
Abstract
Malondialdehyde (MDA) is a product of oxidative damage to lipids, amino acids and DNA, and accumulates with aging and diseases. MDA can possibly react with amines so as to modify proteins and inactivate enzymes; it can also modify nucleosides so as to cause mutagenicity. Brain mitochondrial dysfunction is a major contributor to aging and neurodegenerative diseases. We hypothesize that MDA accumulated during aging targets mitochondrial enzymes so as to cause further mitochondrial dysfunction and additional contributions to aging and neurodegeneration. Herein, we investigated the neuronal mitochondrial toxic effects of MDA on mitochondrial respiration and activities of enzymes (mitochondrial complexes I–V, α-ketoglutarate dehydrogenase (KGDH) and pyruvate dehydrogenase (PDH)), in isolated rat brain mitochondria. MDA depressed mitochondrial membrane potential, and also showed a dose-dependent inhibition of mitochondrial complex I- and complex II-linked respiration. Complex I and II, and PDH activities were depressed by MDA at ≥0.2 μmol/mg; KGDH and complex V were inhibited by ≥0.4 and ≥1.6 μmol MDA/mg, respectively. However, MDA did not have any toxic effects on complex III and IV activities over the range 0–2 μmol/mg. MDA significantly elevated mitochondrial reactive oxygen species (ROS) and protein carbonyls at 0.2 and 0.002 μmol/mg, respectively. As for the antioxidant defense system, a high dose of MDA slightly decreased mitochondrial GSH and superoxide dismutase. These results demonstrate that MDA causes neuronal mitochondrial dysfunction by directly promoting generation of ROS and modifying mitochondrial proteins. The results suggest that MDA-induced neuronal mitochondrial toxicity may be an important contributing factor to brain aging and neurodegenerative diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Increasing evidence supports the free radical theory of aging [1]. Free radical-induced oxidative damage to lipids, proteins and nucleic acids is one of the most important biomarkers of aging [2–4]. Malondialdehyde (MDA), 4-hydroxy-2(E)-nonenal (HNE), acrolein, and other aldehydes are generally considered the end products of lipid peroxidation and are widely measured as indexes of lipid peroxidation. MDA, HNE, and acrolein are all electrophiles and can further react with nucleophilic groups in proteins, nucleic acids, or aminophospholipids in Michael-type 1,4-additions or in more complicated reactions; in this way, they are found to play not only pathological, but physiological roles as well [5].
MDA, in fact, is not only an end-product of lipid peroxidation, but also the oxidative product of amino acids, carbohydrates, and DNA. Both radiation- and iron-induced oxidative damage can cause MDA to be produced from free amino acids [6]. Oxidant-induced DNA strand breakage is accompanied by release of thiobarbituric acid-reactive materials, including MDA [7]. MDA has been shown to be produced by reaction of hydroxyl radicals with deoxyribose moieties [8].
Oxidative mitochondrial damage is a key contributor to aging and age-associated degenerative diseases, such as Parkinson’s and Alzheimer’s disease (AD) [9, 10]. The toxic effects of acrolein and HNE on mitochondria have been examined. Picklo et al. [11] showed that acrolein inhibits state 3 mitochondrial respiration and reduces antioxidant glutathione levels while N-acetylcysteine prevents such inhibition. On the other hand, acrolein does not reduce the activity of complexes I–V, nor does it alter mitochondrial calcium transporter activity and cytochrome c release. Humphries et al. [12] demonstrated that HNE inhibits NADH-linked mitochondrial respiration and selectively inactivates α-ketoglutarate dehydrogenase (KGDH) and pyruvate dehydrogenase (PDH) in rat heart mitochondria. Picklo et al. [11, 13] showed that HNE inhibits complex I- and complex II-linked state 3 respiration and decreases the activities of complexes II and III both in rat brain mitochondria and also in Neuro 2A neuroblastoma cells.
In previous studies, we found that MDA dose-dependently inactivated complexes I, and II, α-ketoglutarate dehydrogenase and PDH in isolated liver mitochondria. These results suggest that MDA not only exists as an end product of lipid peroxidation, but also impairs cellular energy metabolism as a mitochondrial toxin that inhibits oxidative phosphorylation in liver mitochondria [14].
Although representing only ~2% of the weight of the human body, the brain consumes ~20% of the oxygen and consequently a similar proportion of the energy produced by cellular respiration. Therefore, because brain mitochondria operate at high activity to satisfy elevated cellular energy demands, they are subjected to a higher rate of oxidative by-product attack during the process of respiration.
Brain MDA levels vary under different physiological and pathological conditions [15, 16]. Increased levels of plasma MDA were found in patients with AD: values for AD patients versus controls were 0.69 versus 0.57 μmol/l free MDA, and 1.15 versus 1.06 μmol/l total MDA [15, 16]. In a genetic mouse model of aging, the hippocampus MDA level was 11 nmol/mg 6 weeks after birth versus 6 nmol/mg in the controls [17]. The process of ischemia-reperfusion increased rat brain MDA levels to 5.5 nmol/mg from 3.5 nmol/mg in controls [18]. Wang et al. [19] reported that the MDA concentration in brain cortex mitochondria in rat was 8.38 nmol/mg, which was increased to 24.63 nmol/mg by exposure to Fe2+-Cys. In a previous study, we found a large increase in MDA in brain mitochondria of old rats (0.21 μmol/mg in young vs. 0.42 μmol/mg in old) [20].
