
Abstract
It has been shown that synergistic toxic effects of quinolinic acid (QUIN) and glutaric acid (GA), both in isolated nerve endings and in vivo conditions, suggest the contribution of these metabolites to neurodegeneration. However, this synergism still requires a detailed characterization of the mechanisms involved in cell damage during its occurrence. In this study, the effects of subtoxic concentrations of QUIN and/or GA were tested in neuronal cultures, co-cultures (neuronal cells + astrocytes), and mixed cultures (neuronal cells + astrocytes + microglia) from rat cortex and striatum. The exposure of different cortical and striatal cell cultures to QUIN + GA resulted in cell death and stimulated different markers of oxidative stress, including reactive oxygen species (ROS) formation; changes in the activity of antioxidant enzymes such as superoxide dismutase, catalase, and glutathione peroxidase; and depletion of endogenous antioxidants such as -SH groups and glutathione. The co-incubation of neuronal cultures with QUIN + GA plus the N-methyl-d-aspartate antagonist MK-801 prevented cell death but not ROS formation, whereas the antioxidant melatonin reduced both parameters. Our results demonstrated that QUIN and GA can create synergistic scenarios, inducing toxic effects on some parameters of cell viability via the stimulation of oxidative damage. Therefore, it is likely that oxidative stress may play a major causative role in the synergistic actions exerted by QUIN + GA in a variety of cell culture conditions involving the interaction of different neural types.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The metabolic recessive disease glutaric acidemia type I (GA I) is caused by deficiency of the activity of glutaryl-CoA dehydrogenase (GCDH, EC 1.3.99.7), an enzyme that participates in the catabolism of lysine, hydroxylysine, and tryptophan. The lack of activity of this enzyme induces the accumulation of high amounts of glutaric (GA) and 3-hydroxyglutaric (3-HGA) acids in different tissues, as well as in the blood, urine, and cerebrospinal fluid of patients with this disorder [1, 2]. Neurological symptoms are predominant in this cerebral organic acidemia. Macrocephaly and frontotemporal cortical atrophy are features observed at birth. Acute bilateral striatal degeneration occurs between 3 to 36 months of age following encephalopathic episodes often linked to infections, fever, or prolonged fasting; during these events, the concentrations of the accumulating metabolites reach millimolar concentrations [3]. Then, patients progress toward dyskinesia, dystonia, hypotonia, seizures, muscle stiffness, and spasticity [2, 4,5,6].
Neurodegenerative disorders course with different toxic events, one of which is inflammation, a process that contributes to progression of cell and tissue damage. Several parameters of brain energy metabolism are affected by inflammatory processes, further accounting for oxidative stress and excitotoxicity [7,8,9]. Although release of inflammatory cytokines by microglia and macrophages in the CNS is associated with the pathogenesis of neurodegenerative disorders, the precise role of these molecules remains unsolved [10]. One of their known actions is the activation of the kynurenine pathway (KP), the catabolic pathway of tryptophan degradation, that results in the production of intermediate metabolites with neuroactive and/or redox properties. One of these metabolites, quinolinic acid (2,3-pyridine-dicarboxylic acid or QUIN), is necessary to adequate brain functioning when synthesized at normal levels [11, 12]; however, when produced in excess, it plays a toxic role, participating in neurological disorders coursing with excitotoxicity, such as Parkinson’s and Huntington’s diseases, as well as in GA I [12,13,14]. Despite QUIN has been shown to be able of inducing neurotoxic effects by different mechanisms [15], the most important is related with overactivation of N-methyl-d-aspartate receptors (NMDAr) and the subsequent increase of cytosolic Ca2+ concentration, further leading to free radical formation and oxidative damage, mitochondrial dysfunction, cytochrome c release, and ATP exhaustion [15, 16].
Regarding to GA I, several in vitro and in vivo studies have shown that both GA and 3-HGA, the most densely accumulated metabolites in this disorder, are also able to induce excitotoxicity, oxidative stress, and impairment of cellular energy metabolism in the rat brain [17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39]. Although this topic has already been extensively investigated, the toxic mechanisms underlying the acute striatal degeneration observed in GA I patients during metabolic decompensation are not yet well described. It has been suggested that QUIN, a metabolite that is increased during these processes, might potentially contribute to the striatal GA I pathogenesis, promoting severe neurological symptoms [14]. Noteworthy, it is known that the symptomatology of patients suffering from GA I worsens during infection or immunization. Therefore, the hypothesis that high accumulation of organic acids (GA and 3-HGA) during crises associated with increased concentrations of QUIN is potentially relevant to explain the pathophysiology of striatal damage in GA I patients. In turn, this concept suggests that these molecules could exert a toxic synergism to favor a scenario of neuronal cell dysfunction and brain tissue degeneration. In support to this concept, we have recently demonstrated that QUIN can exert such synergism with GA, 3-HGA, and other organic acids typically accumulating in disorders of propionate metabolism, including methylmalonic acid (MMA) and propionic acid (PA) [40]. These effects were evidenced by toxic endpoints such as loss of mitochondrial function, increased production of reactive oxygen species (ROS), and enhanced lipid peroxidation, all of them estimated in freshly prepared rat brain synaptosomal P2/mitochondrial fractions. In addition, we have demonstrated that QUIN is able to enhance toxic endpoints estimated in the striatum of knockout mice for glutaryl-CoA dehydrogenase (Gcdh −/−), an in vivo transgenic mice model of GA I [41]. Among the markers evaluated, it was found that QUIN stimulated lactate release, reduced the activities of complex IV and creatine kinase, increased lipid and protein oxidation, augmented ROS formation, altered the activities of antioxidant enzymes, and recruited pro-inflammatory signals, all in the striatal tissue of the mutant mice compared with the wild type mice (Gcdh +/+). Taken together, the results of these studies support a possible contribution of QUIN to the pathophysiology of GA I during the occurrence of episodes of metabolic decompensation and inflammatory events. However, precise information on how much this toxic synergism affects neuronal cells from the striatum and cortex, and through which mechanisms these damage is produced, is a question that remains unsolved. Therefore, in the present study, we cultured rat neural cells under different experimental conditions (monocultures, co-cultures with astrocytes, and mixed cultures containing neurons + astrocytes + microglia) and exposed them to the presence of QUIN and/or GA in order to establish the precise contribution of this synergic relationship to cell damage and the mechanisms by which the alterations are produced. For this purpose, central components of cell viability [lactate dehydrogenase (LDH) leakage; annexin and propidium iodide (PI) incorporation assays], redox homeostasis [reactive oxygen species (ROS) formation; superoxide dismutase (SOD), catalase (CAT), and glutathione peroxidase (GPx) activities; and the total thiol (-SH) and reduced glutathione (GSH) contents], neuronal plasticity [neurite length and neurite/neuron ratio], and reactive gliosis [glial fibrillary acidic protein (GFAP) increase] were all estimated in the different cultures exposed to QUIN +/− GA for 24 h, thus simulating an acute exposure to these metabolites. Our results reveal that the toxic synergism produced by these molecules augmented cell death, increased ROS formation, and altered antioxidant enzymes activities in different cell cultures, therefore suggesting that oxidative stress might play a major causative role in this new toxic model. These findings are discussed in terms of their relevance for the proposal of a new toxic model for GA I.
