
Abstract
The molecular basis of estrogen-mediated neuroprotection against brain ischemia remains unclear. In the present study, we investigated changes in expression of estrogen receptors (ERs) α and β and excitatory amino acid transporters (EAAT) 1 and 2 in rat organotypic hippocampal slice cultures treated with estradiol and subsequently exposed to oxygen--glucose deprivation (OGD). Pretreatment with 17β-estradiol (10 nM) for 7 days protected the CA1 area of hippocampus against OGD (60 min), reducing cellular injury by 46% compared to the vehicle control group. Levels of ERα protein were significantly reduced by 20% after OGD in both vehicle- and estradiol-treated cultures, whereas ERβ was significantly up-regulated by 25% in the estradiol-treated cultures. In contrast, EAAT1 and EAAT2 levels were unchanged in response to estradiol treatment in this model of OGD. These findings suggest that estrogen-induced neuroprotection against ischemia might involve regulation of ERβ and, consequently, of the genes influenced by this receptor.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Epidemiological [1] and basic science studies [2] underscore the powerful neuroprotective properties of estrogens. Estrogens play this protective role through several routes, including activation of nuclear estrogen receptors (ERs), increased expression of anti-apoptotic proteins, interaction with second messenger cascades, alterations in glutamatergic activation, maintenance of intracellular calcium homeostasis and antioxidant activity [2]. However, the pathways involved in estrogen-induced promotion of neuronal survival remain unclear. Abundant expression of ERs is observed throughout the central nervous system, and estrogens may exert many, though not all, of their neuroprotective effects by binding to two types of these intracellular receptors (ERα and ERβ) and then inducing gene transcription [3, 4]. The relative contribution by either ER subtype to estrogen-induced neuroprotection is still unresolved.
Brain ischemia and several other disease states involve excitotoxic cell death, which is mediated by toxic levels of extracellular glutamate that excessively activate ionotropic glutamate receptors and initiate calcium and/or sodium influx [5]. The removal of glutamate from the extracellular space, permitting normal excitatory transmission and preventing cell death due to excitotoxicity, is accomplished mainly by means of glutamate transporters (also known as excitatory amino acid transporters or EAATs), particularly the glial subtypes, EAAT1 and EAAT2 [6]. Estrogens may exert neuroprotective effects by directly interacting with glutamate receptors [7, 8] and there is evidence of modulation of EAATs by estrogens, although both enhancement [9, 10] and inhibition [11] of Glu uptake have been reported.
We have recently reported that physiological levels of 17β-estradiol (E2) protect against ischemic damage induced by oxygen and glucose deprivation (OGD) in organotypic hippocampal slice cultures, and suggest that this effect could involve phosphatidylinositol 3-kinase signaling [12]. The goal of the present study was to investigate whether the neuroprotection afforded by estradiol involves changes in the abundance of glial glutamate transporters, EAAT1 and EAAT2, and estrogen receptors, ERα and ERβ.
Experimental procedures
Materials
Acrylamide and bisacrylamide used in sodium dodecylsulfate-polyacrylamide gel electrophoresis (SDS-PAGE) were obtained from Sigma (St. Louis, MO, USA). Polyclonal antibodies raised against rabbit EAAT1 and EAAT2 were kindly provided by Dr. D. Pow, University of Newcastle, Australia; ERα was obtained from Santa Cruz Biotechnology (Catalog sc-542) and ERβ was from Calbiochem (Catalog PC168-50UG). A monoclonal antibody raised against mouse β-actin was obtained from Sigma (Catalog A5316). Anti-mouse IgG and anti-rabbit IgG peroxidase-conjugated secondary antibodies and reagents to detect proteins by chemiluminescence were purchased from Amersham International. Millicell culture inserts were obtained from Millipore, culture medium and horse serum heat inactivated were obtained from Gibco. All animal use procedures were approved by local Animal Care Committee and were in accordance with the NIH Guide for Care and Use of Laboratory Animals.
