
Abstract
Purpose
Rap2B, a member of the GTP-binding proteins, is generally up-regulated in numerous types of tumors. Nevertheless, the influence and regulatory mechanisms of Rap2B in gliomas are still not corroborated. Therefore, we analyzed the expression of Rap2B in glioma tissues and cells, and researched its significance in adhesion, proliferation, migration and invasion of the glioma cell line.
Methods
We analyzed the expression of Rap2B in different pathologic grades of glioma tissues by tissue microarray and immunohistochemistry. We assessed the expression of Rap2B in glioma tissue and non-tumor tissue by Western blot. And the expression of Rap2b protein in glioma cells and normal human astrocytes (NHA) was detected by Western blot. In addition, we disclosed the effect of Rap2B knockdown on cell adhesion, proliferation, migration and invasion by using cell attachment assay, CCK-8 assay, cell migration assay and Wound Healing assay, cell invasion assay, respectively. Western blot was used to detect the changes of expression level of NF-kB, MMP-2 and MMP-9 protein when downregulated the expression of Rap2B.
Results
The tissue microarray immunohistochemical results of glioma showed that the expression of Rap2B had no significant correlations between Rap2B expression and the clinicopathologic variables, including patient age (P = 0.352), gender (P = 0.858), WHO Grade (P = 0.693) and histology type (P = 0.877). Western blot analysis showed that the glioma tissue had a dramatically increase of Rap2B expression compared with the non-tumor tissues (P < 0.01). And the expression of Rap2B was markedly up-regulated in all 5 glioma cell lines compared with that in normal human astrocytes (NHA) (P < 0.01). We found that the ability of adhesion, proliferation, migration and invasion of glioma cells were significantly decreased after downregulated Rap2B expression compared with the control group (P < 0.05). In addition, Western blot results showed that the expression levels of NF-kB, MMP-2 and MMP-9 in the interference group were significantly lower than those in the negative control group (P < 0.05).
Conclusions
Rap2B expression is up-regulated in glioma tissues and glioma cell lines. Knockdown of Rap2B inhibits glioma cells’ adhesion and proliferation in vitro. Knockdown of Rap2B inhibits glioma cells’ migration in vitro. Knockdown of Rap2B inhibits glioma cells’ invasion and MMPs activity through NF-kB pathway.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Gliomas represent the most common and aggressive type of tumors in the central nervous system (CNS), which accounts for 50% of primary intracranial tumors [1, 2]. As the most aggressive type of gliomas, glioblastoma multiform (GBM) is the grade IV histological malignancy according to the WHO classification, with a median survival period of less than 1 year [3]. The poor prognosis of glioblastoma is largely attributed to their rapid growth, invasive, migratory, and high rate of recurrence [4]. Therefore, comprehensive understanding of the molecular mechanisms which expedite glioma adhesion, migration and invasion is urgently needed, in order to develop more effective therapies against glioma.
Rap2B was first found when a platelet cDNA library was screened by investigators in 1990 [5, 6]. Rap2B is a member of the Ras family of small GTP-binding proteins, whose expression can be discovered up-regulated in miscellaneous human tumors [7]. Studies have suggested that Ras family members are implicated in a series of biological functions in human cells, such as signal transduction, proliferation and migration [8, 9]. In addition, Rap2B as a novel p53 target takes part in p53-mediated pro-survival function, which also enhances the possibility that targeting Rap2B could sensitize tumor cells to apoptosis in response to DNA damage [10]. Recent studies have showed that miR-342-3p targets Rap2B to inhibit cell proliferation, migration, and invasion of non-small cell lung cancer [11] and Rap2B promotes migration and invasion of human suprarenal epithelioma [12]. Whereas there is little paper research the influences of Rap2B in gliomas.
In this study, we used a tissue microarray (TMA) to estimate the expression of Rap2B in glioma tissues, and the relationship with clinicopathologic features. Meanwhile, we utilized western blot to assay expression of Rap2B in different glioma cell lines. We then demonstrated that knockdown of Rap2B restrained glioma cell adhesion, migration and invasion abilities. Besides, we researched the molecular mechanisms of Rap2B in glioma cells.