Materials and Methods
Reagents
All chemicals were of analytical grade. Phalloidin-fluorescein isothiocynate (FITC), bis-acrylamide, QUIN, acrylamide, polylysine, anti-β III-tubulin, anti-mouse IgG, anti-mouse IgG-FITC, peroxidase-conjugated anti-mouse IgG, and all reagents for cell culture were obtained from Sigma-Aldrich Co. (St. Louis, MO). 4,6-Diamidino-2-phenylindole (DAPI) was from Calbiochem (La Jolla, CA). Polyclonal glial fibrillary acidic protein (GFAP) antibody was from Dako (Glostrup, Denmark). The enhanced chemiluminescence (ECL) detection kit was from Millipore (Billerica, MA). Anti-β-actin and anti-MAP2 antibodies were from Cell Signaling Technology (Boston, MA). Fetal bovine serum (FBS), Dulbecco’s modified Eagle’s medium nutrient mixture F-12 (DMEM/F12), Neurobasal medium, B-27 supplement, fungizone, trypsine, and penicillin/streptomycin were purchased from Life Technologies (Carlsbad, CA). All solutions were prepared on the same day of the experiments and the pH was adjusted to 7.2–7.4 in the appropriate buffer for each method.
Ethics Statement
For animal handling, we followed the experimental protocol described in the “Guide for the Care and Use of Laboratory Animals” (NIH publication 85–23, revised 1985). Our protocol was approved by the Ethics Committee for Animal Research of the Universidade Federal do Rio Grande do Sul (No. 18,266). During the experiments, all efforts were made to minimize the number of animals used and their suffering.
Animals
Pregnant Wistar rats (weighing 200–250 g) were obtained from our breeding stock. All rats were kept in 12 h light-dark cycles at a constant temperature (22 °C) in the colony room, with food and water ad libitum. On gestational day 22, animals were constantly observed. At birth, mothers and pups were euthanized by decapitation, and pups were used for astrocyte and microglial cultures. For neuronal cultures, the embryos from mothers euthanized on gestational day 18 were used.
Cell Cultures
Cell cultures were prepared strictly according to the methods previously described by us [42].
Primary astrocyte cultures were obtained and prepared from the striatum and cerebral cortex of newborn (0–1 day old; P0) Wistar rats. The animals were euthanized by decapitation and their striatum was removed. Dissociated cells were plated in DMEM/F12 supplemented with 10% FBS (pH 7.4), glucose (33 mM), glutamine (2 mM), and sodium bicarbonate (3 mM) in 34.8 and 15.6-mm-diameter wells (six- and 24-well plates; Corning, NY) that had previously been coated with polylysine (1.5 mg/ml; Sigma). Cells were cultured at 37 °C, in a 5% CO2 atmosphere in a humid incubator, replacing the media every 3 days. An approximate count of 2 × 104 astrocytic cells was seeded on polyornitine-treated coverslips and placed on a 24-well plate, or 2 × 105 astrocytic cells were seeded in a six-well plate. Once astrocytes reached semi-confluence (15 days in vitro; DIV), culture medium was removed and cells were incubated for additional 24 h at 37 °C in a 5% CO2 atmosphere in DMEM/F12 without FBS, in the presence of 50 μM QUIN and/or 5 mM GA. Control cultures received the same incubation medium without QUIN or GA. QUIN and GA were prepared prior each treatment and dissolved in DMEM/F12 without FBS. The QUIN concentration employed in this study was established as the half of the concentration needed to alter astrocytic morphology without causing cell death [42]. The GA concentration was established on the basis of previous studies demonstrating moderate or null toxicity in the cerebral cortex of young rats [43].
The striatum and cerebral cortex of 18 Wistar rats at embryonic stage were used to prepare neuronal cell cultures, as previously described [42]. Primary neuronal cell cultures were prepared from the striatum of embryonic (18 days old) Wistar rats, as previously described [44]. For this purpose, pregnant Wistar rats were euthanized, embryos removed, decapitated and their striatum was dissected. Both enzymatic and mechanic digestion consisted of an incubation with 0.05% trypsin for 20 min at 37 °C and further DNase I treatment (0.24 mg/ml in DMEM/F12 supplemented with 10% FBS). Dissociated cells were plated in Neurobasal medium supplemented with 0.5 mM glutamine and B27 supplement (20 ml/l). An approximate number of 5 × 104 neuronal cells were plated on polyornitine-treated coverslips placed on a 24-well plate, or 1.5 × 106 neuronal cells in a six-well plate. Neurobasal medium served to keep neuronal cultures for 24 h; later, culture medium was removed by suction, and cells were incubated for 7 days in a humid incubator (37 °C; 5% CO2). The neuronal culture medium was replaced at 8 DIV by a medium containing 50 μM QUIN and/or 5 mM GA. Control cultures received the same incubation medium without QUIN or GA. QUIN and GA were prepared prior each treatment in Neurobasal medium, and control cultures received no QUIN or GA supplementation. The QUIN dose was established on the basis of previous studies [42, 44], and it represents a dose near the one used to alter neuronal morphology without causing cell death. The GA concentration was established on the basis of previous studies demonstrating moderate or null toxicity in the cerebral cortex of young rats [43], yet this and even higher concentrations are found in post mortem brain of GAcidemic patients during crises of metabolic decompensation frequently occurring during or after inflammatory processes [1, 2].
Primary microglial cultures were obtained from the striatum and cerebral cortex of 1-day-old pups as previously described [42, 45]. Briefly, the striatal tissue of animals was subjected to removal of the meninges and blood vessels, and further triturated. Cells were seeded in DMEM/F12/10% FBS, and 14 DIV after initial seeding, microglial cells were removed from astrocytes monolayer by shaking for 30 min, collected the supernatant, centrifuged, and plated on top of non-related astrocyte monolayers (15 DIV) alone. An estimation of 2 × 104 microglial cells were plated on polyornitine-treated coverslips placed on a 24-well plate, or 2 × 105 microglial cells in a six-well plate.
Freshly obtained and isolated striatal and cortical neurons plated on top of non-related astrocyte monolayers (15 DIV) alone, or non-treated astrocyte monolayers previously associated with microglial cells, served to prepare co-cultures or mixed cultures, respectively. An estimated 5 × 104 neuronal cells were plated on polyornitine-treated coverslips placed on a 24-well plate, or 1.5 × 106 neuronal cells in a six-well plate. Both co-cultures and mixed cultures were kept at 37 °C for 7 days in a humidified 5% CO2 air atmosphere. When neurons were at 7 DIV, the cultures were used for exposure to QUIN and/or GA for additional 24 h. QUIN and GA were freshly prepared prior each treatment and dissolved in DMEM/F12 without FBS. Control cultures received the same incubation medium without QUIN or GA.
Lactate Dehydrogenase Activity Assay
To evaluate plasma membrane integrity, LDH release was determined by monitoring the leakage of the cytosolic LDH to extracellular medium. Briefly, 20 μl of supernatant, 60 μl potassium phosphate buffer 0.1 M, and 10 μl NADH 1.8 mM was incubated during 5 min. Then, 10 μl pyruvate solution (0.6 mM) was added and LDH was measured spectrophotometrically at 340 nm each 15 s during 4 min, following the rate of conversion of reduced nicotinamide adenine dinucleotide to oxidized nicotinamide adenine nucleotide.
Annexin/Propidium Iodide Assay for Cell Death Estimation
The apoptotic detection assay was carried out by surface labeling with the Ca2+-dependent phosphatidylserine-binding protein annexin V. After they had been incubated with the metabolites for 24 h, cells were recovered from the culture plates by 0.05% trypsin-EDTA treatment, centrifuged 1000 rpm for 5 min, and washed once with phosphate-buffered saline (PBS). Cells were labeled by incubation with annexin V-FITC and propidium iodide (PI) in a binding buffer (apoptosis detection kit I-556547; BD Pharmingen) for 15 min at room temperature in the dark, according to the manufacturer’s instruction. Stained cells were acquired (10,000 events) on a FACS Calibur flow cytometer. Analysis was performed in the FlowJo Software.