Hippocampal slice cultures
Organotypic hippocampal slice cultures were prepared as described previously [13, 14]. Transverse hippocampal slices (400 μm) from 6- to 8-day-old male Wistar rat pups were cut using a McIIwain tissue chopper. Slices were transferred to Millicell culture inserts (6 slices per insert) in 6-well plates, and maintained at 37°C in an atmosphere of 5% CO2 for 14 days (div) prior to use. Culture medium, consisting of 50% minimum essential medium (MEM), 25% Hanks balanced salt solution (HBSS), 25% heat-inactivated horse serum, glucose 36 mM, HEPES 25 mM, NaHCO3 4 mM, fungizone 1% and gentamicin 36 μl/100 ml (pH 7.3), was replaced every 3 days.
In vitro model of ischemia
OGD was achieved by combining severe hypoxia with aglycemia, according to the method described by Strasser and Fisher [15], with some modifications [13]. On div 14, slices were washed twice with aglycemic HBSS containing CaCl2 1.26 mM, KCl 5.36 mM, NaCl 136.89 mM, KH2PO4 0.44 mM, Na2HPO4 0.34 mM, MgCl2 0.49 mM, MgSO4 0.44 mM, HEPES 25 mM, fungizone 1% and gentamicin 36 μl/100 ml (pH 7.2), and incubated in this medium for 15 min to deplete glucose from intracellular stores and extracellular space. Medium was then replaced by the same medium, but previously bubbled with N2 for 30 min, and the plate immediately transferred to an anaerobic chamber at 37°C with N2-enriched atmosphere. Cultures were maintained in these conditions for 45 or 60 min (ischemic insult). Following OGD, the slices were washed twice with HBSS and returned to their original culture conditions (see above). Slices in the control groups were treated in parallel to slices in the OGD groups, and incubated for similar periods of time (45 or 60 min) but were washed with glucose fortified medium and exposed to warmed, humidified air with 5% CO2.
Estrogen treatment
In the estradiol group (E2), 17β-estradiol (10 nM in 0.01% dimethylsulfoxide – DMSO) was added to the culture medium on div 7 of culture and maintained throughout the experiment, while in the vehicle group DMSO 0.01% was added to the OGD medium and to the culture medium during the recovery period. On div 14, cultures were exposed to 60 min of OGD followed by a 24-h recovery, as described above [12].
Quantification of cellular death
Cellular damage was assessed by fluorescent image analysis of propidium iodide (PI) uptake. After a recovery period of 22 h, 7.5 μM PI was added to cultures and incubated for 2 h. PI is excluded from healthy cells, but following loss of membrane integrity enters cells, binds to DNA and becomes highly fluorescent [16]. Cultures were observed with an inverted microscope (Nikon Eclipse TE 300) using a standard rhodamine filter set. Images were captured and then analyzed using Scion Image software (www.scioncorp.com). The area where PI fluorescence was detectable above background levels was determined using the “density slice” option of the software and compared to total CA1 area to obtain the percentage of damage [17].
Western blot analysis
After obtaining the fluorescent images, cultured slices were homogenized in lysis buffer (4% SDS, 2.1 mM EDTA, 50 mM Tris), aliquots were taken for protein determination and β-mercaptoethanol was added to a final concentration of 5% [18]. Samples containing 40 μg of protein were resolved by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Then, proteins were electrotransferred to nitrocellulose membranes using a semi-dry transfer apparatus (Bio-Rad Trans-Blot SD). After 2 h incubation at 4°C in blocking solution containing 5% skim milk powder and 0.1% Tween-20 in Tris-buffered saline (TBS; 50 mM Tris–HCl, 1.5% NaCl, pH 7.4), membranes were incubated overnight with the appropriate primary antibody diluted in the same blocking solution. Primary antibodies against the following proteins were used: EAAT1 and EAAT2 (both 1:50,000 dilution, rabbit polyclonal), ERα (1:200 dilution, rabbit polyclonal) and ERβ (1:1,000 dilution, rabbit polyclonal). Subsequently, membranes were washed and incubated for 2 h with horseradish peroxidase-conjugated secondary antibodies recognizing antigens from the same host as the corresponding primary antibody (1:1,000 dilution in blocking solution). Immunoreactive bands were revealed by an enhanced chemiluminescence kit (ECL, Pharmacia), and detected using X-ray films. The same blots were re-probed with β-actin antibody (1:5,000 dilution) as an internal control. The immunoblot films were scanned and the digitized images analyzed with Optiquant software (Packard Instrument). For each experiment, test groups were compared to control groups not exposed to OGD, which were considered as 100%, thus assuring the same signal intensity for all groups.
