
Abstract
Cross-talk between competitive endogenous RNAs (ceRNAs) may play a critical role in revealing potential mechanisms of tumor development and physiology. Glioblastoma is the most common type of malignant primary brain tumor, and the mechanisms of tumor genesis and development in glioblastoma are unclear. Here, to investigate the role of non-coding RNAs and the ceRNA network in glioblastoma, we performed paired-end RNA sequencing and microarray analyses to obtain the expression profiles of mRNAs, lncRNAs, circRNAs and miRNAs. We identified that the expression of 501 lncRNAs, 1999 mRNAs, 2038 circRNAs and 143 miRNAs were often altered between glioblastoma and matched normal brain tissue. Gene ontology and Kyoto Encyclopedia of Genes and Genomes pathway analyses were performed on these differentially expressed mRNAs and miRNA-mediated target genes of lncRNAs and circRNAs. Furthermore, we used a multi-step computational framework and several bioinformatics methods to construct a ceRNA network combining mRNAs, miRNAs, lncRNAs and circRNA, based on co-expression analysis between the differentially expressed RNAs. We identified that plenty of lncRNAs, CircRNAs and their downstream target genes in the ceRNA network are related to glutamatergic synapse, suggesting that glutamate metabolism is involved in glioma biological functions. Our results will accelerate the understanding of tumorigenesis, cancer progression and even therapeutic targeting in glioblastoma.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Background
The central dogma of genomics is a basic rule for gene regulation and cancer genesis. Previous studies of cancer genomes and transcriptomes have identified profound alterations in genes and proteins [1] and also discovered that less than 2% of the total genome encodes protein-coding genes, suggesting that non-coding RNAs represent most of the human transcriptome [2, 3]. Indeed, pre-transcriptional regulation is less well-characterized than transcriptional regulation or post-transcriptional regulation. Recent evidence strongly suggests that noncoding RNAs, including lncRNAs, circular RNAs and miRNAs, are key elements of tumorigenesis and development [4, 5]. Furthermore, lncRNAs and circRNAs could be mediated by micro-RNA-binding sites in 3′ untranslated regions (3′UTRs), suggesting that mRNAs act as competitive endogenous RNAs (ceRNAs) [6]. lncRNAs could also directly regulate the expression of downstream genes [7]. RNA–RNA crosstalk and competing endogenous networks have also been found be important to cancer cells [8,9,10]. For example, Shao identified module biomarkers from a dysregulated ceRNA–ceRNA interaction network in lung adenocarcinoma [11], and Dou found that 24 circRNAs changed during osteoclastogenesis [12]. However, studies within this field are still scarce and in their beginning stages. High-throughput experiments and bioinformatics methods are viable ways to predict changes in non-coding RNAs and construct ceRNA networks in cancers.
Gliomas are the most common type of malignant primary brain tumor, yet no effective treatment for glioblastoma (GBM) currently exists [13]. Patients with GBM multiforme who undergo maximal safe tumor resection and standard radiochemotherapy with temozolomide achieve a median survival of only 14.6 months [14]. To further research and understand the complete pathogenesis of GBM, recent studies have increased their focus on noncoding RNAs. For example, quantitative RT-PCR demonstrated that HOTAIR expression was up-regulated in glioma tissues and cell lines [15]. Han et al. have found that ASLNC22381 and ASLNC2081 might play important roles in the recurrence and malignant progression of GBM [16]. However, comprehensive analyses for noncoding RNAs or ceRNA networks in GBM are still lacking, and studies of RNA–RNA interaction in GBM are also scarce, but the GBM deregulated ceRNA–ceRNA network (GDCCNet) may provide new hope for exploring the pathogenesis of GBM and discovering new, highly accurate diagnostic biomarkers.