Oxidative Stress Parameters and Antioxidant Defenses
ROS Formation Assay by 2′,7′-Dihydrodichlorofluorescein (DCFH) Oxidation
ROS production was measured according to the method of LeBel et al. [46], based on the oxidation of 2′7′-dichlorofluorescein (H2DCF). The samples were incubated in a medium containing 100 μM H2DCF-DA solution. Reaction produces the fluorescent compound dichlorofluorescein (DCF) which is measured at λem = 488 nm and λex = 525 nm.
Superoxide Dismutase Activity Assay
SOD activity assay is based on the capacity of pyrogallol to autoxidize, a process highly dependent on superoxide, which is a substrate for SOD. The inhibitor of pyrogallol autoxidation occurs in the presence of SOD, whose activity can be then indirectly assayed spectrophotometrically at 420 nm. A calibration curve was performed with purified SOD as a standard, in order to calculate the activity of SOD present in the samples. SOD activity was expressed as the amount of enzyme that inhibits the oxidation of epinephrine by 50%, corresponding to one unit.
Catalase Activity Assay
CAT activity was assayed using SpectraMax M5/M5 Microplate Reader (Molecular Devices, MDS Analytical Technologies, Sunnyvale, CA, USA). This method is based on the disappearance of hydrogen peroxide (H2O2) at 240 nm in a reaction medium containing 20 mM H2O2, 0.1% Triton X-100, 10 mM potassium phosphate buffer pH 7.0, and 0.1–0.3 mg protein/ml. One CAT unit is defined as 1 mM of H2O2 consumed per minute.
Glutathione Peroxidase Activity Assay
GPx activity was measured using tert-butyl-hydroperoxide as substrate. NADPH disappearance was monitored at 340 nm using SpectraMax M5/M5 Microplate Reader (Molecular Devices, MDS Analytical Technologies). The medium contained 2 mM GSH, 0.15 U/ml GSH reductase, 0.4 mM sodium azide, 0.5 mM tert-butyl-hydroperoxide, and 0.1 mM NADPH. One GPx unit is defined as 1 mM of NADPH consumed per minute, and the specific activity is represented as GPx units/milligram protein.
Sulfhydryl Content Assay
This assay was performed as described by Aksenov and Markesbery [47], which is based on the reduction of 5,5′-dithio-bis(2-nitrobenzoic acid) (DTNB) by sulfhydryl groups, which in turn, become oxidized (disulfide) generating a yellow derivative (TNB) whose absorption is measured spectrophotometrically at 412 nm. Briefly, 15 μl of homogenate were added to 275 μl of phosphate buffer saline pH 7.4 containing 1 mM EDTA. The reaction was started by the addition of 10 μl of 10 mM DTNB and incubated for 30 min at room temperature in a dark room. The sulfhydryl content is inversely correlated to oxidative damage to the protein.
Reduced Glutathione Levels
GSH levels were estimated based on the method described by Browne and Armstrong [48]. Deproteinized samples were mixed to 100 mM sodium phosphate buffer plus 5 mM EDTA. In a dark room, the fluorescent compound (O-phthaldialdehyde 1 mg/ml diluted in methanol) was added to the media containing the samples. After 15 min, the fluorescence was measured with 420-nm emission and 350-nm excitation. A standard curve was performed using purified GSH. All samples were run in triplicate.
Immunoflorescence Assays for βIII-Tubulin, MAP2, and GFAP Detection
Immunocytochemistry was performed as previously described [42]. Briefly, culture cells had been plated on glass coverslips, treated with metabolites for 24 h, and then fixed with 4% paraformaldehyde for 30 min and permeabilized with 0.1% Triton X-100 in PBS for 5 min at room temperature. After having been blocked, cells were incubated overnight with anti-ΒIII-tubulin and anti-MAP2 (for neurons), and anti-GFAP (for astrocytes) at room temperature, followed by PBS washes and incubation with specific secondary antibody conjugated with Cy3 (sheep anti-rabbit) or with FITC (sheep anti-mouse) for 1 h. In each immunostaining experiment, negative controls were performed by omitting the primary antibody. In controls, no reactivity was observed. The nucleus was stained with 4′,6-diamidino-2-phenylindole (DAPI) (0.25 mg/ml). Cells were visualized with a Nikon inverted microscope, and images were transferred to a computer with a digital camera (Sound Vision).
Morphometric Analysis
For neurons, the neurite/neuron ratio and the neurite length were analyzed in ImageJ (NIH) as previously described [42]. The number of processes originating from the soma was measured manually, and all neurites emerging from neuronal soma were considered. To measure the neurite length, we used an ImageJ plug-in that allows semiautomatic tracing of neuritis for measurements of neurite length. Neurite length was analyzed by considering only the major process of each neuron.
The cytoplasmic area of astrocytes stained for GFAP cytoskeleton was measured in ImageJ and used as a criterion for morphological alteration. An ImageJ plug-in that allows semiautomatic tracing of the area of interest was used to measure the cytoplasmic area. Four independent experiments were performed in triplicate, encompassing 10 fields randomly chosen in each group. The data were stored and morphometric analyses were carried out in GraphPad Prism 5.
Statistical Analysis
Results are presented as mean ± standard deviation. Assays were performed in triplicate and the mean was used for statistical calculations. Quadruplicate experiments were always carried out and the mean used for the calculations. Results were statistically analyzed with two-way ANOVA followed by Bonferroni’s post hoc test. Statistical analysis was performed with the GraphPad 5.0 software. Differences were considered significant when P ≤ 0.05.
Results
Co-administration of QUIN and GA Increased Cell Damage in Striatal and Cortical Cultures and Decreased Viability in Striatal Neurons
We first evaluated the effect of the QUIN + GA co-incubation on LDH release and viability through the annexin and propidium iodide assay in neuronal cultures (Fig. 1a, d), co-cultures (Fig. 1b, e), and mixed cultures (Fig. 1c, f) from the rat striatum (Fig. 1a–c) and cortex (Fig. 1d–f). LDH activity was increased in the QUIN + GA condition (compared to control values) in striatal (Fig. 1a, P < 0.001) and cortical (Fig. 1d, P < 0.01) neuronal cultures, as well as in cortical mixed cultures (P < 0.05).

Effects of QUIN and/or GA on cell viability and damage of neurons, co-cultures (astrocyte and neurons), and mixed cultures (neurons, astrocytes, and microglia). Cells were treated with QUIN (50 μM) and/or GA (5 mM) for 24 h (8 DIV), and cell damage was assessed as LDH activity. Results of striatal cultures are shown in a (neurons), b (co-cultures), and c (mixed cultures). Results of cortical cultures are shown in d (neurons), e (co-cultures), and f (mixed cultures). Viability of striatal (g) and cortical (h) neurons was measured by annexin V-FITC and PI assays. Data are reported as mean ± SD of four independent experiments performed in triplicate. Results were statistically analyzed by two-way ANOVA followed by Bonferroni’s post hoc test. **P < 0.01 and ***P < 0.001, different of control; ### P < 0.001, different of QUIN; & P < 0.05, && P < 0.01, and &&& P < 0.001, different of GA
Interestingly, striatal cultures seem to be more sensitive to GA, since in all cell types there was an increase in LDH release when cultures were treated with this metabolite (Fig. 1a–c, P < 0.001). However, in cortical cultures, additive or synergic effects of both toxins were observed in neuronal and mixed cultures (P < 0.01), except for the case of co-cultures.