Statistical analysis
Data are presented as mean ± SEM of the indicated number of experiments. PI data are expressed as % damage of CA1 region, and Western Blotting data are expressed as % control. One-way analysis of variance (ANOVA) was applied to the means to determine significant differences between experimental groups; Duncan’s multiple range test performed post hoc comparisons.
Results
Estradiol treatment protected against 60 min OGD
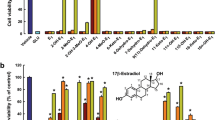
Exposure to OGD for 60 min caused a marked increase in fluorescence in the hippocampal CA1 and CA2 areas, indicative of a high incorporation of PI, as shown in the photomicrograph in Fig. 1a (OGD DMSO group). Quantification of PI fluorescence showed that 60 min OGD resulted in damage to 69% of the CA1 region (Fig. 1b), whereas in the dentate gyrus (DG) area no consistent damage was observed (Fig. 1a). Pretreatment with 10 nM 17β-estradiol for 7 days prior to OGD exposure (OGD E2 group) significantly reduced cellular death in the vulnerable CA1 area by 46% (Fig. 1a, b) compared with vehicle control.

Effect of 17β-estradiol on cellular damage induced by 60 min of OGD in organotypic hippocampal slice cultures. a Representative photomicrographs of cultures show PI fluorescence 24 h after OGD. b Quantitative analysis of CA1 damage in control and OGD slices treated with vehicle (DMSO) or 17β-estradiol (E2). DMSO 0.01% was added during OGD and recovery periods while 17β-estradiol (10 nM) was added 7 days prior to lesion induction and kept throughout the experiment. The area where PI fluorescence was detectable above background levels was determined and compared with the total CA1 area to obtain the percentage of damage. Details on image analysis are given in Materials and methods. Data represent means ± SEM, n = 15 slices per treatment. * Different from vehicle-treated (DMSO) and estradiol-treated (E2) control cultures; # Different from all other groups (one-way ANOVA followed by Duncan’s test, P < 0.001)
Changes in estrogen receptors, ERα and ERβ, in response to estradiol treatment and 60 min OGD
Exposure to OGD for 60 min caused a significant decrease (~20%) in the abundance of ERα protein in cultures treated with both vehicle (81 ± 5% control, n = 10) and estradiol (79 ± 5% control, n = 12) (Fig. 2a). Pretreatment with 10 nM 17β-estradiol for 7 days prior to OGD exposure significantly increased the immunocontent of ERβ by 26% (126 ± 10% control, n = 6) in OGD non-exposed control cultures (Fig. 2b). This increase was sustained even when cultures were exposed to 60 min OGD (122 ± 10% control, n = 6). In contrast, the levels of EAAT1 and EAAT2 in the cultures treated with vehicle or estradiol or OGD were unchanged (Fig. 3a, b).

