
Abstract
Previously, we have shown that aberrant expression of glia maturation factor (GMF), a proinflammatory protein, is associated with the neuropathological conditions underlying diseases suggesting an important role for GMF in neurodegeneration. In the present study, we demonstrate that absence of GMF suppresses dopaminergic (DA) neuron loss, glial activation, and expression of proinflammatory mediators in the substantia nigra pars compacta (SN) and striatum (STR) of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) treated mice. Dopaminergic neuron numbers in the SN and fiber densities in the STR were reduced in wild type (Wt) mice when compared with GMF-deficient (GMF-KO) mice after MPTP treatment. We compared the motor abnormalities caused by MPTP treatment in Wt and GMF-KO mice as measured by Rota rod and grip strength test. Results show that the deficits in motor coordination and decrease in dopamine and its metabolite content were protected significantly in GMF-KO mice after MPTP treatment when compared with control Wt mice under identical experimental conditions. These findings were further supported by the immunohistochemical analysis that showed reduced glial activation in the SN of MPTP-treated GMF-KO mice. Similarly, in MPTP-treated GMF-KO mice, production of inflammatory tumor necrosis factor alpha, interleukine-1 beta, granulocyte macrophage-colony stimulating factor, and the chemokine (C–C motif) ligand 2 MCP-1 was suppressed, findings consistent with a role for GMF in MPTP neurotoxicity. In conclusion, present investigation provides the first evidence that deficiency of GMF protects the DA neuron loss and reduces the inflammatory load following MPTP administration in mice. Thus depletion of endogenous GMF represents an effective and selective strategy to slow down the MPTP-induced neurodegeneration.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Parkinson’s disease (PD) is a progressive disabling neurodegenerative disease. It is estimated that 60,000 new cases are diagnosed each year, joining the one million Americans who currently have PD, and there is no effective treatment for this disease. PD is characterized by the presence of degenerating dopaminergic (DA) neurons, Lewy bodies and activated microglia in the brain. Activated microglia are not only present but also persist in PD brains which facilitate secretion of deleterious inflammatory mediators that contributes to dopaminergic neurodegeneration, and other components of neurodegenerative processes including oxidative stress [9, 12, 14]. The loss of DA neurons in the substantia nigra (SN) and depletion of dopamine in the striatum (STR) results in abnormal motor behavior. Although the underlying cause of PD is not clear exposure to environmental neurotoxin, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) is known to be associated with the pathology of PD in human and in animal models of Parkinsonism. Several studies on experimental models of PD suggest that sustained neuroinflammation exacerbates degeneration of the DA neurons [11, 32]. Post-mortem analysis and in vivo imaging of PD patient brains showed activation of microglia, astrogliosis and infiltration of peripheral immune cells in the SN and STR [11, 27, 35]. These features were also observed in experimental rodents brain after MPTP treatment [17, 21, 26]. The glia maturation factor (GMF), discovered and characterized in our laboratory, is a conserved protein in mammalian brain/central nervous system [22, 23, 40]. We already demonstrated a prominent role for GMF in activation of astrocytes and microglia by various factors and stimuli leading to death of neuronal cells [39]. GMF plays critical role in the pathogenesis of inflammatory and neurodegenerative diseases [38, 42]. It has been previously reported the enhanced expression of GMF in the central nervous system (CNS) of neurodegenerative and autoimmune diseases [40, 41]. Based on proinflammatory functions of GMF [38, 39], we hypothesize that GMF is involved in the progression of PD. This novel hypothesis is based on the recently demonstrated functions of GMF in which we have shown that GMF-deficient (GMF-KO) mouse astrocytes and neuron-glia in culture are resistant to MPP+-induced oxidative and inflammatory damages [18]. In the present study, we compared MPTP-induced DA neuronal loss, glial activation, and behavioral motor deficits in GMF-KO and wild type (Wt) mice.
Materials and Methods
Reagents
Enzyme-linked immunosorbent assay (ELISA) kits for TNF-α, IL-1β and CCL2/MCP-1, were purchased from R&D Systems (Minneapolis, MN). ELISA kit for Dopamine was purchased from GenWay Bioteck Inc, San Diego, CA. Polyclonal anti-NF-κB p65 and cyclo-oxygenase-2 (COX-2) antibodies were obtained from Cell Signaling Technology (Danvers, MA) and Santa Cruz Biotechnology (Santa Cruz, CA). Secondary antibodies conjugated to horseradish peroxidase (Bio-Rad Laboratories, Hercules, CA), Rotamex Rotarod (Columbus Instruments, Columbus, OH). Male C57BL/6 mice (Harlan Sprague-Dawley, Inc., Indianapolis, IN) and MPTP (Sigma, St. Louis, MO) were purchased.
MPTP treatments GMF-KO and Wt mice were maintained at The University of Iowa animal care facility and were used in accordance with the NIH guidelines approved by the Animal Care and Use Committees (IACUC) at the University of Iowa. All experiments using MPTP were performed using previously published safety guidelines [28]. C57BL/6, adult male mice, were treated essentially as previously described regimen of MPTP treatment [17]. Animals were randomly divided into four groups (n = 10–12/group). The first group was saline-treated Wt mice and served as control group; the second group of Wt mice was MPTP-treated (four intraperitoneal injections, at 2 h intervals of 18 mg/kg body weight MPTP in saline); third group has saline-treated GMF-KO mice, served as a control group for fourth group mice and fourth group was MPTP-treated GMF-deficient (GMF-KO) mice.