We hypothesized that the accumulation of MDA with age or the development of brain mitochondrial dysfunction in disease, can further aggravate mitochondrial oxidative damage and cause mitochondrial dysfunction, which may contribute to the development of neurodegeneration and brain aging. In addition, we hypothesized that brain mitochondria, due to their lower electron transfer activities in the mitochondrial respiratory chain, higher levels of lipid peroxidation products and greater mitochondrial fragility, are more sensitive to MDA damage than liver mitochondria [21]. In the present study with isolated rat brain mitochondria, we measured mitochondrial membrane potential, respiration, activities of key enzymes, ROS levels and antioxidant defense system function in the presence of 0–2 μmol MDA/mg, a concentration range comparable to physiological and pathological MDA levels. We found that MDA causes significant mitochondrial dysfunction and ROS production as well as the impairment of antioxidant defense systems in brain mitochondria. More importantly, the brain mitochondria are more susceptible to MDA insult than liver mitochondria.
Materials and Methods
Chemicals
Tris base and NADH were purchased from Amersco Inc. (Palm Harbor, FL); 2,6-dichlorophenol indophenol (DCPIP) from Merck & Co. Inc. (USA); rotenone from Riedel De Haen Seelze (HANNOVE, Germany); tetramethoxypropane, cytochrome C, vitamin C, coenzyme Q1, antimycin A, p-iodonitrotetrazolium violet (INT), dithiothreitol, thiamin pyrophosphate (TPP), 2′,7′-dichlorofluorescein diacetate (DCF-DA), 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide (JC-1) from Sigma Chemical Co. (St. Louis, MO); BCA kit for protein quantification from Shenneng Bocai Ltd (Shanghai, China). Other chemicals were all A. P. reagents from local vendors.
Mitochondrial Preparation and Protein Determination
Male Sprague Dawley rats (8-weeks-old) were purchased from Shanghai Slac Laboratory Animal Co. Ltd (Shanghai, China) and sacrificed by decapitation after an overnight fast. Mitochondria were isolated as described with slight modification [22]. Briefly, tissues were rinsed with saline, weighed, and put into ice-cold isolation buffer containing 0.25 mol/l sucrose, 10 mmol/l Tris base, 0.5 mmol/l EDTA, pH 7.4. Tissues were sheared carefully to mince, and rinsed to get rid of residual blood, then homogenized in 2.5 volume of isolation buffer. The homogenate was adjusted to eight volume with isolation buffer and centrifuged at 1,000g for 4 min; the supernatant fraction was decanted and saved. The pellet was washed once with two volume of isolation buffer. The supernatant fractions were combined and centrifuged at 10,000g for 4 min. The mitochondrial pellet was washed twice with isolation buffer. All above operations were carried out at 4°C. The mitochondrial protein concentration was determined using a BCA method with BSA as standard. An aliquot of freshly prepared mitochondria was used for the assay of mitochondrial respiration, and the rest were frozen at −80°C for enzymatic assays.
MDA Preparation and Incubation with Mitochondria
MDA stock solution was made by adding 3 ml of tetramethoxypropane to 100 ml of 0.01N HCl, stirred 6 h and stored at 4°C, and used from 2 weeks to 2 months. The concentration of MDA was determined at 245 nm using an extinction coefficient of 13,700/mol [23]. In preliminary tests, we found MDA rapidly inhibited the activities of complexes I and II within 5 min at 30°C, and did not produce any further decreases even with incubation times of up to 30 min. In the present study, mitochondrial suspensions were diluted to 0.5 mg/ml with assay buffer containing 100 mmol/l sucrose, 100 mmol/l KCl, 2 mmol/l KH2PO4, 5 mmol/l HEPES, and 0.01 mmol/l EGTA, pH 7.4 [24]. Various concentrations of MDA (or an equal volume of 0.01N HCl as control) from 0 to 2 μmol/mg were exposed to mitochondrial suspensions at 30°C for 5 min. Assays were performed immediately after MDA insult.
Determination of Mitochondrial Membrane Potential and Reactive Oxygen Species (ROS) Generation
Brain mitochondria pretreated with MDA were suspended in assay buffer (final protein concentration 0.05 μg/μl) with 1.5 μg/ml JC-1 and 5 mmol/l succinate. The suspension was incubated at 30°C for 15 min. Mitochondrial membrane potential was measured using flow cytometry as described [25, 26]. The relative mitochondrial membrane potential was represented by the ratio of the fluorescence intensity at 590 nm to that at 530 nm.