Representative Western blots of estrogen receptors a ERα and b ERβ in rat organotypic hippocampal slice cultures treated with vehicle (DMSO) or 10 nM 17β-estradiol (E2), and exposed (OGD) or not (control) to 60 min of OGD. Equal amounts of protein samples were analyzed by polyacrylamide gel electrophoresis and immunoblotting with antibodies against ERα or ERβ and β-actin. The molecular weight of each protein (in kilodalton) is indicated on the left. Densitometric measurements were performed on individual immunoblots obtained from 6 to 12 slices per treatment for each antibody tested. The densitometric values obtained for all antibodies from all treatments were first normalized to their respective β-actin densitometric values and then expressed as percentage of their respective controls (100%). Data represent relative optical density and are expressed as the mean ± SEM. In a * different from vehicle-treated (DMSO; n = 6–10) and estradiol-treated (E2; n = 6–12) control cultures and in b * different from vehicle-treated (DMSO) control and OGD cultures (one-way ANOVA followed by Duncan’s test, P < 0.05)

Representative Western blots of glutamate transporters a EAAT1 and b EAAT2 in rat organotypic hippocampal slice cultures treated with vehicle (DMSO) or 10 nM 17β-estradiol (E2), and exposed (OGD) or not (control) to 60 min of OGD. Equal amounts of protein samples were analyzed by polyacrylamide gel electrophoresis and immunoblotting with antibodies against EAAT1 or EAAT2 and β-actin. The molecular weight of each protein (in kilodalton) is indicated on the left. Densitometric measurements were performed on individual immunoblots obtained from 6 to 12 slices per treatment for each antibody tested. The densitometric values obtained for all antibodies from all treatments were first normalized to their respective β-actin densitometric values and then expressed as percentage of their respective controls (100%). Data represent relative optical density and are expressed as the mean ± SEM different slices from vehicle-treated (DMSO; n = 6–10) and estradiol-treated (E2; n = 6–12) and exposed (OGD) or not (control) to 60 min of OGD
Discussion
Although neuroprotection afforded by estradiol treatment has been extensively studied in a variety of in vivo and in vitro models, the specific mechanisms contributing to this protection remain unknown. In particular, the neuroprotective efficacy of estrogens has been well described in in vitro models of cellular death [12, 19, 20]. We have previously demonstrated the neuroprotective effect of 17β-estradiol in organotypic cultures exposed to OGD and suggest that the neuroprotective mechanisms could involve the recruitment of pathways involving Akt and GSK-3β that reduce apoptosis in this injury model [12]. In the present study, using the same experimental model, we have investigated whether neuroprotection by estradiol treatment involves alteration in the abundance of glial glutamate transporters, EAAT1 and EAAT2, and estrogen receptors, ERα and ERβ.
Our measurements of propidium iodide (PI) uptake are consistent with previous observations of neuroprotection by estradiol treatment in the in vitro model of ischemia [12, 17, 20, 21] used in the present study. The presence of physiological levels of 17β-estradiol for 7 days prior to injury, exerted protective effects against 60 min of OGD consistent with our previous findings.
Many, though not all, of the neuroprotective effects of estrogen are mediated by the activation of estrogen receptors [22, 23]. These receptors, acting as ligand-activated transcription factors, bind to specific DNA sequences and regulate transcription [22]. The differential roles of estrogen receptors subtypes ERα and ERβ for neuroprotection remains unresolved. In the present study, we have observed that the abundance of ERα protein was significantly decreased 24 h after OGD in vehicle-treated and estradiol-treated organotypic cultures. A recent study has shown decreased nuclear ERα expression in hippocampal neurons in Alzheimer patients, suggesting that this neurodegenerative condition may involve ERα-regulated transcription [24]. The present findings are in contrast to a previous report showing that ERα mRNA in the cortical region is dramatically up-regulated with in vivo ischemia [25] and these differences may arise from the experimental models employed in these studies. In the present study, hippocampal slices were exposed to OGD for 45–60 min, followed by a 24 h period of reoxygenation. In contrast, the in vivo study by Dubal et al. utilized a permanent model of focal cerebral ischemia (24 h) without reperfusion, and observed increases in ERα mRNA in cortical tissue. While the MCAO injury model does affect the hippocampus, levels of ERα were not measured in this region. Dubal and colleagues [25], in the same in vivo study, have also shown that injury-induced down-regulation of ERβ mRNA is prevented by estradiol treatment. Although in our in vitro model of OGD we did not observe a down-regulation of the levels of ERβ protein, estradiol treatment induced a significant increase in the abundance of ERβ compared to vehicle-treated cultures, which was sustained even after the lesion induction. Together, these data suggest that ERβ abundance might be involved in the protective effects of estradiol observed in these models of cellular death.