Behavioral Studies
All the behavioral studies were performed at room temperature in a calm room without any outside interference between 09:00 and 17:00 h. The laboratory personnel performing all behavioral tests and data analysis were blinded to treatment paradigm.
Rota Rod
Rotamex Rotarod was used to evaluate the motor coordination skill of mice in each group. The Rota rod unit consists of a rotating rod, which was divided into four parts by compartmentalization to permit the testing of four mice at a time. After twice daily training for two successive days (5 rpm on the first day and 8 rpm on second day) the rotational speed was increased to 10 rpm on the third day in a test session. The time for each mouse remained on the rotating bar was recorded for three trials, at 15 min intervals with maximum trial length of 180 s per trial. The motor deficiency was evaluated as the ability of the mouse to hold the rotating rod.
Grip Strength Test
Grip test was performed as described previously [17]. Briefly, the apparatus consists of a string of 50 cm length, pulled tight between two vertical supports and elevated 40 cm from a flat surface. Mouse was placed on the string at a point midway between supports and evaluated according to the following scale, 0 = fall off, 1 = hangs onto string by two forepaws, 2 = as for 1 but attempts to climb on string, 3 = hangs onto string by two forepaws plus one or both hind paws, 4 = hangs onto string by all forepaws plus tail wrapped around string and 5 = escape.
Dopamine Estimation
Mice form each group were sacrificed and the brains were removed to dissect out the STR. Tissue was homogenised in homogenising buffer provided in the commercial kit. Tissue homogenates were further centrifuged to remove the nuclear debris for the DA estimation by ELISA.
Immunohistochemistry for Tyrosine Hydroxylase (TH) and TH-Positive Cell Counting
Immunohistochemistry for SN and STR was carried out as described previously [17]. Briefly, 20 µm serial coronal brain sections were cut in cryostat (Leica, Germany) and incubated with 0.3 % H2O2 and blocking reagent (5 % normal goat serum, 3 % BSA and 0.3 % Triton-X 100) followed by incubation with the primary rabbit anti-goat TH antibody (Millipore, Temecula, CA) for overnight at 4 °C. The sections were then washed and incubated for 2 h with biotinylated secondary goat anti-rabbit antibody at room temperature followed by ABC standard kit solution and diaminobenzidine as a substrate (Vector Laboratories, Burlingame, CA). All the washings were performed with phosphate buffered saline (PBS). Finally the sections were dehydrated, cover slipped, viewed under a microscope and photomicrograph were taken. The total number of TH-positive neurons in SN was counted from three mice per group by using the optical fractionator, an unbiased stereological technique of cell counting [33, 34, 36]. For the quantification of TH-immunoreactivity in the STR, mean optical densities were obtained from immunostained sections. NIH Image J software was used to quantify the TH positive neurons and fibres in SN and STR. For quantification purposes, at least five randomly selected fields were examined and averaged. Numbers of TH positive cells were represented as percentage of control.
Immunofluorescence for the Detection of Glial Fibrillary Acidic Protein (GFAP), Ionized Calcium-Binding Adaptor Molecule 1 (Iba 1) and COX-2
Immunofluorescence staining was performed to detect expression of astrocytes (GFAP), microglia (Iba1) and COX-2. Serial coronal sections were incubated with blocking reagent followed by primary antibody (rabbit anti-goat GFAP or Iba1 or anti-COX-2) overnight at 4 °C. After washing in PBS, the sections were incubated with Alexa Fluor-488/568 labeled secondary antibody (goat anti-rabbit) at room temperature for 1 h. The sections were then cover-slipped in mounting media and stored in the dark at 4 °C until analysis. Image J software was used to quantify the TH positive neurons and fibres in SN and STR. Number of GFAP/Iba1/COX-2 positive cells is represented as percentage of total positive cells.
Western Blot Analysis
Mid brain were collected from the GMF-KO and Wt mice after MPTP treatments. Samples of nuclear fractions containing equal amounts of protein (35 μg) were separated in 12 % SDS-polyacrylamide gel electrophoresis. Proteins were transferred onto PVDF membrane and incubated overnight at 4 °C with primary rabbit polyclonal antibody against NF-κB p65 (1:1000 dilutions), followed by appropriate secondary antibodies conjugated to horseradish peroxidase (HRP). Proteins recognized by the antibody were visualized by enhanced chemiluminescence Femto kit (Thermo Scientific, Rockford, IL). Blots were stripped and re-probed for β-actin (Sigma) as a loading control. Bands intensity was measured by densitometry and quantified using NIH-Image J software.
ELISA
Tissues were homogenized in tissue lysis buffer (50 mM Tris–Hcl, pH 8.0, 5 mM NaCl, and 1 % Triton X-100). Following centrifugation, supernatants were used for determination of GMF, TNF-α, IL-1β, GM-CSF and MCP-1 by ELISA according to manufacturer’s instruction. Minimum sensitivity of these kits are 10–20 pg/mg protein. The lower detection limit of GMF ELISA is 15–20 pg/mg protein.