Next, we evaluated the type of cell death triggered by QUIN and GA in primary neurons using the annexin-PI assay. We found a decreased number of live striatal neurons in all treated groups (Fig. 1g, P < 0.001) compared to control cultures, where the percent values of cells in early apoptosis, late apoptosis, and necrosis were significantly increased by the QUIN + GA treatment compared to the all other treatments (Fig. 1g, P < 0.001). No changes in these parameters were observed in cortical cultures (Fig. 1h).
QUIN + GA Treatment Increased ROS Formation in Striatal and Cortical Neuronal Cultures, Co-cultures, and Mixed Cultures
The percent levels of ROS formation in neuronal cultures (Fig. 2a, d), co-cultures (Fig. 2b, e), and mixed cultures (Fig. 2c, f) exposed to QUIN + GA are presented in Fig. 2. ROS formation in all striatal (Fig. 2a–c) and cortical (Fig. 2d–f) cultures was significantly increased (P < 0.001) by the QUIN + GA treatment compared to control, QUIN, and GA conditions.

Effects of QUIN and/or GA on 2′,7′-dihydrodichlorofluorescein (DCFH) oxidation (reactive oxygen species (ROS) formation) in neurons, co-cultures (astrocyte and neurons), and mixed cultures (neurons, astrocytes, and microglia). Cells were treated with QUIN (50 μM) and GA (5 mM) for 24 h (8 DIV). Results of striatal cultures are shown in a (neurons), b (co-cultures), and c (mixed cultures). Results of cortical cultures are shown in d (neurons), e (co-cultures), and f (mixed cultures). Data are reported as mean ± SD of four independent experiments performed in triplicate. Results were statistically analyzed by two-way ANOVA followed by Bonferroni’s post hoc test. *P < 0.05, **P < 0.01, and ***P < 0.001, different of control; ### P < 0.001, different of QUIN; & P < 0.05, && P < 0.01, and &&& P < 0.001, different of GA
QUIN + GA Condition Decreased SOD Activity and Increased CAT Activity in Striatal Cultures
Figure 3 depicts the effects of QUIN, GA, and QUIN + GA on SOD activity (Fig. 3a, f, k), CAT activity (Fig. 3b, g, l), GPx activity (Fig. 3c, h, m), -SH groups content (Fig. 3d, i, n), and GSH content (Fig. 3e, j, o) in striatal neuronal cultures (Fig. 3a–e), co-cultures (Fig. 3f–j), and mixed cultures (Fig. 3k–o). The QUIN + GA condition decreased the levels of SOD activity compared to control, QUIN, and GA treatments in neuronal cultures (P < 0.001), co-cultures (P < 0.01), and mixed cultures (P < 0.001). In contrast, CAT activity levels were significantly increased in neuronal cultures (P < 0.001) and co-cultures (P < 0.01) and decreased in mixed cultures (P < 0.001). Significant changes on other parameters induced by QUIN + GA treatment compared to other treatments, included an increase in GPx activity in co-cultures (P < 0.01), a depletion of -SH groups in neuronal cultures and co-cultures (P < 0.001), and a decrease in GSH levels in neuronal cultures (P < 0.01).

Effects of QUIN and/or GA on antioxidant enzyme activity, GSH levels and -SH groups in striatal cell cultures. Cells were treated with QUIN (50 μM) and GA (5 mM) for 24 h (8 DIV). Results of cultured neurons (a–e), co-cultures (f–j), and mixed cultures (k–o) are shown. Data are reported as mean ± SD of four independent experiments performed in triplicate. Results were statistically analyzed by two-way ANOVA followed by Bonferroni’s post hoc test. *P < 0.05, **P < 0.01, and ***P < 0.001, different of control; # P < 0.05, ## P < 0.01, and ### P < 0.001, different of QUIN; & P < 0.05, && P < 0.01, and &&& P < 0.001, different of GA
QUIN + GA Treatment Decreased SOD Activity and GSH Levels in Cortical Cultured Neurons
The effects of QUIN, GA, and QUIN + GA on the cortical SOD activity (Fig. 4a, f, k), CAT activity (Fig. 4b, g, l), GPx activity (Fig. 4c, h, m), -SH groups content (Fig. 4d, i, o), and GSH content (Fig. 4e, j, p) in cortical neuronal cultures (Fig. 4a–e), co-cultures (Fig. 4f–j), and mixed cultures (Fig. 4k–p) are shown in Fig. 4. When cells were treated with QUIN + GA, the levels of SOD were decreased in neuronal cultures (P < 0.001) and co-cultures (P < 0.01) compared to control, QUIN, and GA treatments. In addition, QUIN + GA condition also depleted the levels of GSH in neuronal cultures (P < 0.01) and mixed cultures (P < 0.001) compared to all other treatments. No changes in other parameters were observed.

Effects of QUIN and/or GA on antioxidant enzyme activity, GSH levels and -SH groups content in cortical cell cultures. Cells were treated with QUIN (50 μM) and GA (5 mM) for 24 h (8 DIV). Results of cultured neurons (a–e), co-cultures (f–j), and mixed cultures (k–p) are shown. Data are reported as mean ± SD of four independent experiment performed in triplicate. Results were statistically analyzed by two-way ANOVA followed by Bonferroni’s post hoc test. *P < 0.05, **P < 0.01, and ***P < 0.001, different of control; # P < 0.05, ## P < 0.01, and ### P < 0.001, different of QUIN; & P < 0.05, && P < 0.01, and &&& P < 0.001, different of GA
QUIN + GA Condition Did Not Potentiate the Decrease in Neurite/Neuron Ratio, Neither the Neurite Length in Cortical Cultures Exposed to QUIN or GA
Since neuronal cultures were more susceptible to the QUIN + GA than any other cell culture studied, two endpoints of neurite development were estimated in cortical neurons: neurite/neuron ratio and neurite length after labeling cells with MAP2 (Fig. 5). For both markers, the QUIN + GA treatment did not exert any additive toxic effect compared to the toxins alone. In turn, while QUIN exerted a prominent decrease in both the neurite/neuron ratio and the neurite length (P < 0.001) compared to control, GA produced a less intense effect on both markers (P < 0.01).

Effects of QUIN and/or GA on the neurite length and the neurite/neuron ratio in cultured cortical neurons. Cells were treated with QUIN (50 μM) and GA (5 mM) for 24 h (8 DIV). a Representative images of cells immunostained with anti-MAP2 (green) antibodies and DAPI for nuclei (blue). b Graphical representation of the neurite/neuron ratio. c Graphical representation of neurite length. Data are reported as mean values ± standard error of four independent experiments, considering 10 fields randomly chosen from each group. Results were statistically analyzed by two-way ANOVA followed by Bonferroni’s post hoc test. **P < 0.01 and ***P < 0.001, different of control; ### P < 0.001, different of QUIN; && P < 0.01 and &&& P < 0.001, different of GA
QUIN + GA Condition Did Not Potentiate Reactive Gliosis, Neither Affected the Neurite/Neuron Ratio Nor the Neurite Length in Cortical Co-cultures
Figure 6 depicts the effects of QUIN and/or GA on astrocytic cytoplasmic area as an index of reactive gliosis in cortical co-cultures. The neurite/neuron ratio and the neurite length are also shown as indexes of the physiological status of neuronal cells. Immunocytochemical results (in Fig. 6a) from astrocytes labeled with GFAP and neuronal cells labeled with MAP2 showed no additive effects for the QUIN + GA condition compared to the effects of the toxins alone. QUIN, but not GA, was able to increase reactive gliosis in a significant manner (P < 0.001) compared to control (Fig. 6d). Neither the neurite/neuronal ratio (Fig. 6b) nor the neurite length (Fig. 6c) was affected by QUIN and/or GA in co-cultures.