The differential roles of estrogen receptor subtypes ERα and ERβ in neuroprotection remain unresolved. Recently, Carswell and colleagues [26], using selective estrogen receptor agonists, showed the involvement of ERβ in neuroprotection in a mouse model of global cerebral ischemia. However, Dubal and colleagues [27], using ERα and ERβ knockout mice, found that ERα was required for the protective effects of estradiol against brain injury. Nevertheless, other studies indicate that both estrogen receptor subtypes, ERα and ERβ, can contribute to estrogen-induced neuroprotection [28, 29].
Altered glutamate transport may contribute to the pathogenesis of many neurodegenerative conditions and altered expression or function of EAATs has been identified in various pathologies, including cerebral ischemia [6]. An increasing body of evidence suggests that regulation of glutamate transport may contribute to the development of ischemic tolerance [30, 31]. In the present study, we did not observe any significant changes in the levels of EAAT1 and EAAT2 proteins in response to pretreatment with estradiol or after OGD. Our results are in agreement with the findings of Sato et al. [11] that total expression of EAAT1 is not changed by estradiol treatment. We have recently found that in response to hypoxic preconditioning in the neonatal rat brain, although the immunocontent of EAAT1 and ERβ remain unchanged, EAAT2 and ERα are up-regulated in a similar manner in cortex, while EAAT2 is down-regulated in striatum [32]. However, the abundance of none of these proteins (EAAT1, EAAT2, ERα and ERβ) showed any significant changes in the hippocampus following preconditioning in vivo [12] – findings in agreement with our present observations in this in vitro model. As organotypic cultures might respond similarly to in vivo ischemia, the mechanisms that result in tolerance and the response required for key proteins in particular brain regions could be also similar.
Although in the present studies we were unable to detect any alterations in the abundance of glial EAATs in cultures treated with estradiol, it would seem premature to discount possible modulation of their activity by estrogenic mechanisms. EAAT function is subject to complex modulation and kinase activation is a potent signal regulating the transport of glutamate either by increasing the activity, abundance or cell-surface expression of EAATs [6]. 17β-Estradiol has been reported to enhance Glu uptake in rat synaptosomes treated with amyloid β [9] and in astrocytes from Alzheimer’s patients [10]. In contrast, estrogens were shown to inhibit Glu uptake in cultured astrocytes via ERα [11]. Given that we have shown previously a linkage between neuroprotection induced by 17β-estradiol and phosphatidylinositol 3-kinase [12], which regulates EAAT activity [6], further investigation is warranted into how neuroprotection by estrogen or preconditioning involves modulation of EAAT function.
In summary, the data reported here provide clear evidence that, in organotypic hippocampal slice cultures, ERα is down-regulated by OGD while ERβ is up-regulated by estradiol treatment, suggesting that in vitro, estrogen neuroprotection against ischemia might involve regulation of ER expression and, consequently, of the genes regulated by these receptors. Despite the fact that significant changes in EAAT1 and EAAT2 expression were not detected in this injury model, further work is needed to investigate whether estrogens modulate their function.