Statistical Analysis
Results were expressed as mean ± SEM. Statistical significance was assessed by one-way analysis of variance (ANOVA) followed by the Tukey test. Alternative, Student t test was applied to calculate the statistical significance between the genotypes using GraphPad InStat software. The p value <0.05 was considered statistically significant.
Results
MPTP-Induced Upregulation of GMF Expression in the Substantia Nigra and Striatum of Mouse Brain
To determine the involvement of GMF in the DA neurotoxicity, we administered MPTP or PBS (control) into male Wt mice. The expression of GMF in the microdissected SN and STR, regions known to be involved in PD pathology, were measured by ELISA using a combination of specific affinity purified anti-GMF antibodies. Results from three mice each, at day 1, day 3, and day 5 post MPTP treatments indicated significant up-regulation of GMF protein in the SN and STR (Fig. 1). In the SN, GMF increased from 154 pg/mg of protein to 565 pg/mg of protein (3.7-fold increase), from 137 pg/mg of protein to 730 pg/mg of protein (5.3-fold increase), and from 145 pg/mg of protein to 785 pg/mg of protein (5.4-fold increase) at day 1, day 3, and day 5 following MPTP treatment, respectively. Similarly, in STR following MPTP treatment GMF increased from 107 pg/mg of protein to 338 pg/mg of protein (3-fold increase), from 108 pg/mg of protein to 487 pg/mg of protein (4.5-fold increase), and from 114 pg/mg of protein to 559 pg/mg of protein (4.9-fold increase) at day 1, day 3, and day 5, respectively. These results demonstrate a significant upregulation of GMF is associated with MPTP neurotoxicity in Wt mice.

Enhanced level of GMF in the substantia nigra (SN) and striatum (STR) of mouse brain following MPTP treatments. Wt mice were injected with MPTP or vehicle (saline) and levels of GMF were measured by ELISA at day 1, day 3, and day 5 post injections in microdissected SN and STR regions. The lower detection limit of this GMF ELISA is 15–20 pg/mg protein. Value are expressed as mean ± SEM (n = 6)
GMF-KO Mice Show Reduced Glial Activation Following MPTP Treatment
Activation of microglia and astrocytes, a hallmark of neuroinflammation, has been reported in PD patients and in experimental models of PD. In the present study we analyze astrocyte (GFAP) and microglial (Iba1) activation 5 days post MPTP-intoxication in Wt and GMF-KO mice. For this purpose Wt and GMF-KO mice received 4 s.c injections of saline or MPTP (2 h intervals, day 0), and were sacrificed at day 5. GFAP immunostaining in the mid brain revealed branching as normally observed following astroglial activation and increased number of activated astrocytes after MPTP treatment in Wt mice as compared to PBS injected controls, and was significantly reduced in GMF-KO mice when compared to MPTP-treated animals (Fig. 2). The results also revealed significantly reduced numbers of Iba1-positive activated microglia in GMF-KO mice compared to Wt mice following MPTP-treatment (Fig. 2). The immunofluorescence results were quantitated and presented as GFAP or Iba1-positive cells (% of control) as shown in Fig. 2. There was no significant difference at the basal level of GFAP and Iba1 positive cells in the brain of PBS treated Wt and GMF-KO mice.

MPTP-induced glial activation is inhibited in GMF-KO mice. Representative coronal brain sections from Wt and GMF-KO mice stained for astrocytes (GFAP) and microglia (Iba1) in the mid brain were shown. Wt and GMF-KO mice were sacrificed after 5 days post MPTP or saline injections and evaluated for the glial activations. The profound expression of GFAP (green color) and Iba1 (red color) were observed in MPTP-treated Wt mice as compared to saline treated Wt mice, however; GMF-KO mouse following MPTP treatment showed less glial activation when compared to Wt mice as assessed by fluorescence immunohistochemistry (n = 6) Original magnifications at ×200. GFAP and Iba1 positive cells were counted and presented as % of controls. p < 0.01 (Color figure online)
GMF-KO Mice Show Reduced Expression of NF-κB and COX-2 in the Mid Brain Following MPTP Treatment
In the present study, we have quantified MPTP-induced translocation of NF-κB in the presence and absence of GMF. To detect and quantify NF-κB activation, the translocation of NF-κB p65 subunit to the nucleus was assayed in the nuclear extracts of mid brain using a subunit-specific anti-p65 antibody. MPTP-injected GMF-KO mice displayed significantly (p < 0.05) reduce levels of NF-κB p65 protein in nuclear extracts as compared to Wt mice 5 days post MPTP treatments (Fig. 3a). We further investigated expression of a NF-κB-responsive gene, COX-2, by fluorescence immunohistochemistry. Similar to NF-κB p65, we found significantly reduced number of COX-2 positive neurons (p < 0.01) in MPTP-injected GMF-KO mice as compared to Wt mice (Fig. 3b). Histogram in Fig. 3 shows mean densitometric values of bands after normalizing with β-actin. There was no significant difference at the basal expression level of NF-kB and COX-2 positive cells in the brain of PBS treated Wt and GMF-KO mice.