Here, to study the potential tumorigenic role of this class of miRNA-mediated non-coding RNA interactions, we performed RNA sequencing in GBM and matched normal brain tissues to acquire the expression profiles of mRNAs, circRNAs and lncRNAs. We also used microarrays to detect changes in miRNAs. Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses were performed for mRNAs/lncRNAs–mRNA/circRNAs–miRNA–mRNA interactions with significantly different expression in GBM. Furthermore, we constructed a global deregulated ceRNA–ceRNA network of interactions by integrating miRNA regulation with RNA-seq data and analyzed differences between tumor tissues and normal tissues based on bio-informatic predictions and ceRNA theory. We also constructed that the ceRNA network only contained genes enriched in GBM core pathways (PI3K/AKT/mTOR, p53 and Rb signaling pathways). Our results demonstrate that lncRNAs and circRNAs play an important role in GBM genesis, and our findings may shed new light to the understanding of underlying mechanisms of non-coding RNAs in GBM.
Methods
Sample collection
All patients included in this study were pathologically diagnosed with gliomas, and all surgeries were performed in the West China Hospital at Sichuan University. Samples were immediately numbered, transferred into jars containing liquid nitrogen and stored for future use. All of the tumor samples were confirmed to be GBM by a neuropathologist, and matched normal brain tissues (surrounding tissues) were confirmed to contain no infiltration of tumor cells. The study including all experimental protocols was approved by the Ethics Committee of the west china hospital of Sichuan University and all patients gave pre-operative written informed consent. The methods were carried out in accordance with the approved guidelines.
mRNA library construction and sequencing
Total RNA was extracted using Qiagen (Qiagen, USA) following the manufacturer’s procedure. Total RNA quantity and purity was analyzed using a Bioanalyzer 2100 and an RNA 6000 Nano LabChip Kit (Agilent, CA, USA) with RIN number > 7.0. Using the Epicenter Ribo-Zero Gold Kit (Illumina, San Diego, USA), approximately 5 µg of total RNA from each specific tissue type was used to deplete ribosomal RNA, according to the manufacturer’s instructions. Following purification, the poly(A)− or poly(A)+ RNA fractions were fragmented into small pieces using divalent cations and elevated temperature. Then, the cleaved RNA fragments were reverse-transcribed to create the final cDNA library using the mRNA-seq sample preparation kit (Illumina, San Diego, USA) in accordance with the manufacturer’s protocol. The average insert size for the paired-end libraries was 450 bp (± 50 bp). Then, we performed paired-end sequencing on an Illumina Hiseq2500 (LC Sciences, USA) following the vendor’s recommended protocol.
Sequence and primary analysis
The strand-specific cDNA libraries were constructed from the pooled RNA and were sequenced using the Illumina 2000/2500 sequence platform. Using the Illumina paired-end RNA-seq approach, we sequenced the transcriptome of the brain tissues, generating a total of 28.64 million paired-end reads of 150 bp length. This yielded 85.92 gigabases (Gb) of sequence. The low quality reads (1) reads containing sequencing adaptors; (2) reads containing sequencing primers; (3) nucleotides with a quality score lower than 20 were removed. After read removal, a total of 70.14 G bp of cleaned, paired-end reads were produced. The raw sequencing data have been submitted to the NCBI, the GEO Short Number is GSE86202.
RNA-seq read mapping
We aligned reads from tumors and normal samples to the UCSC (http://genome.ucsc.edu/) human reference genome using the TopHat package. TopHat initially removes a portion of the reads based on the quality information accompanying each read and then maps the reads to the reference genome. TopHat allows for multiple alignments per read (up to 20 by default) and a maximum of two mismatches when mapping the reads to the reference genome. TopHat builds a database of potential splice junctions and confirms the splice junctions by comparing the previously unmapped reads against the database of putative junctions. Transcript abundance estimation and differential expression testing of the aligned read files were processed by Cufflinks, which uses the normalized RNA-seq fragment counts to measure the relative abundances of the transcripts. The unit of measure is fragments per kilobase of exon per million fragments mapped (FPKM). The reference GTF annotation file used in Cufflinks was downloaded from the UCSC database. First, Cufflinks was used to assemble the de novo transcriptome; second, Cuffmerge was used to comerge all transcripts from tumor and normal tissue samples to generate unique transcripts. The UCSC GTF file was passed to Cuffdiff along with the original alignment (SAM) files produced by TopHat. Cuffdiff estimates the abundance of the transcripts listed in the GTF file using alignments from the SAM file and concurrently tests for differential expression.
