrophic factor: findings in schizophrenia. Curr Opin Psychiatry 24:122–127. doi:10.1097/YCO.0b013e3283436eb7
Niitsu T, Shirayama Y, Matsuzawa D, Hasegawa T, Kanahara N, Hashimoto T, Shiraishi T, Shiina A, Fukami G, Fujisaki M, Watanabe H, Nakazato M, Asano M, Kimura S, Hashimoto K, Iyo M (2011) Associations of serum brain-derived neurotrophic factor with cognitive impairments and negative symptoms in schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry 35:1836–1840. doi:10.1016/j.pnpbp.2011.09.004
Asevedo E, Gadelha A, Noto C, Mansur RB, Zugman A, Belangero SI, Berberian AA, Scarpato BS, Leclerc E, Teixeira AL, Gama CS, Bressan RA, Brietzke E (2013) Impact of peripheral levels of chemokines, BDNF and oxidative markers on cognition in individuals with schizophrenia. J Psychiatr Res 47:1376–1382. doi:10.1016/j.jpsychires.2013.05.032
Guo X, Chen ZH, Wang HL, Liu ZC, Wang XP, Zhou BH, Yang C, Zhang XP, Xiao L, Shu C, Chen JX, Wang GH (2015) WSKY, a traditional Chinese decoction, rescues cognitive impairment associated with NMDA receptor antagonism by enhancing BDNF/ERK/CREB signaling. Mol Med Rep 11:2927–2934. doi:10.3892/mmr.2014.3086
Bekinschtein P, Cammarota M, Igaz LM, Bevilaqua LR, Izquierdo I, Medina JH (2007) Persistence of long-term memory storage requires a late protein synthesis- and BDNF-dependent phase in the hippocampus. Neuron 53:261–277. doi:10.1016/j.neuron.2006.11.025
Callaghan CK, Kelly AM (2012) Differential BDNF signaling in dentate gyrus and perirhinal cortex during consolidation of recognition memory in the rat. Hippocampus 22:2127–2135. doi:10.1002/hipo.22033
Almeida RD, Manadas BJ, Melo CV, Gomes JR, Mendes CS, Graos MM, Carvalho RF, Carvalho AP, Duarte CB (2005) Neuroprotection by BDNF against glutamate-induced apoptotic cell death is mediated by ERK and PI3-kinase pathways. Cell Death Differ 12:1329–1343. doi:10.1038/sj.cdd.4401662
Cunha C, Brambilla R, Thomas KL (2010) A simple role for BDNF in learning and memory? Front Mol Neurosci 3:1. doi:10.3389/neuro.02.001.2010
Scott Bitner R (2012) Cyclic AMP response element-binding protein (CREB) phosphorylation: a mechanistic marker in the development of memory enhancing Alzheimer’s disease therapeutics. Biochem Pharmacol 83:705–714. doi:10.1016/j.bcp.2011.11.009
Roels B, Bentley DJ, Coste O, Mercier J, Millet GP (2007) Effects of intermittent hypoxic training on cycling performance in well-trained athletes. Eur J Appl Physiol 101:359–368. doi:10.1007/s00421-007-0506-8
Serebrovskaya TV, Manukhina EB, Smith ML, Downey HF, Mallet RT (2008) Intermittent hypoxia: cause of or therapy for systemic hypertension? Exp Biol Med (Maywood) 233:627–650. doi:10.3181/0710-mr-267
Zhang Y, Yang HT, Zhou ZN (2007) The cardioprotection of intermittent hypoxic adaptation. Sheng Li Xue Bao 59:601–613
Guan Y, Gao L, Ma HJ, Li Q, Zhang H, Yuan F, Zhou ZN, Zhang Y (2010) Chronic intermittent hypobaric hypoxia decreases beta-adrenoceptor activity in right ventricular papillary muscle. Am J Physiol Heart Circ Physiol 298:H1267–H1272. doi:10.1152/ajpheart.00410.2009
Zhu XH, Yan HC, Zhang J, Qu HD, Qiu XS, Chen L, Li SJ, Cao X, Bean JC, Chen LH, Qin XH, Liu JH, Bai XC, Mei L, Gao TM (2010) Intermittent hypoxia promotes hippocampal neurogenesis and produces antidepressant-like effects in adult rats. J Neurosci 30:12653–12663. doi:10.1523/jneurosci.6414-09.2010
Gong SJ, Chen LY, Zhang M, Gong JX, Ma YX, Zhang JM, Wang YJ, Hu YY, Sun XC, Li WB, Zhang Y (2012) Intermittent hypobaric hypoxia preconditioning induced brain ischemic tolerance by up-regulating glial glutamate transporter-1 in rats. Neurochem Res 37:527–537. doi:10.1007/s11064-011-0639-3