Materials and methods
Patients and samples
We purchased a glioma TMA kit from Shanxi Alenabio Bio-technology (Xi’an, Shanxi, China), No: GL2083a. In the light of the WHO criteria, pathologic grades of tumors were divided as follows: 134 cases with Grades I–II and 58 cases with Grades III–IV. The array dot diameter was 1.0 mm, and each dot represented a tissue spot from one individual specimen that was selected and pathologically confirmed. In addition, four paired glioma tissues and adjacent non-tumor tissues from the same patients were obtained from the Department of Neurosurgery, the Affiliated Hospital of Xuzhou Medical University.
Immunohistochemistry of TMA
TMA slices were dewaxed at 62 °C for 2 h, followed by two 30 min washes with xylene. The sections were blended by 5 min washes in 100, 95 and 80% ethanol and distilled water sequentially. With a microwave we heats citrate buffer (pH 6.0) to 95 °C, then put the sections in it for 15 min, and make citrate buffer cool to room temperature after the restoration. Wishing with PBS by three 5 min, we add hydrogen peroxide to slices for 30 min in order to block endogenous peroxidase activity. After incubating the sections for 30 min with 10% goat serum, the sections were incubated with monoclonal rabbit anti-Rap2B antibody (1:500) (Abcam, Cambridge, MA) at 4 °C overnight. Next day remove the slices from 4 °C and rewarming at room temperature 30 min, wishing with PBS by three 5 min. Then, the sections were incubated with a biotinylated secondary antibody (Zhongshan Biotech, Beijing, China) at room temperature for 30 min, followed by the incubation with streptavidin-peroxidase (Zhongshan Biotech, Beijing, China) for an additional 30 min. After rinsing with PBS 3 times for 5 min, the sections were stained using DAB (Zhongshan Biotech, Beijing, China), rinsed in distilled water and counterstained with hematoxylin. Dehydration was then performed in 80, 95, 95 and 100% ethanol and distilled water sequentially. Then the sections were sealed with cover slips. Negative controls were stained with non-immune serum to replace primary antibodies.
Evaluation of immunohistochemical staining
The evaluation of Rap2B staining was blindly and independently examined by the intensity of staining and the proportion of tumor cells showing an unequivocal positive reaction. The Rap2B staining intensity was scored 0–3 (0 = negative; 1 = weak; 2 = moderate; 3 = strong). The proportion of Rap2B-positive stained cells was also scored into four categories: 1 (0–25%), 2 (26–50%), 3 (51–75%) and 4 (76–100%). For statistical analyses, the level of Rap2B staining was evaluated by the immunoreactive score (IRS), which is calculated by multiplying the scores of staining intensity and the proportion of positive cells. According to the IRS, the Rap2B staining pattern was categorized as negative (IRS: 0), weak (IRS: 1–3), moderate (IRS: 4–6) and strong (IRS: 8–12).
Cell culture and transfection
Primary normal human astrocytes (NHA) were bought from the KeyGEN Biotech Company (Nanjing, China) and fostered under the conditions as instructed by the manufacturer. Human glioma cell lines (U251, U87, T98G, SHG44) and Rat glioma cell C6 were bought from the Institute of Biochemistry and Cell Biology, Chinese Academy of Science. Cells were cultivated in high glucose DMEM with 10% fetal bovine serum (FBS) (Invitrogen, Shanghai, China) and in 5% CO2 humidified atmosphere at 37 °C. When cells growed to 50% coalescence, we willed transfected them. Non-specific control siRNA (Qiagen, Mississauga, ON, Canada) or Rap2B siRNA (Qiagen, Mississauga, ON, Canada) was transfected by siLentFect Lipid Reagent (Bio-Rad, Hercules, CA, USA) on the basis of the manufacturer’s instructions. We detached the medium embodying transfection reagents after 24 h. Then the cells were irrigated twice with PBS and maintained in fresh medium. 48 h after transfection, cells were lysed for western blot assay and were used for cell attachment assay, cell migration assay and invasion assay.