lncRNA–mRNA and circRNA/lncRNA–miRNA–mRNA co-expression networks
Using existing miRNA target prediction methods, lncRNA–mRNA and circRNA/lncRNA–miRNA–mRNA interactions were identified [17,18,19,20,21,22,23,24,25]. UTR sequence data were acquired from Ensembl (http://ensemblgenomes.org). TargetScan and miRanda were simultaneously used to predict putative miRNA–mRNA interactions, and only the predicted mRNAs with a context score percentile > 50 in TargetScan and max energy <− 20 in miRanda were included in this study. Based on the ceRNA hypothesis, we also identified putative circRNA/lncRNA–miRNA integrations using TargetScan 7.0 software and miRanda and then filtered mRNAs to include those with a context score percentile > 50 in TargetScan and max energy <− 20 in miRanda. Because lncRNA–mRNA interactions are c
is regulated, lncRNA and mRNA with distances < 100 kb were included. Based on established co-expression data, deregulated ceRNA networks and circRNA/LncRNA/miRNA interactions of interest were mapped using Cytoscape software (V. 3.2.1).
Enrichment analysis
To predict the possible functions of the differentially expressed mRNAs and to explore the pathways in which they participate, differentially expressed mRNAs screened by Volcano Plot filtering were further studied using the GO (http://www.geneontology.org) and KEGG (http://www.genome.jp/kegg) databases. GO and KEGG analyses for differentially expressed lncRNAs and circRNAs identified down-stream genes regulated by the RNAs. The p value < 0.05 and |log2(fold change)|>1 were defined as statistically significant. The calculation of the p value for GO analysis was performed using the following equation:
Results
Expression profiles of mRNAs and miRNAs in GBM
Three matched tumor and normal sample pairs from newly diagnosed GBM patients, whose diagnoses were based on surgical pathology reports and clinical records, were collected. The surrounding tissues contained no infiltrating glioma cells, which was confirmed by HE staining (Fig. 1a). Total RNA and whole transcriptome sequencing data (mRNA, miRNA, lncRNA, circRNA) were acquired from qualified biospecimens (see "Methods"). The Illumina Hiseq2500 platform was used to obtain mRNA, lncRNA, cicrRNA-seq and miRNA data. (Ischemic images shown in Suppl Fig. 1).

Analysis of differentially expressed mRNAs. a Representative MRI and HE staining of a GBM patient (63-year-old male) included in our study. Tu tumor tissue, Cr matched peritumoral normal brain tissue. b Differential mRNA expression profile. c Volcano plot depicting mRNAs. d GO analysis results for differentially expressed mRNAs. e and f KEGG pathway analysis for differentially expressed mRNAs
As a result of this analysis, we identified 46,376 total genes and 1999 genes that were differentially expressed between tumor and normal tissues (log2foldchange > 1, p < 0.05, Fig. 1b), the Volcano plot for mRNAs is showed as Fig. 1c. To analyze the function of these differentially expressed genes in GBM, we next conducted GO (Fig. 1d) and KEGG pathway analysis on these genes (Fig. 1e, f). Genes that were down-regulated were largely involved in glutamatergic synapse pathways, calcium signaling pathways, MAPK signaling pathways and metabolic pathways, all of which are important for neurological excitability and tumor genesis. Gene that were up-regulated were involved in known cancer pathways and metabolic pathways (Fig. 1e).