References
Blanchet PJ, Fang J, Hyland K, Arnold LA, Mouradian MM, Chase TN (1999) Short-term effects of high-dose 17beta-estradiol in postmenopausal PD patients: a crossover study. Neurology 53:91–95
Lee SB, McEwen BS (2001) Neurotrophic and neuroprotective actions of estrogens and their therapeutic implications Annu Rev Pharmacol Toxicol 41:569–591
Aranda A, Pascual A (2001) Nuclear hormone receptors and gene expression. Physiol Rev 81:1269–1304
Hall JM, Couse JF, Korach KS (2001) The multifaceted mechanisms of estradiol and estrogen receptor signaling. J Biol Chem 276:36869–36872
Lipton SA, Rosenberg PA (1994) Excitatory amino acids as a final common pathway for neurologic disorders. New Eng J Med 330:613–622
O’Shea RD (2002) Roles and regulation of glutamate transporters in the central nervous system. Clin Exp Pharmacol Physiol 29:1018–1023
Weaver CE, Park-Chung M, Gibbs TT, Farb DH (1997) 17beta-Estradiol protects against NMDA-induced excitotoxicity by direct inhibition of NMDA receptors. Brain Res 761:338–341
Wong M, Moss RL (1992) Long-term and short-term electrophysiological effects of estrogen on the synaptic properties of hippocampal CA1 neurons. J Neurosci 12:3217–3225
Keller JN, Germeyer A, Begley JG, Mattson MP (1997) 17beta-Estradiol attenuates oxidative impairment of synaptic Na+/K+-ATPase activity, glucose transport, and glutamate transport induced by amyloid beta-peptide and iron. J Neurosci Res 50:522–530
Liang Z, Valla J, Sefidvash-Hockley S, Rogers J, Li R (2002) Effects of estrogen treatment on glutamate uptake in cultured human astrocytes derived from cortex of Alzheimer’s disease patients. J Neurochem 80:807–814
Sato K, Matsuki N, Ohno Y, Nakazawa K (2003) Estrogen inhibit l-glutamate uptake activity of astrocytes via membrane estrogen receptor alpha. J Neurochem 86:1498–1505
Cimarosti H, Zamin LL, Frozza R, Nassif M, Horn AP, Tavares A, Netto CA, Salbego C (2005) Estradiol protects against oxygen and glucose deprivation in rat hippocampal organotypic cultures and activates Akt and inactivates Gsk-3 beta. Neurochem Res 30:191–199
Cimarosti H, Rodnight R, Tavares A, Paiva R, Valentim L, Rocha E, Salbego C (2001) An investigation of the neuroprotective effect of lithium in organotypic slice culture of rat hippocampus exposed to oxygen and glucose deprivation. Neurosci Lett 315:33–36
Stoppini L, Buchs P, Muller DA (1991) Simple method for organotypic cultures of nervous tissue. J Neurosci Methods 37:173–182
Strasser U, Fischer G (1995) Quantitative measurement of neuronal degeneration in organotypic hippocampal cultures after combined oxygen/glucose deprivation. J Neurosci Methods 57:177–186
Noraberg J, Kristensen BW, Zimmer J (1999) Markers for neuronal degeneration in organotypic slice cultures. Brain Res Protocols 3:278–290
Valentim LM, Rodnight R, Geyer AB, Horn AP, Tavares A, Cimarosti H, Netto CA, Salbego CG (2003) Changes in heat shock protein 27 phosphorylation and immunocontent in response to preconditioning to oxygen and glucose deprivation in organotypic hippocampal cultures. Neuroscience 118:379–386
Tavares A, Cimarosti H, Valentim L, Salbego C (2001) Profile of phosphoprotein labelling in organotypic slice cultures of rat hippocampus. Neuroreport 12:2705–2709
Behl C (2002) Oestrogen as a neuroprotective hormone. Nat Rev Neurosci 3:433–442
Wilson ME, Dubal DB, Wise PM (2000) Estradiol protects against injury-induced cell death in cortical explant cultures: a role for estrogen receptors. Brain Res 873:235–242