Western blot analysis
Treated and untreated cells were lysed in radioimmunoprecipitation assay buffer with a freshly added protease inhibitor cocktail (Roche Applied Science, Indianapolis, IN, USA). The protein density was determined by the bicinchoninic acid (BCA) assay (Pierce, Rockford, IL, USA). All protein samples were formulated and denatured, protein was separated by SDS-PAGE in 8–12% gradient polyacrylamide gel and transferred onto polyvinylidene difluoride membranes (Millipore, Billerica, MA, USA). The membranes were blocked in 5% nonfat milk for 3 h at room temperature, and then incubated with primary antibody overnight at 4 °C. The following antibodies were applied: rabbit anti-Rap2B (Abcam, Cambridge, MA), rabbit anti-NF-kB, rabbit anti-MMP2, rabbit anti-MMP9 (all from Cell Signaling Technology, Beverly, MA, USA). The membranes were then incubated with Peroxidase-Conjugated Affinipure Goat Anti-Rabbit IgG secondary antibody (Zhongshan Biotech, Beijing, China) for 2 h at room temperature. Detection was performed by an enhanced chemiluminescence method (Pierce, Rockford, IL, USA).
Cell attachment assay
96-well plates were coated with 1.25 μg/ml fibronectin (BD Biosciences, NJ, USA) in 100 μl PBS overnight at 4 °C. Wells coated with bovine serum albumin (BSA) served as negative control. The plates were blocked with 2.5 mg/ml BSA for 2 h in DMEM at 37 °C. Cells were trypsinized and 2 × 104 cells were seeded in each well for 1 h at 37 °C. Experiments were carried out in triplicate, and three random fields of each well were recorded.
Cell growth assay
Cell growth was analyzed using the Cell Counting Kit-8 (CCK-8; Beyotime, Nantong, China). U251 and U87 cells were cultured at a density of 5 × 103 cells/well and suspended in 100 µl high glucose DMEM containing 10% FBS in 96-well plates and incubated for 1, 2, 3 and 4 days, respectively. Then, 10 µl CCK-8 solution was added to each well and incubated at 37 °C for 1 h. The optical density was measured at 450 nm on an ELX-800 spectrometer reader (Bio-Tek Instruments, Winooski, VT, USA).
Wound-healing assay
Cells were seeded into six-well plates at a density of 5 × 105 cells/well in culture medium and transfects with siRNA. Twenty-four hours after transfection, the cells were scratched on the monolayer with a 10 μl pipette tip, and washed twice with PBS. Then, cells were maintained in high glucose DMEM medium for an additional 24 h. Photographs were taken by an inverted Leica phase-contrast microscope (Leica DFC 300 FX) at 0 and 24 h time points. Wound-healing percentage of the cells was determined by the ratio of healing width at each time point to the wound width at 0 h.
Cell migration assay
Cell migration was determined by using a modified two-chamber plates with a pore size of 8 μm. For the cell migration assay, 1 × 105 U251 and U87 cells were seeded in serum-free medium in the upper chamber. To stimulate migration, 10% of serum containing culture medium was added to the bottom wells. After the cell was incubated for 24 h at 37 °C, cells on the top surface of the insert were carefully removed with a cotton swab and the cells that had traversed the membrane were fixed in methanol, stained with crystal violet to visualize nuclei and the number of migrating cells was counted under 40 × magnification in 5 fields (up, down, median, left, right), and the means for each chamber were determined.
Cell invasion assay
The invasion assay was performed using a modified two-chamber plates with a pore size of 8 μm. The transwell filter inserts were coated with matrigel, 1 × 105 U251 and U87 cells were seeded in serum-free medium in the upper chamber. To stimulate invasion, 10% of serum containing culture medium was added to the bottom wells. After 24 h incubation at 37 °C, cells in the upper chamber were carefully removed with a cotton swab and the cells that had traversed the membrane were fixed in methanol, stained with Giemsa and counted. For counting, five fields (up, down, median, left, right × 40) per filter were counted under a microscope.
Statistical analysis
Statistical analysis was carried out using SPSS 19.0 software (SPSS, Chicago, IL). Values are expressed as the mean ± SD. The relationship between Rap2B staining and the clinicopathologic parameters of the glioma patients, such as age, gender, WHO grade and histologic type, was assessed by χ2 test. For attachment, migration and invasion assay, the results were expressed as the mean ± SD. Each experiment was performed at least three times. Differences were considered significant when P value < 0.05.