Effects of QUIN and/or GA on astrocytic and neuronal morphology. Cells in cortical co-culture were treated with QUIN (50 μM) and GA (5 mM) for 24 h (8 DIV). a Representative images of astrocytes immunostained with anti-GFAP (red), neurons immunostained with MAP2 (green), and DAPI (blue) are shown. Effect of QUIN and GA on neurite per neuron (b), neurite length (c), and cytoplasmic area of astrocytes (d). Data are reported as mean ± standard error of four independent experiments, considering 10 fields randomly chosen from each group. Results were statistically analyzed by two-way ANOVA followed by Bonferroni’s post hoc test. ***P < 0.001, different of control; && P < 0.01, different of GA
MK-801 and Melatonin Decreased the Cell Damage Induced by QUIN + GA in Striatal Neurons, but Only Melatonin Prevented ROS Formation and Loss of Cell Viability
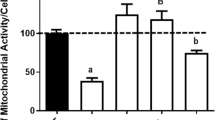
Figure 7 shows the effects of the well-known NMDAr antagonist MK-801, and the broad-spectrum antioxidant melatonin, on the cell damage and loss of viability induced by the QUIN + GA condition in cultured striatal neurons. Exposure of neuronal cells to QUIN + GA provoked a significant increase in LDH leakage (70–98%; P < 0.05–0.01; Fig. 7a, b) and ROS formation (53–121%; P < 0.01–0.001) compared to the control (Fig. 7c,d). MK-801 only reduced the QUIN + GA-induced cell damage (65% below QUIN + GA; P < 0.001; Fig. 7a). In contrast, melatonin decreased both the LDH activity (58% below; P < 0.01) and ROS formation (32% below; P < 0.001) induced by the QUIN + GA condition (Fig. 7b, d, respectively).

Protection against QUIN + GA-induced cell death and ROS production. Striatal neurons were preincubated with the glutamate antagonist MK-801 (a, c) and the antioxidant melatonin (b, d, e) for 1 h before QUIN + GA treatment for 24 h as described in the “Materials and Methods” section. Data are reported as mean ± SD of four independent experiments performed in triplicate. Results were statistically analyzed by two-way ANOVA followed by Bonferroni’s post hoc test. *P < 0.05, **P < 0.01, and ***P < 0.001, different of control; # P < 0.05, ## P < 0.01, and ### P < 0.001, different of QUIN + GA
In regard to the assay to determine the type of cell death produced by the QUIN + GA treatment in primary striatal neurons by the annexin-PI assay (Fig. 7e), for this series of experiments, we found that this treatment decreased the number of living cells by 18% compared to control (P < 0.001) and increased the number of cells in early apoptosis by ~ 20% (P < 0.001), in late apoptosis by ~ 15% (P < 0.001), and in necrosis by ~ 14% (P < 0.01). Melatonin was able to prevent the loss of living cells and the increase in the number of cells in apoptosis and necrosis (P < 0.05). Melatonin per se produced no effects on these parameters.
Discussion
In the present study, we provide evidence demonstrating that simultaneous addition of QUIN and GA to primary striatal and cortical cultures led to alterations in redox homeostasis and cell death. We have also shown a differential susceptibility of neurons, co-cultures, and mixed cultures to these metabolites.
Exposure of primary striatal neurons with either GA or QUIN independently induced neuronal death, as observed in the experiments with annexin-PI. However, when combined, these two metabolites exerted synergistic and differential toxic effects leading to cell death, triggering both apoptotic and necrotic mechanisms. Evidence of this synergistic actions exerted by the combined presence of QUIN + GA has been reported by us both in in vitro—in brain synaptosomes—[40] and in vivo—in glutaryl-CoA dehydrogenase KO mice—[41] conditions.
Certain neurotoxins can induce both apoptosis and necrosis, depending on the toxic conditions and pathways they stimulate, particularly regarding redox levels and mitochondrial integrity. This consideration is consistent with the fact that the signaling pathways involved in the different types of cell death can sometimes overlap and are engaged in response to specific stimuli [49]. In line with this, glutamate can induce either early necrosis or delayed apoptosis in cultures of cerebellar granule cells, wherein the mitochondrial function is a critical factor that determines the mode of neuronal death in excitotoxicity [50]. Moreover, the maintenance of mitochondrial function may therefore be a decisive factor in determining the degree and progression of neuronal injury caused by excitotoxins. Therefore, an increase in cytosolic calcium triggered by excitotoxins like QUIN and GA is critical for mitochondrial dysfunction, causing oxidative stress [51] and nNOS activation and affecting the mitochondrial membrane potential.
Oxidative stress represents a critical event related with QUIN and GA neurotoxicity [15, 20]. Accordingly, we found that ROS generation and depleted antioxidant defenses take part on the QUIN + GA-induced cytotoxicity of QUIN + GA in neuronal cells. When investigating SOD, CAT, and GPx activities, representing the main antioxidant enzymes for the detoxification of reactive species [52], we found diminished SOD and increased CAT activities in striatal neurons, and diminished SOD activity and GSH levels in cortical neurons, together with diminished GSH content and SH groups. In line with this, the antioxidant melatonin, but not the NMDAr antagonist MK-801, was able to prevent both ROS production and LDH release and diminished the number of viable striatal neurons, suggesting that oxidative stress is associated with neuronal death in the combined QUIN + GA toxic pattern.
GSH is the most prevalent cellular thiol that plays an essential role in preserving a reduced intracellular environment, protecting DNA, proteins, and lipids against oxidation. Furthermore, GSH depletion by ROS and RNS is a central signaling event that regulates the activation of cell death pathways in distinct pathologies, such as in common neurodegenerative disorders, cancer, and autoimmunity [53]. Herein, we found depletion in the GSH content in striatal and cortical neurons treated with the toxic metabolites, suggesting that GSH depletion is part of the mechanisms by which QUIN + GA trigger neuronal death. However, it is important to note that, as abovementioned, although melatonin could prevent both cell death and ROS production in striatal neurons, the NMDA antagonist MK-801 was able to prevent only LDH release. These findings suggest that other mechanisms are involved in the neuronal death triggered by QUIN and GA beyond ROS production and excitotoxicity. In line with this, melatonin is known to antagonize both excitotoxicity and mitochondrial dysfunction [54, 55]. Consistently, NMDA-induced calcium rises were diminished by melatonin in cultured mouse striatal neurons [56], also inhibiting nNOS [57]. Moreover, anti-apoptotic effects of melatonin have been described in numerous publications and summarized in several reviews [58,59,60,61], in which it has been demonstrated that this antioxidant acts directly on membrane potential as well as on the mitochondrial permeability transition pore [56]. Therefore, our results suggest that, despite that QUIN + GA model exerts its effect in part by NMDAr overactivation, which would increase the cytosolic calcium, affect mitochondrial function, and trigger mechanisms of apoptotic and necrotic cell death, other mechanisms cannot be discarded. It is important to note that despite QUIN damage to neural cells is greatly ascribed to NMDA-mediated excitotoxicity in several brain areas, our present results suggest that cell death in striatal cultures is through mechanisms not disturbed by NMDA antagonist MK-801. Although further studies with silenced or dysfunctional NMDA receptors will be necessary to assume that QUIN + GA effects are completely dissociated from NMDA receptors, our present findings suggest that, at least in our in vitro conditions, QUIN + GA toxicity is mediated by other mechanisms beyond NMDA receptor. Moreover, studies showed that QUIN actions also involve metabotropic glutamate receptors and Ca2+ release from intracellular stores besides Ca2+ influx through NMDA receptors [44, 62]. In this line, it has been shown that QUIN can produce oxidative damage independent of its activity under NMDA receptor [63]. The evidence collected herein also demonstrates that the QUIN + GA condition induces a synergic action on cell dysfunction and death, supporting a coordinated mechanism of toxicity for these two agents.