GMF-KO mice show reduced expression of NF-κB and COX-2 in the mid brain following MPTP treatment. Wt and GMF-KO mice were treated with MPTP or saline and expression level of NF-kB and COX-2 were assessed by Western blot and immunofluorescence, respectively. Decreased expression level of NF-kB and COX-2 were observed in GMF-KO mice as compared to Wt mice. Histogram shows mean densitometric analysis of bands after normalizing with β-actin as a loading control of each group. Data are mean ± SEM (n = 4)
Diminished Level of Cytokines/Chemokine in the Mid Brain of GMF-KO Mice Following MPTP Treatment
In addition to glial activation, we also assessed the levels of TNF-α, IL-1β, GM-CSF and CCL2/MCP-1 in the mid brain, as a measure of inflammation in the mid brain these mice. MPTP treatment showed consistently decreased levels of TNF-α, IL-1β, GM-CSF and CCL2/MCP-1 in GMF-KO mice when compared with Wt mice (Fig. 4). The levels of TNF-α were 158 ± 21, 753 ± 38, 934 ± 18, and 1144 ± 32 pg/mg protein in Wt mice at 0, 2, 5, and 7 days post MPTP treatment, respectively. TNF-α levels were significantly reduced to 44 ± 14, 212 ± 11, 321 ± 25, and 389 ± 20 pg/mg protein during the same time points in GMF-KO mice (Fig. 4). Similarly, the levels of IL-β (99 ± 18, 557 ± 45, 664 ± 44, and 859 ± 55 pg/mg protein), GM-CSF (111 ± 16, 592 ± 26, 738 ± 31, and 896 ± 30 pg/mg protein), and CCL2/MCP-1 (124 ± 20, 690 ± 51, 821 ± 54, and 1066 ± 69 pg/mg protein) at 0, 2, 5, and 7 days post MPTP treatment, respectively, detected in Wt mice were significantly reduced in GMF-KO mice as follows; IL-β (41 ± 11, 220 ± 18, 240 ± 25, and 307 pg/mg protein), GM-CSF (46 ± 17, 185 ± 11, 259 ± 15, and 298 ± 8 pg/mg protein), and CCL2/MCP-1 (54 ± 14, 267 ± 24, 297 ± 32, and 377 ± 30 pg/mg protein). Thus, these results confirm significantly reduced MPTP-induced neuroinflammation as evidenced by decreased glial activation and significantly reduced production of proinflammatory cytokines/chemokines in GMF-KO mice.

Diminished levels of MPTP-iduced proinflammatory cytokines/chemokine in the mid brain of GMF-KO mice. Wt and GMF-KO mice recieved 4 s.c injections (2 h intervals) of MPTP or PBS (day 0), and were sacrificed at day 2, 5, and 7. Protein level of cytokines TNF-α, IL-1β, GM-CSF and CCL2/MCP were measured in the mid brain by ELISA. GMF-KO mice showed reduced level of cytokines/chemokine when compared to Wt mice following MPTP treatments. Assays had a minimum sensitivity of 10–20 pg/mg protein. Data represent the mean ± SEM (n = 8). p < 0.05
GMF-KO Mice Show Reduced Nigrostriatal Dopaminergic Neuron Loss Following Acute MPTP Treatment
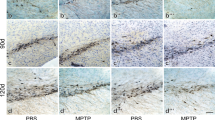
To test the hypothesis that GMF plays a prominent role in dopaminergic neurodegeneration, we investigated DA neurodegeneration in SN and STR following MPTP treatment in WT and GMF-KO mice. For this purpose Wt and GMF-KO mice received 4 injections of PBS or MPTP (2 h intervals, day 0), and were sacrificed at day 2, 5, and 7. The effect of MPTP treatment on Tyrosine hydroxylase (TH)-positive neurons in SN and STR was evaluated (Fig. 5). MPTP treatment caused significant DA neuronal loss (brown color) in the SN (Fig. 5a) and neuronal fibers (brown color) in the STR (Fig. 5b) of Wt mice. In contrast, GMF-KO mice showed diminished dopaminergic neurodegeneration in the SN and the loss of striatal DA fibers was largely attenuated in GMF-KO mice. The number of TH-positive neurons in the SN was counted in three mice per group under microscope and optical density quantification of TH-immunoreactivity in the STR was obtained from immunostained sections with previously described image analysis procedure. In Wt mice the TH-positive neuronal cell counts in the SN 2 days after MPTP treatment were 49 % of the control PBS vehicle treated mice; 5 days, 23 %; and 7 days, 32 % (Fig. 5c). However, in GMF-KO mice neuronal cell counts 2 days after MPTP treatment were 60 % of the control vehicle treated mice; 5 days, 74 %; and 7 days, 85 %. A significant difference in the DA neuron loss between Wt and GMF-KO mice after MPTP treatment, with a greater loss in WT mice occurred at day 5 (Fig. 5c). Thus, quantitation of DA neuron in MPTP-treated Wt and GMF-KO mice confirmed the histological findings (Fig. 5a). The decrease of TH-immunoreactivity occurred in the STR of Wt mice 2 day after MPTP treatment (28 % of vehicle-treated control mice), 5 days (33 % of control), and 7 days (31 % of control), whereas in GMF-KO mice the MPTP-induced decrease of TH-immunoreactivity was markedly attenuated (2 day of PBS-treated control mice 87 %, 5 days 89 %, and 7 days 92 % of PBS-treated control). Densitometric estimation of TH-immunoreactivity levels in the STR confirmed that MPTP induces a greater loss of dopaminergic neurons in the presence of GMF in Wt mice when compared with in the absence of GMF in GMF-KO mice (Fig. 5d). These results demonstrated a significant difference in TH neuronal loss at each time point after MPTP treatment in the SN as well as STR between Wt and GMF-KO mice suggesting an involvement of GMF in MPTP-induced dopaminergic neurotoxicity. There was no significant difference at the basal level of TH-positive cells in SN of PBS treated Wt and GMF-KO mice.