Expression profiles of lncRNAs, GO and KEGG pathway analysis for differentially expressed lncRNAs
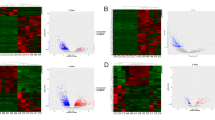
To study the potential biological functions of lncRNAs and circRNAs in GBM, we used RNA sequencing in human GBM and matched normal brain tissues and identified lncRNA and circRNA expression profiles. Total RNA was extracted from the matched tumor and normal tissues as described in the "Methods" section. Using authoritative data sources (see "Methods"), we acquired the lncRNA expression profiles of the GBM and normal brain tissues. A total of 33,318 lncRNAs were annotated in this study, and among these lncRNAs, 501 lncRNAs were identified to be differentially expressed between GBM and normal tissues (p < 0.05, Fig. 2a, h). the Volcano plot for lncRNAs is showed as Fig. 2b. Of these lncRNAs, 247 were identified to be up-regulated in GBM compared to the normal brain tissue, while 254 lncRNAs were identified to be down-regulated (Fig. 2c, d, h). The functions and pathways of these non-coding RNAs were further identified using GO (http://www.geneontology.org) and KEGG (http://www.genome.jp/kegg) databases (p < 0.05, Fig. 2e, f). Differentially expressed lncRNAs were classified as biological processes (BP), cellular components (CC) and molecular functions (MF) using GO analysis. The most enriched and meaningful BP terms were related to apoptotic processes, metabolic processes, transcription, DNA-templated glutathione metabolic processes (GO:0006749), astrocyte development (GO:0014002) and DNA-templated transcription initiation (GO:0006352). The most enriched GG term was cytoplasm, and the most enriched term under the MF classification was protein binding. We next conducted KEGG analysis on the differentially expressed lncRNAs (Fig. 2g), including a sub-analysis for up-regulated and down-regulated lncRNAs. The top 20 up-regulated lncRNAs, down-regulated lncRNAs and KEGG analysis-identified pathways are listed (Tables S1–S3). The Venn diagram for up and down regulated lncRNAs is displayed on Fig. 2i, j and hierarchical clustering (Fig. 2k) showing up and down regulated lncRNAs among the three groups from I and J.

Analysis of differentially expressed lncRNAs. a Circos plots showing all of the lncRNAs from GBM and matched normal brain tissue. b Volcano plot depicting lncRNAs. c and d Expression levels of all biuniquely annotated and reliably quantifiable lncRNAs in three matched normal and GBM samples; normalized log2 values were averaged among the samples. e GO enrichment analysis of differentially expressed lncRNAs. f and g Significant results from KEGG pathway analysis of differentially expressed lncRNAs. h Clustered heat map of differential lncRNA expression. i and j Venn diagram depicting up- and down-regulated lncRNAs. k Hierarchical clustering of up- and down-regulated lncRNAs among the three groups from i and j
Expression profiles miRNAs and circRNAs in GBM, GO and KEGG pathway analysis for differentially expressed circRNAs
From the microarray, a total of 1449 miRNAs were identified, and we found 143 differentially expressed miRNAs with p < 0.05 (Fig. 3a).

Analysis for differentially expressed miRNAs and circRNAs. a Clustered heat map of miRNAs differentially expressed between GBM and matched normal brain tissue as determined by microarray (p < 0.05). b Circos plots for all circRNAs. c Volcano plot depicting all circRNAs. d and e Expression levels of all biuniquely annotated and reliably quantifiable circRNAs in three matched normal and GBM samples; normalized log2 values were averaged among the samples. f GO enrichment analysis for the predicted target genes of the differentially expressed circRNAs. g and h Significant results of KEGG pathway analysis for differentially expressed circRNAs. i Clustered heat map of differential circRNA expression. j and k Venn diagram depicting up- and down-regulated circRNAs. l Hierarchical clustering of up- and down-regulated circRNAs among the three groups from j and k
In total, 19,665 circRNAs were detected in normal tissues, and 6779 circRNAs were detected in tumor tissues. The differential expression profile is presented (Fig. 3b, i), and expression levels of all biuniquely annotatable and reliably quantifiable circRNAs in three matched normal and GBM samples (Fig. 3c, d). Further more, differentially expressed circRNAs were presented using Volcano Plot filtering (Fig. 3e). GO analysis of genes downstream from differentially expressed circRNAs was also conducted (p < 0.05. Fig. 3f). The three most common BP terms were transcription, DNA-templated signal transduction and regulation of transcription. The most enriched GG term was cytoplasm, and the most enriched MF term was protein binding. KEGG pathway analysis revealed that the differentially expressed (both up-regulated and down-regulated, Fig. 3g, h) circRNAs in GBMs are related to many pathways important in cancer pathogenesis, such as the MAPK pathway, the ErbB pathway and the calcium signaling pathway, etc. The top 20 pathways identified are also listed (Table S4). To explore the possible biological functions of circRNAs, based on ceRNA hypothesis, we used bioinformatics methods to predict circRNAs–miRNA–mRNA linkages, and then we conducted GO and KEGG analyses on the target mRNAs. Our functional analysis showed that in GBM, circRNAs are not only involved in important cancer-related pathways such as the Notch signaling pathway and the MAPK signaling pathway, but they also participate in many biological processes, such as the glutamatergic synapse and calcium signaling (Fig. 3f, g, h).Among these differentially expressed circRNAs, 2002 circRNAs were down-regulated and 36 circRNAs were up-regulated (log2-fold change > 1, p < 0.05, Fig. 3j, k, l).
