
Abstract
Studies have demonstrated that oxidative stress is associated with amphetamine-induced neurotoxicity, but little is known about the adaptations of antioxidant enzymes in the brain after amphetamine exposure. We studied the effects of acute and chronic amphetamine administration on superoxide dismutase (SOD) and catalase (CAT) activity, in a rodent model of mania. Male Wistar rats received either a single IP injection of d-amphetamine (1 mg/kg, 2 mg/kg, or 4 mg/kg) or vehicle (acute treatment). In the chronic treatment rats received a daily IP injection of either d-amphetamine (1 mg/kg, 2 mg/kg, or 4 mg/kg) or vehicle for 7 days. Locomotor behavior was assessed using the open field test. SOD and CAT activities were measured in the prefrontal cortex, hippocampus, and striatum. Acute and to a greater extent chronic amphetamine treatment increased locomotor behavior and affected SOD and CAT activities in the prefrontal cortex, hippocampus and striatum. Our findings suggest that amphetamine exposure is associated with an imbalance between SOD and CAT activity in the prefrontal cortex, hippocampus and striatum.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
There is an emerging body of data indicating that major neuropsychiatric disorders, such as bipolar disorder (BD) and schizophrenia, are associated with increased oxidative stress and changes in antioxidant enzymatic defense [1–3]. A recent genetic study found that BD was significantly associated with a single-nucleotide polymorphism in the TRPM2 gene, which is involved in intracellular calcium homeostasis in response to oxidative stress [4]. In addition, chronic administration of lithium and valproate (first-line mood stabilizers) demonstrated robust antioxidant properties against glutamate-induced oxidative stress in vitro [5].
It has long been recognized that the administration of dopaminergic drugs induces manic symptoms in individuals with BD [6, 7]. Further, the use of amphetamine in healthy volunteers induced manic symptoms, such as enhanced mood, racing thoughts, high energy and restlessness [8]. Considering the difficulty of modeling the highly complex mood swinging nature of BD, the psychostimulant-induced hyperactivity is the best established animal model of mania [9–11]. We have recently found that longer administration of amphetamine was associated with increased protein and lipid oxidative damage in rat brain [12]. As an extension of this latter study, we assessed the effects of acute and chronic amphetamine administration on superoxide dismutase and catalase activity (two major antioxidant enzymes) in a rodent model of mania.
Experimental procedure
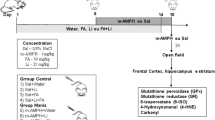
Adult male Wistar rats, obtained from our breeding colony, were maintained on a 12-h light/dark cycle, with free access to food and water. All experimental procedures involving animals were performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals and the Brazilian Society for Neuroscience and Behavior (SBNeC) recommendations for animal care. Rats received 1 mg/kg, 2 mg/kg, or 4 mg/kg IP injections of d-amphetamine (Sigma, St Louis, USA) either as an acute treatment (single injection) or chronic treatment (once daily injection for 7 days). Locomotor activity was measured 2 h after the last injection, and the rats were sacrificed by decapitation right after the behavioral experiment. The prefrontal cortex, hippocampus and striatum were dissected, rapidly frozen, and stored at −80°C until assayed.
Locomotor activity was assessed using the open-field task. This task was carried out in 40 × 60 cm open field surrounded by 50 cm high walls made of brown plywood with a frontal glass wall. The floor of the open field was divided into 12 equal rectangles by black lines. The animals were gently placed on the left rear rectangle, and allowed to explore the arena. Crossings of the black lines and rearings performed were counted for 5 min. To determine CAT activity, the brain tissue was sonicated in 50 mM phosphate buffer and the resulting suspension was centrifuged at 3,000g for 10 min. The supernatant was used for enzyme assay. CAT activity was measured by the rate of decrease in hydrogen peroxide absorbance at 240 nm [13]. SOD activity was assayed by measuring the inhibition of adrenaline auto-oxidation, as previously described [14]. Differences among groups were performed using one-way ANOVA and multiple comparisons were performed by a Newman-Keuls test. Behavioral data are presented as mean ± SEM. and biochemical data are presented as mean ± SD. In all comparisons, P < 0.05 was considered to indicate statistical significance.