Significant suppression of MPTP-induced nigrostriatal DA neuron loss in GMF-KO mice. Wt and GMF-KO mice recieved 4 s.c injections (2 h intervals) of saline or MPTP (day 0), and were sacrificed at day 2, 5, and 7. Tyrosine hydroxylase (TH)-positive neurons in SN (a) and neuronal fibers in the STR (b) were evaluated by immunohistochemistry. Representative photomicrographs, at ×10, ×20 and ×40 magnification, show more loss of TH-positive neurons in SN and fibres in STR (brown color) in Wt mice as compared to barely detectable degeneration in GMF-KO mice at each time point after MPTP treatment. Time course change in the number of TH-positive neurons in the SN (c) and TH-immunoreactive fibers density in the STR (d) following MPTP treatment show significant difference in TH neuronal loss between Wt and GMF-KO mice at each time point examined. Value are expressed as mean ± SEM (Color figure online)
Attenuation of MPTP-Induced Dopamine Depletion And Motor Behavioral Impairments in GMF-KO Mice
A reduction in tyrosine hydroxylase, dopamine, and DA metabolites concentration is hallmark of Parkinson’s disease and the main cause of behavioral disorders. To explore the effect of lack of GMF, we examined the concentration of dopamine in brain tissues from Wt and GMF-KO mice 5 days post MPTP treatments. Results (Fig. 6) show that the MPTP treatment significantly (p < 0.01) decreases the level of dopamine in Wt mice (32.84 ± 6.57 ng/mg protein) as compared with the saline treated Wt mice (127.31 ± 18.58 ng/mg protein). GMF-KO mice showed significant (p < 0.05) suppression of dopamine loss (60.55 ± 13.96 ng/mg protein) as compared to Wt mice (32.84 ± 6.57 ng/mg protein) following MPTP treatments (Fig. 6). Next, we compared the motor abnormalities caused by MPTP treatment in Wt and GMF-KO mice. Mice were subjected to behavioral tests 5 days after MPTP treatment. In both Rotarod and Grip strength test, GMF-KO mice performed better than Wt mice (Fig. 7). For this purpose, the mice were pre trained on the Rotarod and tested at a series of increasing speeds, recording the time that the animal remains on the rod at each speed. The Grip strength was rated on a scale of 0–5. On the Rotarod test, the latency of GMF-KO mice to fall was significantly (p < 0.01) reduced compared with Wt mice followed by MPTP treatments (Fig. 7a). GMF-KO mice also displayed a significant (p < 0.01) grip strength compared with Wt mice after MPTP treatment (Fig. 7b). Wt mice treated with PBS only did not display differences in latency to fall and grip strength test. These results show that the MPTP-induced dopamine depletion was attenuated in GMF-KO mice along with improved motor behavior when compared with Wt mice following MPTP-treatment. We didn’t find any significance difference at the basal level of dopamine and motor skills of PBS treated Wt and GMF-KO mice.