ROS in mitochondria were estimated by determining hydrogen peroxide which can oxidize dichlorofluorescein (DCFH, derived from DCF-DA after cleavage of the acetyl groups) to its fluorescent form. Brain mitochondria pretreated with MDA were suspended in assay buffer with 5 μmol/l DCF-DA and 5 mmol/l succinate. The hydrogen peroxide so generated was monitored fluorescently at 30°C for 15 min using a plate reader with excitation at 480 nm and emission at 530 nm. The ROS levels were calculated as relative fluorescence units (RFU) per minute [27].
Determination of Mitochondrial Respiratory Activity
Mitochondrial respiration was determined using a Clark oxygen electrode (Hansatech, UK) [28]. A mitochondrial suspension containing 0.5 mg was added to 1 ml of assay buffer at 30°C. Oxygen consumption was measured in the absence (state 4 activity) or presence of 0.25 mmol/l ADP (state 3 activity) and succinate (final concentration 10 mmol/l) or malate/glutamate (2.5 mmol/l) as substrate.
Assays of Mitochondrial Enzyme Activities
NADH-CoQ oxidoreductase (complex I) activity was assayed by monitoring the reduction of DCPIP (ε = 21 mmol−1 cm−1) at 600 nm upon addition of assay buffer (10× buffer containing 0.5 mol/l Tris–HCl, pH 8.1, 1% BSA, 10 μmol/l antimycin A, 3 mmol/l KCN, 0.5 mmol/l coenzyme Q1) [29]. The reaction was started by adding 200 μmol/l NADH and scanned at 600 nm for 2 min; 3 μmol/l rotenone was added into the above system as the blank.
Assays of succinate-CoQ oxidoreductase (complex II), CoQ-cytochrome c reductase (complex III), and cytochrome c oxidase (complex IV) activities were performed as follows [11, 12, 28]. Briefly, complex II activity was measured in assay buffer (10× buffer containing 0.5 mol/l phosphate buffer, pH 8.1, 1% BSA, 10 μmol/l antimycin A, 3 mmol/l KCN, 0.5 mmol/l coenzyme Q1) with mitochondria at a final concentration of 20 μg/ml and 60 μmol/l DCPIP; the reaction was started with 10 mmol/l succinate and scanned at 600 nm for 2 min at 30°C. Complex III activity was assayed by monitoring the reduction of cytochrome c at 550 nm (ε = 18.5 mmol−1 cm−1) upon the addition of assay buffer (10× buffer containing 1 mol/l phosphate buffer, pH 6.5, 3 mmol/l KCN, 0.8% tween-20, 2 mmol/l decylubiquinol) with 60 μmol/l cytochrome c; the reaction was started with 2 μg/ml mitochondria and scanned at 550 nm with the reference at 540 nm for 2 min. For the analysis of complex IV activity, assay buffer contained 50 mmol/l phosphate buffer, 0.1% BSA, 0.2% tween-20 and 60 μmol/l reduced cytochrome c. The reaction was scanned at 550 nm with the reference at 540 nm.
Complex V activity was measured as oligomycin-sensitive, Mg2+-ATPase activity. The process was performed by measuring the increase in NADPH absorption at 340 nm upon the addition of 25 μl of 10 mmol/l ADP to 225 μl of a mixture of 10 mmol/l HEPES pH 8.0, 20 mmol/l succinate, 20 mmol/l glucose, 3 mmol/l MgCl2, 11 mmol/l AMP, 0.75 mmol/l NADP+, 10 mmol/l K2HPO4, 4 μ/ml hexokinase, 2 μ/ml glucose-6-phosphate dehydrogenase and 60 μg/ml mitochondria [30].
The activity of KGDH was assayed by measuring the reduction of NAD+ at 340 nm [12]. Briefly, a mitochondrial suspension was diluted with water to 0.5 mg/ml. After adding different concentrations of MDA (or an equal volume of 0.01N HCl as control), the mixture was incubated at 30°C for 5 min. Then an aliquot was added to an assay mixture containing 200 μmol/l TPP, 0.5 mmol/l NAD+, 130 μmol/l coenzyme A, 2.5 μmol/l rotenone in plate wells (to make a final mitochondrial concentration of 50 μg/ml). The reaction was started with 2 mmol/l α-ketoglutarate and scanned at 340 nm (SPECTRAmax 190, Molecular Devices Corporation, USA) for 2 min to measure NADH generation.
The PDH assay was carried out according to Hinman’s method [31]. Briefly, MDA was reacted with a mitochondrial suspension; 100 μl of the mixture was then added to an assay buffer containing 2.5 mmol/l NAD+, 0.1 mmol/l coenzyme A, 0.2 mmol/l TPP, 0.3 mmol/l dithiothreitol, 1 mmol/l MgCl2, 1 mg/ml BSA, 0.05 mol/l phosphate buffer, pH 7.8, 0.6 mmol/l INT, 0.1 mg/ml dihydrolipoamide dehydrogenase; the reaction was started with 5 mmol/l pyruvate and scanned at 500 nm for 5 min. All assays were performed at 30°C.