Results
Correlation of Rap2B expression with clinicopathological parameters
The association between Rap2B expression and the clinicopathological data was summarized in Table 1. WHO grade and histologic type are known to be important prognostic markers for the patients with glioma. But we did not find any significant correlations between Rap2B expression and the clinicopathologic variables, including patient age (P = 0.352), gender (P = 0.858), WHO Grade (P = 0.693) and histology type (P = 0.877).
Rap2B expression is up-regulated in glioma tissues and glioma cell lines
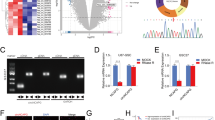
For the sake of investigate whether Rap2B expression is changed in glioma, we used a TMA to estimate the Rap2B expression in 192 gliomas The representative pictures presented in (Fig. 1a) revealed that Rap2B protein mainly localized in cytoplasm was situated in brown. Western blot assay was done applying four glioma tissues and paired non-tumor tissues. It was drastically increase of Rap2B expression in glioma tissues compared with the non-tumor tissues (Fig. 1b). In addition, Western blot analyses showed that expression of Rap2B was significantly higher in all five analyzed glioma cell lines, including SHG44, C6, U251, T98G, U87, in comparison with normal human astrocytes (NHA) (Fig. 1c). Collectively, our results suggest that Rap2B is up-regulated in gliomas.

Expression of Rap2B in glioma tissues and glioma cell lines. a Negative and positive staining in glioma tissue (magnification × 400). b Whole-cell protein extracts were further prepared from four paired tumor adjacent normal brain tissues and glioma tissues. The Rap2B protein level was determined by Western blot analysis. c Western blot analysis of Rap2B expression in normal human astrocytes NHA and glioma cell lines, including SHG44, C6, U251, T98G, U87. All experiments were carried out in triplicate. Data are shown as mean ± SE. **P < 0.01, ***P < 0.001
Knockdown of Rap2B inhibits glioma cells’ adhesion in vitro
Tumor cell adhesion to the extracellular matrix is implicated in tumor cell motility and metastasis. We then utilized the attachment assay to test whether Rap2B influence cell adhesiveness. We discovered that cell attachment ability was reduced by 63% and 52% in down-regulation of Rap2B in U251 and U87 cells (Fig. 2a, b).

Knockdown of Rap2B inhibits glioma cells adhesion and proliferation ability. a, b Cell attachment assay after knockdown of Rap2B in U251 and U87 cells. The graph shows the percentage of attached cells compared with the control group. c, d CCK-8 cell proliferation assay was performed after Rap2B knockdown in U251 and U87 cells. All experiments were carried out in triplicate. Data are shown as mean ± SE. **P < 0.01, ***P < 0.001
Knockdown of Rap2B inhibits glioma cells’ proliferation in vitro
Owing to the fact that Rap2B expression is significantly increased in glioma tissues compared with adjacent non-tumor tissues, Rap2B may play vital roles in tumor progression. U251 and U87 cells were transfected with siRNA targeting Rap2B to knockdown Rap2B expression. The results of the CCK-8 cell proliferation assays revealed decreased growth rates in the U251 and U87 cells depleted of Rap2B (Fig. 2c and d).
Knockdown of Rap2B inhibits glioma cells’ migration in vitro
To investigate the role of Rap2B in U251 and U87 cell migration ability, the wound healing and transwell migration assays were used. We observed that wound recovery was markedly decreased by 36.99% and 34.78% after Rap2B knockdown (Fig. 3a, b). And the number of migrated cells transfected with Rap2B-siRNA was obviously decreased in contrast to the negative control group (Fig. 3c, d). These results suggested that the silencing of Rap2B inhibited the migration ability of U251 and U87 cell lines.