Results
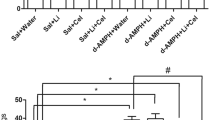
As expected, both acute and repeated amphetamine administration significantly increased locomotor activity (Figs. 1, 2). Figures 3 and 4 illustrate that a single amphetamine injection increased SOD activity in the prefrontal cortex, whereas repeated amphetamine exposure increased SOD activity in the hippocampus and decreased striatal SOD activity. No effects on CAT activity were observed after a single amphetamine injection in any brain region (Fig. 5). Repeated amphetamine administration increased CAT activity in the prefrontal cortex and decreased striatal CAT activity (Fig. 6). Higher amphetamine dosage (4 mg/kg) increased CAT activity, whereas a lower dosage (2 mg/kg) decreased CAT activity in the hippocampus after repeated amphetamine exposure.

Numbers of crossings in the acute treatment

Numbers of crossings in the chronic treatment

SOD activity in the acute treatment SOD: superoxide dismutase

SOD activity in the chronic treatment SOD: superoxide dismutase

Catalase activity in the acute treatment

Catalase activity in the chronic treatment
Discussion
In the present study, we demonstrated that acute and, to a greater extent, repeated amphetamine exposure modified SOD and CAT activities in the prefrontal cortex, hippocampus and striatum. Using the same animal model, we have recently reported that repeated amphetamine administration increased oxidative stress in a greater extent than single amphetamine use [12]. SOD acts by metabolizing the excess of superoxide anion (O ·−2 ) generation and by producing hydrogen peroxide (H2O2). CAT metabolizes the excess of H2O2 producing O2 + H2O, thereby decreasing the intracellular redox status. The brain is particularly prone to oxidative damage due to its relative high content of peroxidizable fatty acids and limited antioxidant capacity [15]. Previous studies have demonstrated that alterations on the redox state can lead to an imbalance between SOD and CAT activities and to oxidative stress [16, 17]. In situations which SOD levels are increased without a concomitant CAT increase, the intermediate product H2O2 may accumulate and generate hydroxyl radicals, which may lead to lipid and protein oxidation (damage).
Even though there is compelling data indicating that oxidative stress plays a major role in amphetamine-induced neurotoxicity [18, 19], there is a paucity of data assessing the effects of amphetamine on antioxidant enzymes. In a model of neurotoxicity (4 × 10 mg/kg), Jayanthi et al. [20] found that methamphetamine exposure decreased SOD activity in the frontal cortex and decreased CAT activity in the striatum of CD-1 mice. Using a different model (20 mg/kg for 14 days), Carvalho et al. [21] showed that amphetamine decreased SOD activity in the striatum and increased CAT activity in the prefrontal cortex of Wistar rats. On the other hand, D’Almeida et al. [22] found no changes in SOD and CAT activity after chronic methamphetamine treatment (2.5 mg/kg for 5 months) in Wistar rats. The profound methodological differences between studies make it difficult to draw conclusions, and therefore further studies are necessary to clarify the importance of these antioxidant changes. Interestingly, it has been reported that the dopamine D2 receptor agonist ropinirole protected mouse striatal neurons against 6-hydroxydopamine (6-OHDA) toxicity, by increasing SOD, CAT, and glutathione activity [23], whereas the dopamine agonist cabergoline demonstrated robust antioxidant effects against 6-OHDA-neurotoxicity [24]. In addition, the stimulation of D2 presynaptic autoreceptors may exert neuroprotective effects by a negative feedback mechanism, reducing the release of dopamine for oxidation by monoamine oxidase [25]. Studies assessing the effect of D2 blockers on amphetamine-induced oxidative stress would help to increase our knowledge on the dopamine-mediated neuroprotection and neurotoxicity.