Significant suppression of MPTP-induced dopamine depletion in GMF-KO mice. GMF-KO mice showed significant suppression of dopamine level in striatum as compared to Wt mice following MPTP treatments. There was no significant difference in dopamine level between PBS treated Wt and GMF-KO mice. Value are expressed as mean ± SEM (n = 6)

Attenuation of MPTP-induced motor behavioral impairments in GMF-KO mice We assessed the Rotarod performance and grip strength in MPTP-intoxicated Wt and GMF-KO mice at 5 days post MPTP-intoxication. Wt mice treated with MPTP showed decreased motor skill as compared to saline treated Wt mice. In contrast, GMF-KO mice showed significant improved motor skill as compared to Wt mice after MPTP treatments as measured by Rotarod and grip strength tests. Value are expressed as mean ± SEM (n = 10)
Discussion
The present study demonstrates that deficiency of GMF rescues the DA neuronal loss and reduces the inflammatory load following MPTP administration in mice. Absence of endogenous GMF represents an effective and selective strategy to reduce the MPTP-induced neurodegeneration. PD, one of the most prevalent and progressive neurodegenerative diseases, characterized by tremor and motor impairments due to the loss of DA neurons in the SN [2, 7, 20]. However, the cause of the DA neuron loss in PD has not been fully understood. Parkinsonian neurotoxin MPTP is known to produce PD pathology in both humans and mice by inducing the DA neuron loss [29]. Recent studies have indicated that GMF is involved in the pathogenesis of neurodegenerative diseases [41, 42]. However, the exact association and underlying mechanism of GMF in the loss of DA neurons in PD is not clear. In the present study, we tested the hypothesis that deficiency of GMF would rescue the DA neurons loss and improve the motor deficit through the downregulation of the glial mediated neuroinflammatory cascade. For this purpose, we first measured the GMF level in the brain of Wt mice after MPTP injections. We found enhanced level of GMF in Wt mice followed by MPTP injection as compared to saline treated Wt mice. Our finding on enhanced GMF level following neurotoxin insult was consistent with previous reports where enhanced level of GMF in the brain has been documented in many conditions associated with inflammation and neurodegeneration [38, 42]. Since an enhanced level of GMF is a prominent mediator of inflammatory signal transduction in the brain leading to the neuronal death, we determined how the lack of GMF affects the regulation of inflammation and loss of DA neurons in MPTP model of mouse. To explore the vulnerability of DA neurons in mice, we treated Wt and GMF-KO mice with acute MPTP challenge. Data on GMF-KO mice indicate improved motor skills, reduced loss of TH-positive DA neurons and fibers along with dopamine, and diminished level of proinflammatory cytokines compared to control Wt mice. The reduction in MPTP-induced neuroinflammation was quantified by decreased number of astrocyte and microglia as well as downregulation of NF-kB and proinflammatory cytokines/chemokine in GMF-KO mice. The decrease in glial activation in the GMF-KO mice was also associated with increased TH-positive neurons following MPTP challenge, suggesting that suppression or inhibition of endogenous GMF may be an effective and selective strategy to slow down the inflammation associated neurodegeneration in the PD pathogenesis. Our findings are in agreement with the earlier studies carried out by us and others, where neurotoxins are known to potentiate inflammatory responses in presence of GMF and down-regulation of GMF reduced the inflammatory responses and suppressed neurodegeneration [15, 16, 18, 41]. These data provide support to our hypothesis that increased levels of GMF in the PD brain contributes to neuroinflammatory and neurodegenerative processes.
The most prominent biochemical changes in the mid brain of PD patients and MPTP-treated mice were decreased levels of dopamine [4, 13]. Such deficits in striatal dopamine in MPTP-treated mice led to a decreased latency to fall on an accelerating Rotarod, reflecting diminished coordination and balance [16, 31]. Another study has hypothesized that preservation of the DA neurons may contribute to improved functional outcome in PD [5]. The hypothesis is consistent with the present finding that GMF deficiency improves MPTP-induced behavioral changes (Rotarod and grip strength performances) and protects against MPTP-induced DA neurotoxicity. It is worthy to note whether neuroprotection afforded by lack of GMF results in the restoration or prevention of further decrease of DA neurons in the mid brain of mice. Our TH immunostaining revealed that absence of GMF reduced the loss TH-positive neurons and density of striatal TH-positive fibers following MPTP treatment. Then, it is likely that that absence of GMF play a key role in improvement of MPTP-induced motor dysfunction and suppression of DA neurodegeneration.
The importance of glial involvement and upregulation of proinflammatory mediators in PD have been supported by the analysis of PD patients post mortem brains that provided clear evidence of glial activation in the SN region [6, 25, 27]. We have previously shown that overexpressing GMF in astrocytes promotes the production and secretion of GM-CSF which in turn activates microglia and play critical role in mediating neurotoxicity [39, 42]. In the present study, we report that absence of GMF suppressed the glial cell activation-induced inflammatory response and rescued DA neurons from MPTP-induced toxicity. Our data are in agreement with other studies where blockade of microglia activation by selective inhibitors prevented MPTP-induced neurotoxicity [3, 24, 37]. Therefore, we assume that GMF antagonist might well serve as a novel compound that could be used as an anti-inflammatory agent for PD.
Activated microglia upregulates NF-κB nuclear translocation and releases various inflammatory mediators, including TNF-α, IL-1β, and chemokines [1, 8, 19]. The transcription factor, NF-κB plays an important role in the regulation of several proinflammatory mediators that are directly involved in neuroinflammation and neurodegeneration. Selective inhibition of NF-kB can prevent DA neuronal loss in a MPTP-induced mouse model of PD [10]. Astrocytes and microglia are the primary sources of cytokines and chemokines that play a role in the neuroinflammatory responses [30]. From these observations, it is clear that cytokines and chemokines produced by glial cells may play a key role in neurodegeneration after MPTP treatments. Similarly we also found increased expression of NF-kB and proinflammatory cytokines/chemokine TNF-α, IL-1β, GM-CSF and CCL2/MCP-1 following MPTP treatment and the absence of GMF suppressed the glial cell activation-induced upregulation of NF-kB and proinflammatory cytokines/chemokine and rescues DA neuron from MPTP-induced toxicity.
Conclusion
In summary, our data demonstrate that MPTP-induced overexpression of GMF in glial cells lead to activation of inflammatory signaling pathways resulting in neuronal cell, target cell population in PD, injury and motor behavioral dysfunction (Fig. 8). Conversely, in GMF-KO mice activation of NF-κB was significantly suppressed along with reduced expression and secretion of inflammatory mediators resulting in attenuation of motor deficit. This may have important implications in neurological disorders such as PD. To the best of our knowledge this is the first report in which deficiency of GMF is demonstrated to rescue the dopaminergic neuronal loss via down regulation of inflammatory responses in an experimental model of PD, thus emphasizing the therapeutic potential of GMF in inflammatory disorders.