Detection of Protein Carbonyls
Protein carbonyls in brain mitochondria were assayed with the Oxyblot protein oxidation detection kit (Chemicon, CA, USA). Carbonyl groups on the protein side chains were derivatized to 2,4,-dinitrophenylhydrazones ((DNP)-hydrazones), then separated by polyacrylamide gel electrophoresis followed by western blotting [4]. Another polyacrylamide resolving gel (12%, w/v) loaded with the same amount of samples was electrophoresed and stained with Coomassie Brilliant Blue R250 as loading control.
Measurement of Reduced Glutathione (GSH) in Rat Brain Mitochondria
The GSH level was evaluated by determining total thiols in brain mitochondria. 5, 5′-dithiobis (2-nitrobenzoic acid (DTNB) reacts with thiols to form a yellow product. The optical density, measured at 412 nm, is directly proportional to the glutathione concentration in the sample using the GSH Detection Kit (Bioassay Systems, CA).
Assays for Superoxide Dismutase and Catalase in Rat Brain Mitochondria
Superoxide dismutase (SOD; E.C.: 1.15.1.1) activity was assayed using the SOD Detection Kit (Sigma, St. Louis, MO). The kit utilizes Dojindo’s highly water-soluble tetrazolium salt, WST-1 (2-(4-iodophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)- 2H-tetrazolium, monosodium salt) that produces a water-soluble formazan dye upon reduction with superoxide anion. The rate of the reduction with superoxide anion is linearly related to xanthine oxidase (XO) activity, and is inhibited by SOD; therefore, the inhibitory activity of SOD or SOD-like materials can be determined colorimetrically. In the present study, SOD activities were examined in brain mitochondria following MDA exposure without adding any mitochondrial substrates.
Data Analysis
Data were expressed as mean ± SD and statistical differences were calculated by one-way analysis of variance (ANOVA) and Tukey post hoc comparisons, using SPSS statistical software.
Results
Effects of MDA on Mitochondrial Respiration
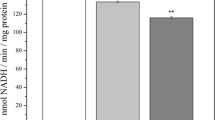
Respiratory activities related to complex I or to complex II were, respectively, measured with malate/glutamate or succinate as substrates (Fig. 1). With malate/glutamate as respiration substrates, state 3 respiration decreased with MDA concentrations as low as 0.2 μmol/mg. With succinate applied as substrate, state 3 respiration significantly declined with MDA concentrations of ≥0.4 μmol MDA/mg (Fig. 1), and the state 4 rate was mildly decreased by 1.6 and 2 μmol MDA/mg (data not shown). At the highest MDA dose of 2 μmol/mg, both complex I-linked and complex II-linked respiration significantly dropped by 40% and 54%, respectively, compared to control. The decline in mitochondrial respiration indicates that MDA causes severe impairment of the integrity of both the outer and inner brain mitochondrial membranes.

Inhibitory effects of MDA on the state 3 respiration of isolated rat brain mitochondria. Respiration unit values in nmol O2 min−1 mg−1 protein display the mean ± SD of three independent experiments, each experiment performed in triplicate. *Significantly different versus absence of MDA, P < 0.05
Effects of MDA on Mitochondrial Membrane Potential
As shown in Fig. 2, MDA dose-dependently inhibited brain mitochondrial potential. At 2 μmol MDA/mg, the potential exhibited a 50% drop compared to the control without MDA, which was consistent with the dysfunction of mitochondrial respiration induced by MDA. This implies that mitochondrial morphology was quickly destroyed by MDA exposure.

Inhibitory effect of MDA on the membrane potential of rat brain mitochondria. Mitochondrial suspensions were exposed to various concentrations of MDA (or an equal volume of 0.01N HCl as control) from 0 to 2 μmol/mg at 30°C for 5 min. The assay of membrane potential was performed immediately after insult using flow cytometry and JC-1 fluorescence detection. Values are mean ± SD of three experiments, each experiment performed in triplicate. *Significantly different versus absence of MDA, P < 0.05
Effects on Respiratory Chain Complex Activities
Respiration and oxidative phosphorylation in mitochondria are carried out by five complexes (I–V). We examined the effects of MDA on the activity of each complex. We found complex I and II activities decreased significantly at MDA concentrations ≥0.2 μmol/mg, progressively decreasing with increasing MDA dose (Fig. 3). However, complexes III and IV exhibited resistance to MDA insult over the entire range of 0–2 μmol/mg (data not shown). The activity of complex V was not significantly inhibited at MDA concentrations less than 1.6 μmol/mg (Fig. 3).

Inhibitory effects of MDA on complex I, II and V activities in isolated mitochondria of rat brain. Mitochondrial suspensions were exposed to various concentrations of MDA (or an equal volume of 0.01N HCl as control) from 0 to 2 μmol/mg at 30°C for 5 min. The complex I, II and V activities were determined immediately after insult. Values in percent relative to control are mean ± SD of three experiments, each experiment performed in triplicate. The control activities of complexes I, II, and V in brain mitochondria without insult were 86.49 ± 3.08 nmol DCPIP min−1 mg−1, 76.55 ± 6.42 nmol DCPIP min−1 mg−1 and 412.54 ± 22.57 nmol NADPH min−1 mg−1, respectively, set to 100% in each case. *Significantly different versus absence of MDA, P < 0.05
Effects on PDH and KGDH Activity
PDH and KGDH are the key dehydrogenases that provide NADH as substrate for the respiratory chain. PDH activity was significantly decreased when 0.1 μmol MDA/mg was present, and slightly decreased further with increasing MDA doses (Fig. 4). KGDH activity was significantly decreased at 0.4 μmol MDA/mg. Compared with control, 50% of KGDH activity was lost at 2 μmol MDA/mg (Fig. 4).