Rybnikova E, Mironova V, Pivina S, Tulkova E, Ordyan N, Nalivaeva N, Turner A, Samoilov M (2007) Involvement of the hypothalamic–pituitary–adrenal axis in the antidepressant-like effects of mild hypoxic preconditioning in rats. Psychoneuroendocrinology 32:813–823. doi:10.1016/j.psyneuen.2007.05.010
Yang Y, Zhang X, Cui H, Zhang C, Zhu C, Li L (2014) Apelin-13 protects the brain against ischemia/reperfusion injury through activating PI3K/Akt and ERK1/2 signaling pathways. Neurosci Lett 568:44–49. doi:10.1016/j.neulet.2014.03.037
Wang H (2014) Establishment of an animal model of vascular dementia. Exp Ther Med 8:1599–1603. doi:10.3892/etm.2014.1926
Kato H, Liu Y, Araki T, Kogure K (1991) Temporal profile of the effects of pretreatment with brief cerebral ischemia on the neuronal damage following secondary ischemic insult in the gerbil: cumulative damage and protective effects. Brain Res 553:238–242
Kirino T, Tamura A, Sano K (1986) A reversible type of neuronal injury following ischemia in the gerbil hippocampus. Stroke 17:455–459
Levine BD (2002) Intermittent hypoxic training: fact and fancy. High Alt Med Biol 3:177–193. doi:10.1089/15270290260131911
Zhuang J, Zhou Z (1999) Protective effects of intermittent hypoxic adaptation on myocardium and its mechanisms. Biol Signals Recept 8:316–322
Zhang Y, Zhou ZN (2012) Beneficial effects of intermittent hypobaric hypoxia on the body. Zhongguo Ying Yong Sheng Li Xue Za Zhi 28:504–9
Udayabanu M, Kumaran D, Nair RU, Srinivas P, Bhagat N, Aneja R, Katyal A (2008) Nitric oxide associated with iNOS expression inhibits acetylcholinesterase activity and induces memory impairment during acute hypobaric hypoxia. Brain Res 1230:138–149. doi:10.1016/j.brainres.2008.06.081
Wu L, Feng XT, Hu YQ, Tang N, Zhao QS, Li TW, Li HY, Wang QB, Bi XY, Cai XK (2015) Global gene expression profile of the hippocampus in a rat model of vascular dementia. Tohoku J Exp Med 237:57–67. doi:10.1620/tjem.237.57
Treves A, Tashiro A, Witter MP, Moser EI (2008) What is the mammalian dentate gyrus good for? Neuroscience 154:1155–1172. doi:10.1016/j.neuroscience.2008.04.073
Lee KY, Jeong EJ, Huh J, Cho N, Kim TB, Jeon BJ, Kim SH, Kim HP, Sung SH (2012) Cognition-enhancing and neuroprotective activities of the standardized extract of Betula platyphylla bark and its major diarylheptanoids. Phytomedicine 19:1315–1320. doi:10.1016/j.phymed.2012.09.012
Debette S (2013) Vascular risk factors and cognitive disorders. Rev Neurol (Paris) 169:757–764. doi:10.1016/j.neurol.2013.07.022
Mou L, Heldt SA, Ressler KJ (2011) Rapid brain-derived neurotrophic factor-dependent sequestration of amygdala and hippocampal GABA(A) receptors via different tyrosine receptor kinase B-mediated phosphorylation pathways. Neuroscience 176:72–85. doi:10.1016/j.neuroscience.2010.12.041
Aleisa AM, Alzoubi KH, Gerges NZ, Alkadhi KA (2006) Chronic psychosocial stress-induced impairment of hippocampal LTP: possible role of BDNF. Neurobiol Dis 22:453–462. doi:10.1016/j.nbd.2005.12.005
Kim J, Kwon JT, Kim HS, Josselyn SA, Han JH (2014) Memory recall and modifications by activating neurons with elevated CREB. Nat Neurosci 17:65–72. doi:10.1038/nn.3592
Kim DH, Kim JM, Park SJ, Cai M, Liu X, Lee S, Shin CY, Ryu JH (2012) GABA(A) receptor blockade enhances memory consolidation by increasing hippocampal BDNF levels. Neuropsychopharmacology 37:422–433. doi:10.1038/npp.2011.189
Kim HJ, Kim W, Kong SY (2013) Antidepressants for neuro-regeneration: from depression to Alzheimer’s disease. Arch Pharm Res 36:1279–1290. doi:10.1007/s12272-013-0238-8
Nagahara AH, Merrill DA, Coppola G, Tsukada S, Schroeder BE, Shaked GM, Wang L, Blesch A, Kim A, Conner JM, Rockenstein E, Chao MV, Koo EH, Geschwind D, Masliah E, Chiba AA, Tuszynski MH (2009) Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer’s disease. Nat Med 15:331–337. doi:10.1038/nm.1912