Sequence of events following MPTP treatment in mice. MPTP-induced GMF-overexpression in glial cells leads to the activation and translocation of NF-κB resulting in release of inflammatory mediators such as TNF-α, IL-1β, and COX2, which in turn cause neuroinflammation and neurodegeneration. Furthermore, GMF-dependent depletion of dopamine and loss of DA neurons finally results in motor behavioral dysfunction
References
Aoki E, Yano R, Yokoyama H, Kato H, Araki T (2009) Role of nuclear transcription factor kappa B (NF-kappaB) for MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahyropyridine)-induced apoptosis in nigral neurons of mice. Exp Mol Pathol 86:57–64
Chung KK, Dawson VL, Dawson TM (2003) New insights into Parkinson’s disease. J Neurol 250(Suppl 3):III15–III24
Chung YC, Bok E, Huh SH, Park JY, Yoon SH, Kim SR, Jin BK et al (2011) Cannabinoid receptor type 1 protects nigrostriatal dopaminergic neurons against MPTP neurotoxicity by inhibiting microglial activation. J Immunol 187:6508–6517
Chung YC, Kim SR, Park JY, Chung ES, Park KW, Won SY, Jin BK et al (2011) Fluoxetine prevents MPTP-induced loss of dopaminergic neurons by inhibiting microglial activation. Neuropharmacology 60:963–974
Crocker SJ, Smith PD, Jackson-Lewis V, Lamba WR, Hayley SP, Grimm E, Park DS et al (2003) Inhibition of calpains prevents neuronal and behavioral deficits in an MPTP mouse model of Parkinson’s disease. J Neurosci 23:4081–4091
Damier P, Hirsch EC, Zhang P, Agid Y, Javoy-Agid F (1993) Glutathione peroxidase, glial cells and Parkinson’s disease. Neuroscience 52:1–6
Dawson TM, Dawson VL (2003) Molecular pathways of neurodegeneration in Parkinson’s disease. Science 302:819–822
Dheen ST, Jun Y, Yan Z, Tay SS, Ling EA (2005) Retinoic acid inhibits expression of TNF-alpha and iNOS in activated rat microglia. Glia 50:21–31
Dias V, Junn E, Mouradian MM (2013) The role of oxidative stress in Parkinson’s disease. J Parkinsons Dis 3:461–491
Ghosh A, Roy A, Liu X, Kordower JH, Mufson EJ, Hartley DM, Pahan K et al (2007) Selective inhibition of NF-kappaB activation prevents dopaminergic neuronal loss in a mouse model of Parkinson’s disease. Proc Natl Acad Sci USA 104:18754–18759
Hirsch EC, Hunot S, Damier P, Faucheux B (1998) Glial cells and inflammation in Parkinson’s disease: a role in neurodegeneration? Ann Neurol 44:S115–S120
Hirsch EC, Vyas S, Hunot S (2012) Neuroinflammation in Parkinson’s disease. Parkinsonism Relat Disord 18(Suppl 1):S210–S212
Jackson-Lewis V, Przedborski S (2007) Protocol for the MPTP mouse model of Parkinson’s disease. Nat Protoc 2:141–151
Jenner P (2007) Oxidative stress and Parkinson’s disease. Handb Clin Neurol 83:507–520
Kaimori JY, Takenaka M, Nakajima H, Hamano T, Horio M, Sugaya T, Imai E et al (2003) Induction of glia maturation factor-beta in proximal tubular cells leads to vulnerability to oxidative injury through the p38 pathway and changes in antioxidant enzyme activities. J Biol Chem 278:33519–33527
Kawasaki T, Ishihara K, Ago Y, Baba A, Matsuda T (2007) Edaravone (3-methyl-1-phenyl-2-pyrazolin-5-one), a radical scavenger, prevents 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurotoxicity in the substantia nigra but not the striatum. J Pharmacol Exp Ther 322:274–281
Khan MM, Kempuraj D, Thangavel R, Zaheer A (2013) Protection of MPTP-induced neuroinflammation and neurodegeneration by Pycnogenol. Neurochem Int 62:379–388
Khan MM, Kempuraj D, Zaheer S, Zaheer A (2014) Glia maturation factor deficiency suppresses 1-Methyl-4-Phenylpyridinium-induced oxidative stress in astrocytes. J Mol Neurosci 53:590–599
Kim JS, Ryu SY, Yun I, Kim WJ, Lee KS, Park JW, Kim YI (2006) 1alpha,25-Dihydroxyvitamin D(3) Protects Dopaminergic Neurons in Rodent Models of Parkinson’s Disease through Inhibition of Microglial Activation. J Clin Neurol 2:252–257
Lang AE, Lozano AM (1998) Parkinson’s disease. First of two parts. N Engl J Med 339:1044–1053
Lee E, Park HR, Ji ST, Lee Y, Lee J (2014) Baicalein attenuates astroglial activation in the 1-methyl-4-phenyl-1,2,3,4-tetrahydropyridine-induced Parkinson’s disease model by downregulating the activations of nuclear factor-kappaB, ERK, and JNK. J Neurosci Res 92:130–139
Lim R, Miller JF, Zaheer A (1989) Purification and characterization of glia maturation factor beta: a growth regulator for neurons and glia. Proc Natl Acad Sci USA 86:3901–3905