Of note, in co-cultures, QUIN + GA did not exert any synergistic effect on LDH release as compared to GA alone. Indeed, GA alone exerted a prominent increase in LDH release in striatal co-culture, while QUIN + GA had no effect on this parameter. Although both metabolites are able to alter the redox homeostasis and increase ROS production in striatal and cortical co-cultures, these effects seem not to be associated with the cell death observed.
Our results showing no cell death in striatal co-cultures suggest that the neuron-astrocyte interaction could prevent cell death caused by the metabolites. Accordingly, a recent study with striatal co-cultures treated with QUIN showed that the metabolite activates FGF2/Erk signaling, preventing neuronal and astrocytic cytoskeletal disruption and ROS production [42]. This effect might be inherent also to GA. On the other hand, in cortical co-cultures, QUIN + GA caused a decrease in SOD and GPx activity, with an increase in ROS production and concomitant cell death. In turn, this effect highlights different mechanisms triggered by the metabolites on striatal or cortical cells, supporting regional susceptibilities to toxic insults.
Contrariwise, when mixed cultures were treated with QUIN + GA, a potentiating effect of the two metabolites was observed. The results showed a diminished SOD and CAT in striatal mixed cultures and diminished SOD, GPx activities, and GSH content in cortical mixed cultures. This imbalance resulted in increased ROS production and cell death in both striatal and cortical cells. These results demonstrate a role of microglial cells on the effects triggered by the metabolites, especially in striatal cultures, since we did not observe cell death when microglial cells are not present.
Microglia can respond to injury, ischemia, infection, or inflammation in the CNS with morphological modifications, activation and expression of membrane receptors, and secretion of pro-inflammatory cytokines or mediators like ROS and RNS [64, 65]. These findings could explain, at least in part, why the symptomatology of patients with GA I is worsened during infections or immunization. During these scenarios, microglia can be activated and secrete QUIN, which in turn induces more microglial activation, with the consequent ROS production and cell death. According to our study, the role of microglia in the cell death triggered by the metabolites seems to be more prominent in striatal cells. This is in line with striatal damage observed in GA I patients and could explain, at least in part, the degeneration associated with infections, fever, or prolonged fasting.
Compared with glutamate or NMDA, the extent of neuronal damage caused by GA alone is weak. Therefore, it is suggested that additional amplifying factors are necessary to explain the neurodegeneration seen in the disease. The hypothesis that QUIN may be involved in the neuropathology of GA I started in 1987 [13]. This hypothesis stated that deficiency of glutaryl-CoA dehydrogenase blocks the glutarate pathway, and this could activate the first enzyme of kynurenine pathway in macrophages and microglia, especially during inflammatory processes, leading to overproduction of QUIN. Consequently, synergistic effect of these two metabolites, which alone does not have sufficient toxic potency, may be involved in the pathogenesis of striatal degeneration.
In summary, our results, together with previous reports in literature demonstrating synergic toxic actions of QUIN + GA in rat brain synaptosomes [40], as well as in the mutant mice model of GA I produced by deletion of GCDH [41], support the existence of cooperative mechanisms elicited by these two metabolites to potentiate neuronal cell damage in a process that is not only relevant for GA I but also for other neurological disorders with excitotoxic, pro-oxidant, pro-inflammatory, and degenerative components. Finally, Fig. 8 summarizes the mechanisms proposed to occur in cells exposed to the QUIN + GA condition and the role that NMDAr blockade and the induction of oxidative damage play in this model.

Schematic representation of hypothetical mechanisms involved in the toxic pattern triggered by quinolinic acid (QUIN) + glutaric acid (GA) in striatal and cortical neuronal cell cultures, co-cultures (neuronal cells plus astrocytes), and mixed cultures (neuronal cells plus astrocytes plus microglia) from the rat brain. In the center, neuronal cells are directly attacked by QUIN + GA via overstimulation of N-methyl-d-aspartate receptor (NMDAr) and/or oxidative stress. These effects will trigger damaging cell pathways while silencing survival signals, leading to cell death produced by toxic synergism. When neuronal cells are co-incubated with astrocytes (right box), the defense systems of the later help to protect neuronal cells by providing them antioxidant and anti-inflammatory assistance, reducing excitability, and stimulating survival signaling pathways. In turn, when neuronal cells are co-incubated with astrocytes and activated microglia (left box), the toxic responses affecting neuronal cells exposed to QUIN + GA prevail and are stimulated by damaging signals produced by microglia, including pro-inflammatory responses, loss of antioxidant potency, and activation of deadly cascades; thus, microglia could silence the positive effects of astrocytes in the QUIN + GA toxic paradigm
References
Brismar J, Ozand PT (1995) CT and MR of the brain in glutaric aciduria type I: a review of 59 published cases and a report of 5 new patients. Am J Neuroradiol 16:675–683
Goodman S, Frerman F (2001) Organic acidemias due to defects in lysine oxidation: 2-ketoadipic academia and glutaric acidemia. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) The metabolic and molecular bases of inherited disease. McGraw-Hill Inc., New York, pp. 2195–2204
Goodman SI, Norenberg MD, Shikes RH, Breslich DJ, Moe PG (1977) Glutaric aciduria: biochemical and morphologic considerations. J Pediatr 90:746–750
Heringer J, Boy SP, Ensenauer R, Assmann B, Zschocke J, Harting I, Lücke T, Maier EM et al (2010) Use of guidelines improves the neurological outcome in glutaric aciduria type I. Ann Neurol 68:743–752
Hoffmann GF, Zschocke J (1999) Glutaric aciduria type I: from clinical, biochemical and molecular diversity to successful therapy. J Inherit Metab Dis 22:381–391
Kölker S, Sauer SW, Hoffmann GF, Müller I, Morath MA, Okun JG (2008) Pathogenesis of CNS involvement in disorders of amino and organic acid metabolism. J Inherit Metab Dis 31:194–204