Inactivating effect of MDA on PDH and KGDH activities in isolated mitochondria of rat brain. Mitochondrial suspensions were exposed to various concentrations of MDA (or an equal volume of 0.01N HCl as control) from 0 to 2 μmol/mg at 30°C for 5 min. The PDH and KGDH activities were determined immediately after insult. Values are mean ± SD of three experiments, each experiment performed in triplicate. The control activities of PDH and KGDH in brain mitochondria without insult were 12.52 ± 0.53 nmol INT min−1 mg−1 and 80.63 ± 10.20 nmol NAD+ min−1 mg−1, respectively, set to 100% in each case. *Significantly different versus absence of MDA, P < 0.05
Effects on Protein Carbonyls
Brain mitochondria were significantly modified by MDA and showed higher levels of protein carbonyls at concentrations as low as 0.002 μmol MDA/mg. Compared with the vehicle-treated control, protein carbonyls increased more than nine times when brain mitochondria were exposed to 2 μmol MDA/mg (Fig. 5a–c).

Immunochemical detection of carbonylated protein isolated from rat brain mitochondria treated with MDA. DNP hydrazine-treated proteins were resolved on an SDS/polyacrylamide gel, transferred to a PVDF membrane, and the carbonylated proteins were detected by reaction with anti-DNP polyclonal antibody. Carbonyl band intensities were normalized with Coomassie Blue R250. a: immunoblot of protein carbonyls; brain mitochondria were exposed to 0–2 μmol/mg MDA as indicated. b: proteins were stained with Coomassie Blue R250 as loading control. c: quantitative results of ratio of intensity of protein carbonyls to intensity of protein. Values are mean ± SD of three experiments. The blot density of the brain mitochondria without insult is set to 100%. *Significantly different versus absence of MDA, P < 0.05
Effects on Oxidant Generation and the Antioxidant Defense System
MDA itself is an oxidant that can directly cause oxidative damage such as protein modification; it can also cause generation of more ROS to aggravate oxidative damage. As shown in Fig. 6, MDA exposure remarkably increased ROS in brain mitochondria at 0.2 μmol MDA/mg. At 2 μmol MDA/mg, ROS production increased more than three times compared to the control.

Effect of MDA on ROS (reactive oxygen species) generation in isolated rat brain mitochondria. Mitochondrial suspensions were exposed to various concentrations of MDA (or an equal volume of 0.01N HCl as control) from 0 to 2 μmol/mg at 30°C for 5 min. The oxidant level was determined immediately after insult by DCFH fluorescence using a plate reader with excitation at 480 nm and emission at 530 nm. ROS levels are represented in relative fluorescence units (RFU) per minute. Values are mean ± SD of three experiments, each experiment performed in triplicate. The fluorescence of brain mitochondria without insult is set to 100%. *Significantly different versus absence of MDA, P < 0.05
SOD and GSH, which serve as a mitochondrial antioxidant enzyme and antioxidant, respectively, were measured to evaluate the responses of the mitochondrial antioxidant defense system when brain mitochondria were subjected to an MDA insult. Both were found not very sensitive to MDA. SOD activity in brain mitochondria did not change until 2 μmol MDA/mg were present (Fig. 7); as shown in Fig. 8, GSH levels did not decrease until MDA reached 1.6 μmol/mg.

Inhibitory effect of MDA on the SOD activity of isolated rat brain mitochondria. Values are mean ± SD of three experiments, each experiment performed in triplicate. The SOD activity of brain mitochondria without insult is set to 100%. *Significantly different versus absence of MDA, P < 0.05

Inhibitory effect of MDA on GSH levels in isolated rat brain mitochondria. Values are mean ± SD of three experiments, each experiment performed in triplicate. The GSH level of mitochondria without insult is set to 100%. *Significantly different versus absence of MDA, P < 0.05
Discussion
In the present study, we found that MDA caused an inhibition of both complex I- and complex II-linked respiration, an inhibition of mitochondrial membrane potential, and inhibition of mitochondrial enzyme activities, including the activities of complexes I, II, V, PDH and KGDH. In addition, MDA also caused increases in mitochondrial protein carbonyls and ROS generation, and slight decreases in SOD activity and GSH levels.