Lim R, Zaheer A, Lane WS (1990) Complete amino acid sequence of bovine glia maturation factor beta. Proc Natl Acad Sci USA 87:5233–5237
Liu Y, Hu J, Wu J, Zhu C, Hui Y, Han Y, Fan W et al (2012) alpha7 nicotinic acetylcholine receptor-mediated neuroprotection against dopaminergic neuron loss in an MPTP mouse model via inhibition of astrocyte activation. J Neuroinflammation 9:98
McGeer PL, Itagaki S, Boyes BE, McGeer EG (1988) Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 38:1285–1291
Noelker C, Stuckenholz V, Reese JP, Alvarez-Fischer D, Sankowski R, Rausch T, Bacher M et al (2013) CNI-1493 attenuates neuroinflammation and dopaminergic neurodegeneration in the acute MPTP mouse model of Parkinson’s disease. Neurodegener Dis 12:103–110
Ouchi Y, Yagi S, Yokokura M, Sakamoto M (2009) Neuroinflammation in the living brain of Parkinson’s disease. Parkinsonism Relat Disord 15(Suppl 3):S200–S204
Przedborski S, Jackson-Lewis V, Naini AB, Jakowec M, Petzinger G, Miller R, Akram M (2001) The parkinsonian toxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP): a technical review of its utility and safety. J Neurochem 76:1265–1274
Przedborski S, Vila M (2003) The 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model: a tool to explore the pathogenesis of Parkinson’s disease. Ann N Y Acad Sci 991:189–198
Ransohoff RM, Glabinski A, Tani M (1996) Chemokines in immune-mediated inflammation of the central nervous system. Cytokine Growth Factor Rev 7:35–46
Riederer P, Wuketich S (1976) Time course of nigrostriatal degeneration in parkinson’s disease. A detailed study of influential factors in human brain amine analysis. J Neural Transm 38:277–301
Sanchez-Guajardo V, Barnum CJ, Tansey MG, Romero-Ramos M (2013) Neuroimmunological processes in Parkinson’s disease and their relation to alpha-synuclein: microglia as the referee between neuronal processes and peripheral immunity. ASN Neuro 5:113–139
Sugama S, Cho BP, Degiorgio LA, Shimizu Y, Kim SS, Kim YS, Joh TH et al (2003) Temporal and sequential analysis of microglia in the substantia nigra following medial forebrain bundle axotomy in rat. Neuroscience 116:925–933
Sugama S, Yang L, Cho BP, DeGiorgio LA, Lorenzl S, Albers DS, Joh TH et al (2003) Age-related microglial activation in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced dopaminergic neurodegeneration in C57BL/6 mice. Brain Res 964:288–294
Tansey MG, Goldberg MS (2010) Neuroinflammation in Parkinson’s disease: its role in neuronal death and implications for therapeutic intervention. Neurobiol Dis 37:510–518
West MJ (1999) Stereological methods for estimating the total number of neurons and synapses: issues of precision and bias. Trends Neurosci 22:51–61
Wu DC, Jackson-Lewis V, Vila M, Tieu K, Teismann P, Vadseth C, Przedborski S (2002) Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. J Neurosci 22:1763–1771
Zaheer A, Sahu SK, Wu Y, Zaheer A, Haas J, Lee K, Yang B (2007) Diminished cytokine and chemokine expression in the central nervous system of GMF-deficient mice with experimental autoimmune encephalomyelitis. Brain Res 1144:239–247
Zaheer A, Zaheer S, Sahu SK, Knight S, Khosravi H, Mathur SN, Lim R (2007) A novel role of glia maturation factor: induction of granulocyte-macrophage colony-stimulating factor and pro-inflammatory cytokines. J Neurochem 101:364–376
Zaheer S, Thangavel R, Sahu SK, Zaheer A (2011) Augmented expression of glia maturation factor in Alzheimer’s disease. Neuroscience 194:227–233
Zaheer S, Wu Y, Sahu SK, Zaheer A (2011) Suppression of neuro inflammation in experimental autoimmune encephalomyelitis by glia maturation factor antibody. Brain Res 1373:230–239
Zaheer S, Thangavel R, Wu Y, Khan MM, Kempuraj D, Zaheer A (2013) Enhanced expression of glia maturation factor correlates with glial activation in the brain of triple transgenic Alzheimer’s disease mice. Neurochem Res 38:218–225
Acknowledgments
We would like to thank John Nehman and Hayate Javed for excellent technical help. This work was supported by the National Institute of Neurological Disorders and Stroke Grants NS073670 and VA Merit Review Award.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Khan, M.M., Zaheer, S., Thangavel, R. et al. Absence of Glia Maturation Factor Protects Dopaminergic Neurons and Improves Motor Behavior in Mouse Model of Parkinsonism. Neurochem Res 40, 980–990 (2015). https://doi.org/10.1007/s11064-015-1553-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-015-1553-x