Tao X, Finkbeiner S, Arnold DB, Shaywitz AJ, Greenberg ME (1998) Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron 20:709–726
Lonze BE, Ginty DD (2002) Function and regulation of CREB family transcription factors in the nervous system. Neuron 35:605–623
Sakamoto K, Karelina K, Obrietan K (2011) CREB: a multifaceted regulator of neuronal plasticity and protection. J Neurochem 116:1–9. doi:10.1111/j.1471-4159.2010.07080.x
Shakil H, Saleem S (2013) Genetic deletion of prostacyclin IP receptor exacerbates transient global cerebral ischemia in aging mice. Brain Sci 3:1095–108
Wang J, Ming H, Chen R, Ju JM, Peng WD, Zhang GX, Liu CF (2015) CIH-induced neurocognitive impairments are associated with hippocampal Ca(2+) overload, apoptosis, and dephosphorylation of ERK1/2 and CREB that are mediated by overactivation of NMDARs. Brain Res 1625:64–72. doi:10.1016/j.brainres.2015.08.012
Alonso M, Medina JH, Pozzo-Miller L (2004) ERK1/2 activation is necessary for BDNF to increase dendritic spine density in hippocampal CA1 pyramidal neurons. Learn Mem 11:172–178. doi:10.1101/lm.67804
Choi YS, Cho HY, Hoyt KR, Naegele JR, Obrietan K (2008) IGF-1 receptor-mediated ERK/MAPK signaling couples status epilepticus to progenitor cell proliferation in the subgranular layer of the dentate gyrus. Glia 56:791–800. doi:10.1002/glia.20653
Iida N, Namikawa K, Kiyama H, Ueno H, Nakamura S, Hattori S (2001) Requirement of Ras for the activation of mitogen-activated protein kinase by calcium influx, cAMP, and neurotrophin in hippocampal neurons. J Neurosci 21:6459–6466
Ying SW, Futter M, Rosenblum K, Webber MJ, Hunt SP, Bliss TV, Bramham CR (2002) Brain-derived neurotrophic factor induces long-term potentiation in intact adult hippocampus: requirement for ERK activation coupled to CREB and upregulation of Arc synthesis. J Neurosci 22:1532–1540
Jeon SJ, Rhee SY, Seo JE, Bak HR, Lee SH, Ryu JH, Cheong JH, Shin CY, Kim GH, Lee YS, Ko KH (2011) Oroxylin A increases BDNF production by activation of MAPK-CREB pathway in rat primary cortical neuronal culture. Neurosci Res 69:214–222. doi:10.1016/j.neures.2010.11.008
Bath KG, Akins MR, Lee FS (2012) BDNF control of adult SVZ neurogenesis. Dev Psychobiol 54:578–589. doi:10.1002/dev.20546
Arany I, Megyesi JK, Reusch JE, Safirstein RL (2005) CREB mediates ERK-induced survival of mouse renal tubular cells after oxidant stress. Kidney Int 68:1573–1582. doi:10.1111/j.1523-1755.2005.00569.x
Schulte JH, Schramm A, Klein-Hitpass L, Klenk M, Wessels H, Hauffa BP, Eils J, Eils R, Brodeur GM, Schweigerer L, Havers W, Eggert A (2005) Microarray analysis reveals differential gene expression patterns and regulation of single target genes contributing to the opposing phenotype of TrkA- and TrkB-expressing neuroblastomas. Oncogene 24:165–177. doi:10.1038/sj.onc.1208000
Acknowledgements
This work was supported by Natural Science Foundation of Hebei Province, China (C2014206363) and Hebei Province Students innovative and entrepreneurial project (201510089016).
Author Contributions
Designed experiment and wrote manuscript: YL. Performed experiment: JW, ZL, XW. Analyzed data: HuijuanMa. Wrote manuscript: SZ. Reviewed and approved the final version of the manuscript: SW, YZ.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Authors have no conflict of interest in this study.
Ethical Approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
Rights and permissions
About this article

Cite this article
Wang, J., Zhang, S., Ma, H. et al. Chronic Intermittent Hypobaric Hypoxia Pretreatment Ameliorates Ischemia-Induced Cognitive Dysfunction Through Activation of ERK1/2-CREB-BDNF Pathway in Anesthetized Mice. Neurochem Res 42, 501–512 (2017). https://doi.org/10.1007/s11064-016-2097-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-016-2097-4