The effective concentrations of MDA for inhibiting mitochondrial function are comparable to those of HNE and acrolein in rat brain mitochondria [11]. Acrolein decreases state 3 respiration of brain mitochondria over the range of 0.1–1.0 μmol/mg with the IC50 being 0.4 μmol/mg, and has no reduction in activity of complexes I–V [12, 32]. In rat brain mitochondria, HNE decreases state 3 respiration in the range 20–300 μmol/l (0.2–3 μmol/mg) with an IC50 value of 200 μmol/l (2 μmol/mg), and inhibits complexes II and III in the range of 100–300 μmol/l (1–3 μmol/mg) [11]. In rat brain cortex synaptosomes, HNE concentrations from 1 to 100 μmol/l (but not MDA, hexanal, nonaldehyde, or 2-t-nonenal) impair glutamate transport and mitochondrial function measured by MTT reduction, and increase thiobarbituric acid-reactive substances [32]. That effective concentrations of MDA are similar to those of HNE and acrolein indicates that MDA is no less active in causing mitochondrial dysfunction than either HNE or acrolein, although both of the latter were shown more potent in causing other biological toxicity [5].
Previously, we studied the inhibitory effects of MDA on respiration and mitochondrial enzyme activities in liver mitochondria [14]. Comparing results for complex I-linked respiration, we find that MDA exposure has a similar effective concentration (0.2 μmol MDA/mg) in both brain and liver mitochondria. However, for complex II-linked respiration, brain mitochondria exhibit greater vulnerability (0.4 μmol MDA/mg in brain vs. 0.8 μmol MDA/mg in liver). With respect to the activities of individual enzyme complexes, both complexes I and II in brain exhibited higher susceptibility to MDA than in liver mitochondria. In brain, concentrations of MDA as low as 0.2 μmol/mg inhibited these complexes; while in liver, four times or eight times higher MDA doses (0.8 and 1.6 μmol/mg) were required to cause loss of complex I or complex II activity, respectively. This suggests that brain mitochondria are more vulnerable than liver mitochondria to MDA toxicity. Several potential reasons may underlie the differences in susceptibility to MDA insult between liver and brain mitochondria. First, in contrast to hepatocytes that are continuously subjected to protein and phospholipid turnover and to the elimination of dysfunctional organelles, most brain cells age at the same rate as the animal ages, while ongoing neurogenesis in the adult is spatially restricted and quantitatively negligible [33]. Thus most neurons are long-living cells and exhibit slow turnover of their mitochondria and mitochondrial components, which contributes to the cellular accumulation of dysfunctional mitochondria. Therefore, the higher levels of lipid peroxidation products, and the increased fragility and water permeability of brain mitochondria cause vulnerability to any extra MDA insult [21]. Second, the antioxidant enzyme SOD exhibits lower activity in brain mitochondria than in liver mitochondria [21], which reduces the defense capacity of brain mitochondria against damage induced by external MDA and its product oxidants. Another possibility is that the electron transfer activities of liver mitochondria are much higher than those of brain [34], suggesting more active enzymes per mg are available to contribute to the increased resistance to MDA insult observed in liver mitochondria.
As an electrophilic reagent, MDA at 10 mmol/l at 37°C for 24 h can modify 26 lysine residues out of the total 56 present in BSA [35]. Besides lysine residues, MDA can also modify His, Tyr, Arg, and Met residues to different extents [36]. We hypothesize that the inactivation of enzymes induced by MDA is mainly due to the modification of nucleophilic groups in proteins. As expected, we found that protein carbonyl levels were significantly enhanced by MDA (at concentrations as low as 0.002 μmol/mg) in brain mitochondria, and rose remarkably with increasing MDA.
It is interesting to note that MDA, besides being an oxidative agent that causes direct oxidative damage (such as by oxidatively modifying proteins), also induces more ROS in brain mitochondria. Though not as sensitive in brain mitochondria as ROS induction, antioxidant defense levels, as measured by SOD activity and GSH concentrations, were decreased, but only at high MDA concentrations. These results suggest that MDA can cause brain oxidative damage by two mechanisms: both as a direct oxidant and also as an oxidant inducer.
The results suggest MDA, rather than being merely an end product of oxidative damage to lipids, amino acids, and DNA, is also a mitochondrial toxin which can further impair mitochondrial respiration, membrane potential, mitochondrial complexes I, II, and V, PDH, and KGDH. Our results measuring the effects of MDA on mitochondrial function, along with other studies with HNE and acrolein, suggest that mitochondrial dysfunction is not only caused by ROS directly, but also by oxidative damage by-products as an indirect oxidative injury. Because the mechanism of this indirect injury differs from that of direct free radical damage, the by-product-caused mitochondrial dysfunction associated with aging and age-related degenerative diseases needs to be ameliorated by different strategies, such as by protecting mitochondrial enzyme active sites and reducing MDA, rather than by simply supplying antioxidants. For example, the MDA-caused modification of enzymes may be prevented or ameliorated by dietary supplementation of mitochondrial cofactors and substrates or their precursors [9, 37, 38].
In conclusion, we have revealed that MDA can cause brain mitochondrial toxicity in a number of ways. These include inhibition of mitochondrial respiration, reduction of both membrane potential and enzyme activities, protein modification, ROS induction, and by decreasing the antioxidant defense system. In order to effectively counteract such injury secondary to oxidative damage, a different strategy should be taken to prevent and ameliorate the mitochondrial toxicity associated with aging and neurodegenerative diseases, especially the toxicity to brain mitochondria caused by the by-products of oxidation, such as MDA, HNE and acrolein.