Hensley K, Mhatre M, Mou S, Pye QN, Stewart C, West M, Williamson KS (2006) On the relation of oxidative stress to neuroinflammation: lessons learned from the G93A-SOD1 mouse model of amyotrophic lateral sclerosis. Antioxid Redox Signal 8:2075–2087
Hunter RL, Dragicevic N, Seifert K, Choi DY, Liu M, Kim HC, Cass WA, Sullivan PG et al (2007) Inflammation induces mitochondrial dysfunction and dopaminergic neurodegeneration in the nigrostriatal system. J Neurochem 100:1375–1386
Tilleux S, Hermans E (2007) Neuroinflammation and regulation of glial glutamate uptake in neurological disorders. J Neurosci Res 85:2059–2070
Muhl H, Pfeilschifter J (2003) Endothelial nitric oxide synthase: a determinant of TNFalpha production by human monocytes/macrophages. Biochem Biophys Res Commun 310:677–680
Maddison DC, Giorgini F (2015) The kynurenine pathway and neurodegenerative disease. Semin Cell Dev Biol 40:134–141
Schwarcz R, Bruno JP, Muchowski PJ, Wu HQ (2012) Kynurenines in the mammalian brain: when physiology meets pathology. Nat Rev Neurosci 13:465–477
Heyes MP (1987) Hypothesis: a role for quinolinic acid in the neuropathology of glutaric aciduria type I. Can J Neurol Sci 14:441–443
Varadkar S, Surtees R (2004) Glutaric aciduria type I and kynurenine pathway metabolites: a modified hypothesis. J Inherit Metab Dis 27:835–842
Pérez-De La Cruz V, Carrillo-Mora P, Santamaría A (2012) Quinolinic acid, an endogenous molecule combining excitotoxicity, oxidative stress and other toxic mechanisms. Int J Tryptophan Res 5:1–8
Ribeiro CAJ, Grando V, Dutra Filho CS, Wannmacher CM, Wajner M (2006) Evidence that quinolinic acid severely impairs energy metabolism through activation of NMDA receptors in striatum from developing rats. J Neurochem 99:1531–1542
Dalcin KB, Rosa RB, Schmidt AL, Winter JS, Leipnitz G, Dutra-Filho CS, Wannmacher CM, Porciúncula LO et al (2007) Age and brain structural related effects of glutaric and 3-hydroxyglutaric acids on glutamate binding to plasma membranes during rat brain development. Cell Mol Neurobiol 27:805–818
de Oliveira MF, Hagen ME, Pederzolli CD, Sgaravatti AM, Durigon K, Testa CG, Wannmacher CM, de Souza Wyse AT et al (2003) Glutaric acid induces oxidative stress in brain of young rats. Brain Res 964:153–158
Fighera MR, Royes LF, Furian AF, Oliveira MS, Fiorenza NG, Frussa-Filho R, Petry JC, Coelho RC et al (2006) GM1 ganglioside prevents seizures, Na+,K+-ATPase activity inhibition and oxidative stress induced by glutaric acid and pentylenetetrazole. Neurobiol Dis 22:611–623
Jafari P, Braissant O, Bonafé L, Ballhausen D (2011) The unsolved puzzle of neuropathogenesis in glutaric aciduria type I. Mol Genet Metab 104:425–437
Kölker S, Ahlemeyer B, Krieglstein J, Hoffmann GF (1999) 3-Hydroxyglutaric and glutaric acids are neurotoxic through NMDA receptors in vitro. J Inherit Metab Dis 22:259–262
Kölker S, Ahlemeyer B, Krieglstein J, Hoffmann GF (2001) Contribution of reactive oxygen species to 3-hydroxyglutarate neurotoxicity in primary neuronal cultures from chick embryo telencephalons. Pediatr Res 50:76–82
Kölker S, Ahlemeyer B, Hühne R, Mayatepek E, Krieglstein J, Hoffmann GF (2001) Potentiation of 3-hydroxyglutarate neurotoxicity following induction of astrocytic iNOS in neonatal rat hippocampal cultures. Eur J Neurosci 13:2115–2122
Kölker S, Okun JG, Ahlemeyer B, Wyse AT, Hörster F, Wajner M, Kohlmüller D, Mayatepek E et al (2002) Chronic treatment with glutaric acid induces partial tolerance to excitotoxicity in neuronal cultures from chick embryo telencephalons. J Neurosci Res 68:424–431
Kölker S, Köhr G, Ahlemeyer B, Okun JG, Pawlak V, Hörster F, Mayatepek E, Krieglstein J et al (2002) Ca(2+) and Na(+) dependence of 3-hydroxyglutarate-induced excitotoxicity in primary neuronal cultures from chick embryo telencephalons. Pediatr Res 52:199–206
Kölker S, Koeller DM, Sauer S, Hörster F, Schwab MA, Hoffmann GF, Ullrich K, Okun JG (2004) Excitotoxicity and bioenergetics in glutaryl-CoA dehydrogenase deficiency. J Inherit Metab Dis 27:805–812
Kölker S, Sauer SW, Okun JG, Hoffmann GF, Koeller DM (2006) Lysine intake and neurotoxicity in glutaric aciduria type I: towards a rationale for therapy? Brain 129:e54
Latini A, Borba Rosa R, Scussiato K, Llesuy S, Belló-Klein A, Wajner M (2002) 3-Hydroxyglutaric acid induces oxidative stress and decreases the antioxidant defenses in cerebral cortex of young rats. Brain Res 956:367–373
Latini A, Scussiato K, Leipnitz G, Dutra-Filho CS, Wajner M (2005) Promotion of oxidative stress by 3-hydroxyglutaric acid in rat striatum. J Inherit Metab Dis 28:57–67
Latini A, Ferreira GC, Scussiato K, Schuck PF, Solano AF, Dutra-Filho CS, Vargas CR, Wajner M (2007) Induction of oxidative stress by chronic and acute glutaric acid administration to rats. Cell Mol Neurobiol 27:423–438
Magni DV, Furian AF, Oliveira MS, Souza MA, Lunardi F, Ferreira J, Mello CF, Royes LF et al (2009) Kinetic characterization of l-[(3)H]glutamate uptake inhibition and increase oxidative damage induced by glutaric acid in striatal synaptosomes of rats. Int J Dev Neurosci 27:65–72
Olivera-Bravo S, Fernández A, Sarlabós MN, Rosillo JC, Casanova G, Jiménez M, Barbeito L (2011) Neonatal astrocyte damage is sufficient to trigger progressive striatal degeneration in a rat model of glutaric acidemia-I. PLoS One 6:e20831
Olivera S, Fernandez A, Latini A, Rosillo JC, Casanova G, Wajner M, Cassina P, Barbeito L (2008) Astrocytic proliferation and mitochondrial dysfunction induced by accumulated glutaric acidemia I (GAI) metabolites: possible implications for GAI pathogenesis. Neurobiol Dis 32:528–534
Porciúncula LO, Dal-Pizzol A Jr, Coitinho AS, Emanuelli T, Souza DO, Wajner M (2000) Inhibition of synaptosomal [3H]glutamate uptake and [3H]glutamate binding to plasma membranes from brain of young rats by glutaric acid in vitro. J Neurol Sci 173:93–96
Porciúncula LO, Emanuelli T, Tavares RG, Schwarzbold C, Frizzo ME, Souza DO, Wajner M (2004) Glutaric acid stimulates glutamate binding and astrocytic uptake and inhibits vesicular glutamate uptake in forebrain from young rats. Neurochem Int 45:1075–1086
Rosa RB, Schwarzbold C, Dalcin KB, Ghisleni GC, Ribeiro CA, Moretto MB, Frizzo ME, Hoffmann GF et al (2004) Evidence that 3-hydroxyglutaric acid interacts with NMDA receptors in synaptic plasma membranes from cerebral cortex of young rats. Neurochem Int 45:1087–1094
Rosa RB, Dalcin KB, Schmidt AL, Gerhardt D, Ribeiro CA, Ferreira GC, Schuck PF, Wyse AT et al (2007) Evidence that glutaric acid reduces glutamate uptake by cerebral cortex of infant rats. Life Sci 81:1668–1676
Stokke O, Goodman SI, Moe PG (1976) Inhibition of brain glutamate decarboxylase by glutarate, glutaconate, and beta-hydroxyglutarate: explanation of the symptoms in glutaric aciduria? Clin Chim Acta 66:411–415