Acute and chronic amphetamine exposure modulated SOD and CAT activity with a distinct pattern for each brain region and dosage regimen. These findings may explain, in part, why different areas of the brain are differentially susceptible to the toxic effects of amphetamine [26, 27]. One reason for these discrepancies may be fact the basal activities of various antioxidant enzymes, such as SOD, CAT, glutathione reductase, glutathione peroxidase, and glutathione-S-transferase are highly variable across brain regions [21]. It has been recently demonstrated that amphetamine regulates the expression of SOD mRNA via c-fos/c-jun activation in the hypothalamus [28]. Although speculative, it is possible that the regulation of antioxidant’s gene expression by amphetamine in other brain regions may be relevant to the differences observed. Future studies addressing the genes regulated by amphetamine exposure may help us clarify this issue.
We also found that during acute treatment, 4 mg/kg of amphetamine increased the locomotor behavior to a lesser extent than 2 mg/kg. During chronic treatment all of the dosages induced the same level of locomotor activation. This is in line with previous reports showing that the locomotor activity after acute amphetamine challenge reduces with increasing dosage, due to the emergence of stereotypic behavior [29, 30]. In conclusion, our findings suggest that acute and, to a greater extent, chronic amphetamine administrations are associated with an imbalance between SOD and CAT activities. This is possibly due to changes in the intracellular redox state, in the prefrontal cortex, hippocampus, and striatum. Such an imbalance may increase the predisposition to the generation of free radicals and therefore increase the susceptibility to oxidative damage. Given the recent evidence that oxidative stress may play a role in the pathophysiology of BD [1–4], this animal model may be a useful tool to further test the molecular underpinnings underlying amphetamine-induced oxidative stress.
References
Kuloglu M, Ustundag B, Atmaca M et al (2002) Lipid peroxidation and antioxidant enzyme levels in patients with schizophrenia and bipolar disorder. Cell Biochem Funct 20:171–175
Ozcan ME, Gulec M, Ozerol E et al (2004) Antioxidant enzyme activities and oxidative stress in affective disorders. Int Clin Psychopharmacol 19:89–95
Ranjekar PK, Hinge A, Hegde MV et al (2003) Decreased antioxidant enzymes and membrane essential polyunsaturated fatty acids in schizophrenic and bipolar mood disorder patients. Psychiatry Res 121:109–122
McQuillin A, Bass NJ, Kalsi G et al (2006) Fine mapping of a susceptibility locus for bipolar and genetically related unipolar affective disorders, to a region containing the C21ORF29 and TRPM2 genes on chromosome 21q22.3. Mol Psychiatry 11:134–142
Shao L, Young LT, Wang JF (2005) Chronic treatment with mood stabilizers lithium and valproate prevents excitotoxicity by inhibiting oxidative stress in rat cerebral cortical cells. Biol Psychiatry 58:879–884
Murphy DL, Brodie HK, Goodwin FK et al (1971) Regular induction of hypomania by L-dopa in “bipolar” manic-depressive patients. Nature 229:135–136
Gerner RH, Post RM, Bunney WE Jr (1976) A dopaminergic mechanism in mania. Am J Psychiatry 133:1177–1180
Jacobs D, Silverstone T (1986) Dextroamphetamine-induced arousal in human subjects as a model for mania. Psychol Med 16:323–329
Einat H, Kofman O, Belmaker RH (2000) Animal models of bipolar disorder: From A single episode to progressive cycling models. In: Myslobodsky MS, Weiner I (eds) Contemporary issues in modeling psychopathology. Kluwer Academic, Boston, pp 165–179
Machado-Vieira R, Kapczinski F, Soares JC (2004) Perspectives for the development of animal models of bipolar disorder. Prog. Neuropsychopharmacol. Biol. Psychiatry 28:209–224