References
Harman D (1961) Prolongation of the normal lifespan and inhibition of spontaneous cancer by antioxidants. J Gerontol 16:247–254
Floyd RA, West M, Hensley K (2001) Oxidative biochemical markers; clues to understanding aging in long- lived species. Exp Gerontol 36:619–640. doi:10.1016/S0531-5565(00)00231-X
Liu J, Mori A (1999) Stress, aging, and brain oxidative damage. Neurochem Res 24:1479–1497. doi:10.1023/A:1022597010078
Levine RL, Garland D, Oliver CN et al (1990) Determination of carbonyl content in oxidatively modified proteins. Methods Enzymol 186:464–478. doi:10.1016/0076-6879(90)86141-H
Esterbauer H, Schaur RJ, Zollner H (1991) Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med 11:81–128. doi:10.1016/0891-5849(91)90192-6
Gutteridge JMC (1981) Thiobarbituric acid-reactivity following iron-dependent free-radicals damage to amino acids and carbohydrates. FEBS Lett 128:343–346. doi:10.1016/0014-5793(81)80113-5
Burger RM, Berkowitz AR, Peisach J, Horwitz SB (1980) Origin of malondialdehyde from DNA degraded by Fe(II) x bleomycin. J Biol Chem 255:11832–11838
Cheeseman KH, Emery S, Maddix SP, Slater TF, Burton GW, Ingold KU (1988) Studies on lipid peroxidation in normal and tumour tissues. The Yoshida rat liver tumour. Biochem J 250:247–252
Liu J, Ames BN (2005) Reducing mitochondrial decay with mitochondrial nutrients to delay and treat cognitive dysfunction, Alzheimer’s disease, and Parkinson’s disease. Nutr Neurosci 8:67–89. doi:10.1080/10284150500047161
Liu J, Atamna H, Hirohiko K, Ames BN (2002) Delaying brain mitochondrial decay and aging with mitochondrial antioxidants and metabolites. Ann N Y Acad Sci 959:133–166
Picklo MJ, Amarnath V, McIntyre JO, Graham DG, Montine TJ (1999) 4-Hydroxy-2(E)-nonenal inhibits CNS mitochondrial respiration at multiple sites. J Neurochem 72:1617–1624. doi:10.1046/j.1471-4159.1999.721617.x
Humphries KM, Szweda LI (1998) Selective inactivation of alpha-ketoglutarate dehydrogenase and pyruvate dehydrogenase: reaction of lipoic acid with 4-hydroxy-2-nonenal. Biochemistry 37:15835–15841. doi:10.1021/bi981512h
Neely MD, Zimmerman L, Picklo MJ et al (2000) Congeners of N(alpha)-acetyl-l-cysteine but not aminoguanidine act as neuroprotectants from the lipid peroxidation product 4-hydroxy-2-nonenal. Free Radic Biol Med 29:1028–1036. doi:10.1016/S0891-5849(00)00411-1
Long J, Wang X, Gao H et al (2006) Malonaldehyde acts as a mitochondrial toxin: inhibitory effects on respiratory function and enzyme activities in isolated rat liver mitochondria. Life Sci 79:1466–1472. doi:10.1016/j.lfs.2006.04.024
Greilberger J, Koidl C, Greilberger M et al (2008) Malondialdehyde, carbonyl proteins and albumin-disulphide as useful oxidative markers in mild cognitive impairment and Alzheimer’s disease. Free Radic Res 42:633–638. doi:10.1080/10715760802255764
Bourdel-Marchasson I, Delmas-Beauvieux MC, Peuchant E et al (2001) Antioxidant defences and oxidative stress markers in erythrocytes and plasma from normally nourished elderly Alzheimer patients. Age Ageing 30:235–241. doi:10.1093/ageing/30.3.235
Nagai T, Yamada K, Kim HC et al (2003) Cognition impairment in the genetic model of aging klotho gene mutant mice: a role of oxidative stress. FASEB J 17:50–52
Zhou XM, Cao YL, Dou DQ (2006) Protective effect of ginsenoside-Re against cerebral ischemia/reperfusion damage in rats. Biol Pharm Bull 29:2502–2505. doi:10.1248/bpb.29.2502
Wang QL, Lin M, Liu GT (2001) Antioxidative activity of natural isorhapontigenin. Jpn J Pharmacol 87:61–66. doi:10.1254/jjp.87.61
Long J, Gao F, Tong L, Cotman CW, Ames BN, Liu J (2008) Mitochondrial decay in the brains of old rats: ameliorating effect of alpha-lipoic acid and acetyl-l-carnitine. Neurochem Res. doi:10.1007/s11064-008-9850-2
Navarro A, Boveris A (2004) Rat brain and liver mitochondria develop oxidative stress and lose enzymatic activities on aging. Am J Physiol Regul Integr Comp Physiol 287:R1244–R1249. doi:10.1152/ajpregu.00226.2004
Stephan K, Chang M, Brass EP, Hoppel CL (1991) Decreased activities of ubiquinol: ferricytochrome c oxidoreductase(complex and ferrocytochrome c:oxygen oxidoreductase (in liver mitochondria from rats with hydroxycobalamin-induced methylmalonic aciduria. J Biol Chem 266:20998–21003