Knockdown of Rap2B inhibits glioma cells motility. a, b Wound-healing assay was performed after Rap2B knockdown in U251 and U87 cells. There was significant delay in wound closure after Rap2B knockdown compared with the control cells. c, d Cell migration assay was performed after Rap2B knockdown in U251 and U87 cells. U251-siRap2B cells and U87-siRap2B cells decreased the ability to migrate through Boyden chamber compared with the control cells. All experiments were carried out in triplicate. Data are shown as mean ± SE. **P < 0.01, ***P < 0.001
Knockdown of Rap2B inhibits glioma cells’ invasion and MMPs activity through NF-kB pathway
To investigate the function of Rap2B in U251 and U87 cell invasion ability, the transwell invasion assays were utilized. The transwell invasion assay suggested that lower cells pierced through the matrigel-coated chambers in siRNA-transfected cells (Fig. 4a, b). These results revealed that the silencing of Rap2B inhibited the invasiveness of U251 and U87 cell lines. Since MMPs have been seen as intimately associated with metastasis in many tumor cells, we then investigated whether Rap2B knockdown in U251 and U87 cells had an effect on the expression of MMP-2 and MMP-9. Western blot assay was utilized and the results revealed that the knockdown of Rap2B markedly decreased the protein levels of MMP-2 and MMP-9 in both cell lines (Fig. 4c, d). Furthermore, NF-kB is familiar to stimulate the MMPs bringing out invasion. Our reports discovered that the NF-kB expression was markedly increased in U251 and U87 cells after down-regulation of Rap2B (Fig. 4c, d).

Knockdown of Rap2B inhibits glioma cells invasion ability. a, b Matrigel cell invasion assay was performed after the knockdown of Rap2B in U251 and U87 cells. c Western blot analysis of the relative protein levels of NF-kB, MMP-2 and MMP-9 in down-regulation of Rap2B and control group of U251 and U87 cells. d Quantitative analysis of relative protein level of Rap2B, NF-kB, MMP-2 and MMP-9 in glioma U251 and U87 cells. All experiments were carried out in triplicate. Data are shown as mean ± SE. **P < 0.01, ***P < 0.001
Discussion
The Rap family of small GTP-binding proteins is made up of five members, namely, Rap1A, Rap1B, Rap2A, Rap2B and Rap2C, which are divided into two subfamilies, namely, Rap1 and Rap2 [13]. All share probably 50% amino acid sequence identity with the ras proteins, with the level of similarity within the Rap group being more enormous than 60% [14]. Rap2B gene is situated in 3q25.2 of human chromosome which is the hot field of cancer research, and its cDNA includes an open reading frame of 552 bp [12]. Rap2B can stimulate the NF-kappaB signaling pathway [15] and adjust p53-mediated pro-survival function to facilitate carcinogenesis [10]. It was justified that Rap2B was up-regulation in human primary lung squamous cell carcinoma and Rap2B was seen as a new candidate oncogene of lung cancer due to its ability of virulent transformation of Rat1 cell in vitro [10, 15]. Though Rap2B includes conserved domain and was part of the Ras superfamily, and previous researches indicated that the expression of Rap2B was up-regulation in suprarenal epithelioma and breast cancer [12], but little is known about the expression and function of Rap2B in gliomas. In this study, our data revealed that Rap2B is markedly increased in clinical glioma tissues and glioma cells compared to tumor adjacent normal brain tissue and NHA by Western blot technique. In a word, our data strongly suggest that Rap2B may play a vital role in the glioma development and progression. Our results are consistent with previous findings.
A vital role of invasive tumor cells is the ability to pass through the extracellular matrix (ECM) tissue borders, a process that can be accomplished by stimulating metalloproteases(MMPs) [16]. NF-KB is known to regulate the expression of genes involved in cancer cell invasion and metastasis, and it can promote the expression of MMP-2 and MMP-9 [17, 18]. MMP-2 and MMP-9 are massive expressed in glioma, and the expression of MMPs in glioma has been disclosed to be linked with invasion capacity and the risk of recurrence [19]. In the meantime, we found that knockdown of Rap2B significantly reduced cell invasiveness of U251 and U87 cells, which might be caused by the down-regulated expression of MMP-2 and MMP-9.