Frey BN, Andreazza AC, Ceresér KMM et al (In press) Effects of mood stabilizers on hippocampus BDNF levels in an animal model of mania. Life Sci
Frey BN, Martins MR, Petronilho FC et al (In press) Increased oxidative stress after repeated amphetamine exposure: possible relevance as an animal model of acute mania. Bipolar Disord
Aebi H (1984) Catalase in vitro. Methods Enzymol 105:121–126
Bannister JV, Calabrese L (1987) Assays for superoxide dismutase. Methods Biochem Anal 32:279–312
Floyd RA (1999) Antioxidants, oxidative stress, and degenerative neurological disorders. Proc Soc Exp Biol Med 222:236–245
Klamt F, Dal-Pizzol F, Conte da Frota ML et al (2001) Imbalance of antioxidant defense in mice lacking cellular prion protein. Free Radic Biol Med 30:1137–1144
Andrades M, Ritter C, Moreira JC et al (2005) Oxidative parameters differences during non-lethal and lethal sepsis development. J Surg Res 125:68–72
Cadet JL, Brannock C (1998) Free radicals and the pathobiology of brain dopamine systems. Neurochem Int 32:117–131
Brown JM, Yamamoto BK (2003) Effects of amphetamines on mitochondrial function: Role of free radicals and oxidative stress. Pharmacol Ther 99:45–53
Jayanthi S, Ladenheim B, Cadet JL (1998) Methamphetamine-induced changes in antioxidant enzymes and lipid peroxidation in copper/zinc-superoxide dismutase transgenic mice. Ann N Y Acad Sci 844:92–102
Carvalho F, Fernandes E, Remiao F et al (2001) Adaptative response of antioxidant enzymes in different areas of rat brain after repeated d-amphetamine administration. Addict Biol 6:213–221
D’Almeida V, Camarini R, Azzalis LA et al (1995) Antioxidant defense in rat brain after chronic treatment with anorectic drugs. Toxicol Lett 81:101–105
Iida M, Miyazaki I, Tanaka K et al (1999) Dopamine D2 receptor-mediated antioxidant and neuroprotective effects of ropinirole, a dopamine agonist. Brain Res 838:51–59
Yoshioka M, Tanaka K, Miyazaki I et al (2002) The dopanime agonist cabergoline provides neuroprotection by activation of the glutathione system and scavenging free radicals. Neurosci Res 43:259–67
O’Neill MJ, Hicks CA, Ward MA et al (1998) Dopamine D2 receptor agonists protect against ischaemia-induced hippocampal neurodegeneration in global cerebral ischaemia. Eur J Pharmacol 352:37–46
Ryan LJ, Linder JC, Martone ME et al (1990) Histological and ultrastructural evidence that D-amphetamine causes degeneration in neustriatum and frontal cortex of rats. Brain Res 518:67–77
Eisch AJ, Gaffney M, Weihmuller FB et al (1992) Striatal subregions are differentially vulnerable to the neurotoxic effects of methamphetamine. Brain Res 598:321–326
Hsieh YS, Yang SF, Chiou HL et al (in press) Activations of c-fos/c-jun signaling are involved in the modulation of hypothalamic superoxide dismutase (SOD) and neuropeptide Y (NPY) gene expression in amphetamine-mediated appetite suppression. Toxicol Appl Pharmacol
Antoniou K, Kafetzopoulos E, Papadopoulou-Daifoti Z et al (1998) d-amphetamine, cocaine and caffeine: a comparative study of acute effects on locomotor activity and behavioural patterns in rats. Neurosci Biobehav Rev 23:189–196
Ott DA, Mandel RJ (1995) Amphetamine sensitivity in open-field activity vs. the prepulse inhibition paradigm. Brain Res Bull 37:219–222
Acknowledgements
This study was partly supported by CNPq, FAPESC, UNESC, and CAPES Foundation (Brazil).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Frey, B.N., Valvassori, S.S., Réus, G.Z. et al. Changes in Antioxidant Defense Enzymes after d-amphetamine Exposure: Implications as an Animal Model of Mania. Neurochem Res 31, 699–703 (2006). https://doi.org/10.1007/s11064-006-9070-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-006-9070-6