Xu G-P, Dave KR, Vivero R, Schmidt-Kastner R, Sick TJ, Pérez-Pinzón MA (2002) Improvement in neuronal survival after ischemic preconditioning in hippocampal slice cultures. Brain Res 952:153–158
Kumar V, Chambon P (1988) The estrogen receptor binds tightly to its responsive element as a ligand-induced homodimer. Cell 55:145–156
Pike CJ (1999) Estrogen modulates neuronal Bcl-xL expression and – amyloid-induced apoptosis: relevance to Alzheimer’s disease. J Neurochem 72:1552–1563
Hu X-Y, Qin S, Lu Y-P, Ravid R, Swaab DF, Zhou JN (2003) Decreased estrogen receptor-alpha expression in hippocampal neurons in relation to hyperphosphorylated tau in Alzheimer patients. Acta Neuropathol 106:213–220
Dubal DB, Shughrue PJ, Wilson ME, Merchenthaler I, Wise PM (1999) Estradiol modulates Bcl-2 in cerebral ischemia: a potential role for estrogen receptors. J Neurosci 19:6385–6393
Carswell HVO, Macrae IM, Gallagher L, Harrop E, Horsburgh KJ (2004) Neuroprotection by a selective estrogen receptor agonist in a mouse model of global ischemia. Am J Physiol Heart Circ Physiol 287:H1501–H1504
Dubal DB, Zhu H, Yu J, Rau SW, Shughrue PJ, Merchenthaler I, Kindy MS, Wise PM (2001) Estrogen receptor alpha, not beta, is a critical link in estradiol-mediated protection against brain injury. Proc Natl Acad Sci USA 98:1952–1957
Fitzpatrick JL, Mize AL, Wade CB, Harris JA, Shapiro RA, Dorsa DM (2002) Estrogen-mediated neuroprotection against beta-amyloid toxicity requires expression of estrogen receptor alpha or beta and activation of the MAPK pathway. J Neurochem 82:674–682
Zhao L, Wu T-W, Brinton RD (2004) Estrogen receptor subtypes alpha and beta contribute to neuroprotection and increased Bcl-2 expression in primary hippocampal neurons. Brain Res 1010:22–34
Douen AG, Akiyama K, Hogan MJ, Wang F, Dong L, Chow AK, Hakim A (2000) Preconditioning with cortical spreading depression decreases intraischemic cerebral glutamate levels and down-regulates excitatory amino acid transporters EAAT1 and EAAT2 from rat cerebral cortex plasma membranes. J Neurochem 75:812–818
Romera C, Hurtado O, Botella SH, Lizasoain I, Cárdenas A, Fernández-Tomé P, Leza JC, Lorenzo P, Moro MA (2004) In vitro ischemic tolerance involves upregulation of glutamate transport partly mediated by the TACE/ADAM17-tumor necrosis factor-alpha pathway. J Neurosci 24:1350–1357
Cimarosti H, Jones NM, O’Shea RD, Pow DV, Salbego C, Beart PM (2005) Hypoxic preconditioning in neonatal rat brain involves regulation of excitatory amino acid transporter 2 and estrogen receptor alpha. Neurosci Lett 385:52–57
Acknowledgements
Supported in part by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq – Brazil), which provided HC a scholarship for research towards Ph.D. in Australia, and by Neurosciences Victoria (NMJ) and a Program Grant (#236805) from the NH&MRC (Australia), of which PMB is a Research Fellow. We thank Dr. D. Pow (University of Queensland) for the gifts of glutamate transporters antibodies.
Author information
Authors and Affiliations
Corresponding author
Additional information
Helena Cimarosti and Ross D. O’Shea, equal first authors.
Rights and permissions
About this article
Cite this article
Cimarosti, H., O’Shea, R.D., Jones, N.M. et al. The Effects of Estradiol on Estrogen Receptor and Glutamate Transporter Expression in Organotypic Hippocampal Cultures Exposed to Oxygen--Glucose Deprivation. Neurochem Res 31, 483–490 (2006). https://doi.org/10.1007/s11064-006-9043-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-006-9043-9