Wajner M, Kölker S, Souza DO, Hoffmann GF, de Mello CF (2004) Modulation of glutamatergic and GABAergic neurotransmission in glutaryl-CoA dehydrogenase deficiency. J Inherit Metab Dis 27:825–828
Colín-González AL, Paz-Loyola AL, Serratos I, Seminotti B, Ribeiro CA, Leipnitz G, Souza DO, Wajner M et al (2015) Toxic synergism between quinolinic acid and organic acids accumulating in glutaric acidemia type I and in disorders of propionate metabolism in rat brain synaptosomes: relevance for metabolic acidemias. Neuroscience 308:64–74
Seminotti B, Amaral AU, Ribeiro RT, Rodrigues MD, Colín-González AL, Leipnitz G, Santamaría A, Wajner M (2016) Oxidative stress, disrupted energy metabolism, and altered signaling pathways in glutaryl-CoA dehydrogenase knockout mice: potential implications of quinolinic acid toxicity in the neuropathology of glutaric acidemia type I. Mol Neurobiol 53:6459–6475
Pierozan P, Biasibetti H, Schmitz F, Ávila H, Parisi MM, Barbe-Tuana F, Wyse AT, Pessoa-Pureur R (2016) Quinolinic acid neurotoxicity: differential roles of astrocytes and microglia via FGF-2-mediated signaling in redox-linked cytoskeletal changes. Biochim Biophys Acta 1863:3001–3014
Ferreira GC, Tonin A, Schuck PF, Viegas CM, Ceolato PC, Latini A, Perry ML, Wyse AT et al (2007) Evidence for a synergistic action of glutaric and 3-hydroxyglutaric acids disturbing rat brain energy metabolism. Int J Dev Neurosci 25:391–398
Pierozan P, Ferreira F, de Lima BO, Pessoa-Pureur R (2015) Quinolinic acid induces disrupts cytoskeletal homeostasis in striatal neurons. Protective role of astrocyte-neuron interaction. J Neurosci Res 93:268–284
Hu X, Zhang D, Pang H, Caudle WM, Li Y, Gao H, Liu Y, Qian L et al (2008) Macrophage antigen complex-1 mediates reactive microgliosis and progressive dopaminergic neurodegeneration in the MPTP model of Parkinson’s disease. J Immunol 181:7194–7204
LeBel CP, Ali SF, McKee M, Bondy SC (1990) Organometal-induced increases in oxygen reactive species: the potential of 2′,7′-dichlorofluorescin diacetate as an index of neurotoxic damage. Toxicol Appl Pharmacol 104:17–24
Aksenov MY, Markesbery WR (2001) Changes in thiol content and expression of glutathione redox system genes in the hippocampus and cerebellum in Alzheimer’s disease. Neurosci Lett 302:141–145
Browne RW, Armstrong D (1998) Reduced glutathione and glutathione disulfide. Methods Mol Biol 108:347–352
Maiuri MC, Zalckvar E, Kimchi A, Kroemer G (2007) Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol 8:741–752
Ankarcrona M, Dypbukt JM, Bonfoco E, Zhivotovsky B, Orrenius S, Lipton SA, Nicotera P (1995) Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron 15:961–973
Peng TI, Jou MJ (2010) Oxidative stress caused by mitochondrial calcium overload. Ann N Y Acad Sci 1201:183–188
Gutteridge JM, Halliwell B (2000) Free radicals and antioxidants in the year 2000. A historical look to the future. Ann N Y Acad Sci 899:136–147
Circu ML, Aw TY (2012) Glutathione and modulation of cell apoptosis. Biochim Biophys Acta 1823:1767–1777
Hardeland R (2009) Neuroprotection by radical avoidance: search for suitable agents. Molecules 14:5054–5102
Hardeland R, Coto-Montes A, Poeggeler B (2003) Circadian rhythms, oxidative stress, and antioxidative defense mechanisms. Chronobiol Int 20:921–962
Andrabi SA, Sayeed I, Siemen D, Wolf G, Horn TF (2004) Direct inhibition of the mitochondrial permeability transition pore: a possible mechanism responsible for anti-apoptotic effects of melatonin. FASEB J 18:869–871
Entrena A, Camacho ME, Carrión MD, López-Cara LC, Velasco G, León J, Escames G, Acuña-Castroviejo D et al (2005) Kynurenamines as neural nitric oxide synthase inhibitors. J Med Chem 48:8174–8181
Sainz RM, Mayo JC, Rodriguez C, Tan DX, Lopez-Burillo S, Reiter RJ (2003) Melatonin and cell death: differential actions on apoptosis in normal and cancer cells. Cell Mol Life Sci 60:1407–1426
Pandi-Perumal SR, Srinivasan V, Maestroni GJ, Cardinali DP, Poeggeler B, Hardeland R (2006) Melatonin: nature’s most versatile biological signal? FEBS J 273:2813–2838
Acuña-Castroviejo D, Escames G, Rodriguez MI, Lopez LC (2007) Melatonin role in the mitochondrial function. Front Biosci 12:947–963
Wang X (2009) The antiapoptotic activity of melatonin in neurodegenerative diseases. CNS Neurosci Ther 15:345–357
Pierozan P, Zamoner A, Soska ÂK, de Lima BO, Reis KP, Zamboni F, Wajner M, Pessoa-Pureur R (2012) Signaling mechanisms downstream of quinolinic acid targeting the cytoskeleton of rat striatal neurons and astrocytes. Exp Neurol 233:391–399
Stípek S, Stastný F, Pláteník J, Crkovská J, Zima T (1997) The effect of quinolinate on rat brain lipid peroxidation is dependent on iron. Neurochem Int 30:233–237
von Bernhardi R, Eugenín-von Bernhardi L, Eugenín J (2015) Microglial cell dysregulation in brain aging and neurodegeneration. Front Aging Neurosci 7:124
Syed Mortadza SA, Wang L, Li D, Jiang LH (2015) TRPM2 channel-mediated ROS-sensitive Ca(2+) signaling mechanisms in immune cells. Front Immunol 6:407
Acknowledgements
We are grateful to the financial support of CNPq, PROPESq/UFRGS, FAPERGS, PRONEX, FINEP Rede Instituto Brasileiro de Neurociência (IBN-Net) # 01.06.0842-00, and Instituto Nacional de Ciência e Tecnologia-Excitotoxicidade e Neuroproteção (INCT-EN).
Funding
This work was supported by CNPq, PROPESq/UFRGS, FAPERGS, PRONEX, FINEP Rede Instituto Brasileiro de Neurociência (IBN-Net) # 01.06.0842-00, and Instituto Nacional de Ciência e Tecnologia-Excitotoxicidade e Neuroproteção (INCT-EN).
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: PP, ALCG, AW, MW, and AS. Performed the experiments and analyzed the data: PP, ALCG, HB, and JCS. Wrote the paper: PP, MW, and AS.
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Research Involving Animals
For animal handling, we followed the experimental protocol described in the “Guide for the Care and Use of Laboratory Animals” (NIH publication 85–23, revised 1985). Our protocol was approved by the Ethics Committee for Animal Research of the Universidade Federal do Rio Grande do Sul (No. 18,266). During the experiments, all efforts were made to minimize the number of animals used and their suffering.
Electronic supplementary material
ESM 1
(DOCX 424 kb)
Rights and permissions
About this article

Cite this article
Pierozan, P., Colín-González, A.L., Biasibetti, H. et al. Toxic Synergism Between Quinolinic Acid and Glutaric Acid in Neuronal Cells Is Mediated by Oxidative Stress: Insights to a New Toxic Model. Mol Neurobiol 55, 5362–5376 (2018). https://doi.org/10.1007/s12035-017-0761-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-017-0761-6