In summary, our results demonstrates that Rap2B play a vital role in human glioma pathogenesis. Overexpression of Rap2B influences glioma progression by enhancing cell adhesion, migration and invasion. Our results imply that Rap2B may be an important marker and a therapeutic target for gliomas, so hoping that we can provide an available therapeutic strategy to help control the progression of gliomas.
References
Hamza MA, Gilbert M (2014) Targeted therapy in gliomas. Curr Oncol Rep 16(4):379
Hu S, Xu L, Li L et al (2019) Overexpression of lncRNA PTENP1 suppresses glioma cell proliferation and metastasis in vitro. Oncol Targets Ther 12:147–156
Wen PY, Kesari S (2008) Malignant gliomas in adults. New Engl J Med 359(5):492–507
Zhen Y, Nan Y, Guo S et al (2018) Knockdown of NEAT1 repressed the malignant progression of glioma through sponging miR-107 and inhibiting CDK14. J Cell Physiol 1:1. https://doi.org/10.1002/jcp.27727
Ohmstede CA, Farrell FX, Reep BR, Clemetson KJ, Lapetina EG (1990) RAP2B: a RAS-related GTP-binding protein from platelets. Proc Natl Acad Sci USA 87(17):6527–6531
Farrell FX, Ohmstede CA, Reep BR, Lapetina EG (1990) cDNA sequence of a new ras-related gene (rap2b) isolated from human platelets with sequence homology to rap2. Nucleic Acids Res 18(14):4281
Zhang L, Duan HB (2017) Knockdown of Rap2B Inhibits the Proliferation and Invasion in Hepatocellular Carcinoma Cells. Oncol Res 25:19–27
Itoh M, Nelson CM, Myers CA, Bissell MJ (2007) Rap1 integrates tissue polarity, lumen formation, and tumorigenic potential in human breast epithelial cells. Can Res 67(10):4759–4766
Kooistra MR, Dube N, Bos JL (2007) Rap1: a key regulator in cell-cell junction formation. J Cell Sci 120(Pt 1):17–22
Di J, Tang J, Qian H et al (2017) p53 upregulates PLCε-IP3-Ca pathway and inhibits autophagy through its target gene Rap2B. Oncotarget 8:64657–64669
Peng YG, Zhang ZQ, Chen YB (2016) Rap2b promotes proliferation, migration, and invasion of lung cancer cells. J Recept Signal Transduct Res 36:459–464
Di J, Huang H, Qu D et al (2015) Rap2B promotes proliferation, migration, and invasion of human breast cancer through calcium-related ERK1/2 signaling pathway. Sci Rep 5:12363
Torti M, Lapetina EG (1994) Structure and function of rap proteins in human platelets. Thromb Haemost 71(5):533–543
Campa MJ, Farrell FX, Lapetina EG, Chang KJ (1993) Microinjection of Rap2B protein or RNA induces rearrangement of pigment granules in Xenopus oocytes. Biochem J 292(Pt 1):231–236
Xie X, Liu H, Wang M et al (2015) miR-342-3p targets RAP2B to suppress proliferation and invasion of non-small cell lung cancer cells. Tumour Biol 36:5031–5038
Che F, Xie X, Wang L et al (2018) B7-H6 expression is induced by lipopolysaccharide and facilitates cancer invasion and metastasis in human gliomas. Int Immunopharmacol 59:318–327
Mei P, Bai J, Shi M, Liu Q, Li Z, Fan Y et al (2014) BRMS1 suppresses glioma progression by regulating invasion, migration and adhesion of glioma cells. PLoS ONE 9(5):e98544
Cicek M, Fukuyama R, Welch DR, Sizemore N, Casey G (2005) Breast cancer metastasis suppressor 1 inhibits gene expression by targeting nuclear factor-kappaB activity. Cancer Res 65(9):3586–3595
Fan YC, Zhu YS, Mei PJ, Sun SG, Zhang H, Chen HF et al (2014) Cullin1 regulates proliferation, migration and invasion of glioma cells. Med Oncol 31(10):227
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Miao, F., Cui, C., Zuo, D. et al. Rap2B promotes cell adhesion, proliferation, migration and invasion of human glioma. J Neurooncol 143, 221–229 (2019). https://doi.org/10.1007/s11060-019-03163-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-019-03163-6