Liu J, Yeo HC, Doniger SJ, Ames BN (1997) Assay of aldehydes from lipid peroxidation: gas chromatography-mass spectrometry compared to thiobarbituric acid. Anal Biochem 245:161–166. doi:10.1006/abio.1996.9990
Moreira PI, Santos MS, Moreno AM, Seica R, Oliveira CR (2003) Increased vulnerability of brain mitochondria in diabetic (Goto-Kakizaki) rats with aging and amyloid-beta exposure. Diabetes 52:1449–1456. doi:10.2337/diabetes.52.6.1449
Cossarizza A, Ceccarelli D, Masini A (1996) Functional heterogeneity of an isolated mitochondrial population revealed by cytofluorometric analysis at the single organelle level. Exp Cell Res 222:84–94. doi:10.1006/excr.1996.0011
Zamzami N, Metivier D, Kroemer G (2000) Quantitation of mitochondrial transmembrane potential in cells and in isolated mitochondria. Methods Enzymol 322:208–213. doi:10.1016/S0076-6879(00)22021-1
Hagen TM, Liu J, Lykkesfeldt J et al (2002) Feeding acetyl-l-carnitine and lipoic acid to old rats significantly improves metabolic function while decreasing oxidative stress. Proc Natl Acad Sci USA 99:1870–1875. doi:10.1073/pnas.261708898
Yang S, Tan TMC, Wee A, Leow CK (2004) Mitochondrial respiratory function and antioxidant capacity in normal and cirrhotic livers following partial hepatectomy. CMLS, Cell Mol Life Sci 61:220–229. doi:10.1007/s00018-003-3357-4
Janssen AJ, Trijbels FJ, Sengers RC et al (2007) Spectrophotometric assay for complex I of the respiratory chain in tissue samples and cultured fibroblasts. Clin Chem 53:729–734. doi:10.1373/clinchem.2006.078873
Zheng J, Ramirez VD (2000) Inhibition of mitochondrial proton F0F1-ATPase/ATP synthase by polyphenolic phytochemicals. Br J Pharmacol 130:1115–1123. doi:10.1038/sj.bjp.0703397
Hinman LM, Blass JP (1981) An NADH-linked spectrophotometric assay for pyruvate dehydrogenase complex in crude tissue homogenates. J Biol Chem 256:6583–6586
Keller JN, Mark RJ, Bruce AJ et al (1997) 4-Hydroxynonenal, an aldehydic product of membrane lipid peroxidation, impairs glutamate transport and mitochondrial function in synaptosomes. Neuroscience 80:685–696. doi:10.1016/S0306-4522(97)00065-1
Gould E, McEwen BS (1993) Neuronal birth and death. Curr Opin Neurobiol 3:676–682. doi:10.1016/0959-4388(93)90138-O
Long J, Wang X, Gao H et al (2007) d-Galactose toxicity in mice is associated with mitochondrial dysfunction: protecting effects of mitochondrial nutrient r-alpha-lipoic acid. Biogerontology 8:373–381. doi:10.1007/s10522-007-9081-y
Beppu M, Fukata Y, Kikugawa K (1988) Interaction of malondialdehyde-modified bovine serum-albumin and mouse peritoneal-macrophages. Chem Pharm Bull (Tokyo) 36:4519–4526
Buttkus H (1967) The reaction of myosin with malonaldehyde. J Food Sci 32:432–434. doi:10.1111/j.1365-2621.1967.tb09703.x
Ames BN, Elson SI, Silver EA (2002) High-dose vitamin therapy stimulates variant enzymes with decreased coenzyme binding affinity (increased K(m)): relevance to genetic disease and polymorphisms. Am J Clin Nutr 75:616–658
Ames BN, Liu J, Atamna H, Hagen TM (2003) Delaying the mitochondrial decay of aging in the brain. Clin Neurosci Res 2:331–338. doi:10.1016/S1566-2772(03)00010-0
Acknowledgments
The authors thank Dr. Edward Sharman for his critical reading and careful editing of this manuscript. This study was supported by an Outstanding Oversea Scholars Award from the Chinese Academy of Sciences, Shanghai Pujiang Talent Award, and a Hi-Sun Science and Technology Prize from Zhejiang Hi-Sun Pharmaceuticals, Inc.
Author information
Authors and Affiliations
Corresponding author
Additional information
Special issue article in honor of Dr. Akitane Mori.
Rights and permissions
About this article
Cite this article
Long, J., Liu, C., Sun, L. et al. Neuronal Mitochondrial Toxicity of Malondialdehyde: Inhibitory Effects on Respiratory Function and Enzyme Activities in Rat Brain Mitochondria. Neurochem Res 34, 786–794 (2009). https://doi.org/10.1007/s11064-008-9882-7
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-008-9882-7