Construction of a lncRNAs/circRNAs–miRNA–mRNA GBM co-expression network
To systematically explore the influence of dynamic changes in ceRNA regulation on gene expression in GBM, we constructed a GBM ceRNA network, integrating matched expression profiles of lncRNAs, circRNAs, miRNAs and mRNAs. We used Targetscan 7.0 (http://www.targetscan.org/) and miRanda (http://www.microrna.org/microrna/home.do) to identify circRNA–miRNA interactions. We used the STRING Protein database (http://string-db.org/) (see "Methods") to analyze lncRNA–miRNA interactions.
Target genes were selected according to the results of GO or KEGG pathway analysis or according to previous reports of glioma-associated genes, and then the global miRNA-mediated ceRNA network was constructed (Fig. 4, Table 1). Of the differentially expressed lncRNAs, 296 were identified as interacting with 1798 mRNAs. The network contained 302 deregulated genes, 53 miRNAs, 779 circRNAs and 33 lncRNAs. We next filtered the key genes that were enriched in core GBM pathways (p53, PI3K-AKT-mTOR and RB pathways) as sub-group analyses (Fig. 5a). These sub-groups contained 28 deregulated genes, 34 miRNAs, 644 circRNAs, 311 lncRNAs and 1670 edges. Then, we filtered key miRNAs that had opposite expression levels when compared to mRNAs. The criteria for mRNA filtering included an mRNA expression log2fold change > 1, a p value < 0.05 and the presence of reversed expression levels with miRNAs. This network map contained nine deregulated genes, five miRNAs, 128 circRNAs and 78 lncRNAs (Fig. 5b). From this co-expression network, we can see that both lncRNAs and circRNAs are involved in the process of gliomagenesis, and many miRNAs or genes are regulated by both lncRNAs and circRNAs.

Representation of a global ceRNA–ceRNA network. The yellow nodes represent mRNAs, the red nodes represent miRNAs, the green nodes represent lncRNAs, and the blue nodes represent circRNAs. There were 53 miRNAs, 339 lncRNAs, 779 circRNAs, 282 coding genes, 302 mRNAs and 2506 edges in the network. The right panels are the profiles of the mRNAs and ncRNAs included in the network

a Representation of a ceRNA network of core pathways in GBM. This network contains differentially expressed genes belonging to the PI3K/AKT pathway, p53 pathway or Rb pathway. The yellow nodes represent mRNAs, the red nodes represent miRNAs, the green nodes represent lncRNAs, and the blue nodes represent circRNAs. b The filtered ceRNA network extracted from the global ceRNA network
Discussion
MicroRNAs (miRNAs) are a major class of small non-coding RNAs (ncRNAs) that are 20–22 nucleotides long [26]. In recent decades, it has become recognized that miRNAs could negatively regulate gene expression through guiding the RNA-induced silencing complex (RISC) to target mRNAs, leading to RNA degradation or translational repression at the post-transcriptional level [27]. Salmena et al. first proposed the “ceRNA hypothesis” describing how messenger RNAs, transcribed pseudogenes and long noncoding RNAs ‘‘talk’’ to each other using microRNA response elements (MREs) as letters of a new language. Based on this hypothesis, miRNAs are more likely to be an RNA bridge between non-coding RNAs and mRNAs [28]. The long non-coding RNAs (lncRNAs) are RNA transcripts that can vary in length from 200 nucleotides to 100 kilobases [28], and expression profiles of lncRNAs reveal their tissue-specific characteristics and alterations across various stages of cell differentiation. In the central nervous system, lncRNAs Ube3a-as, DGCR5 and NRON were shown to be critical for the lineage differentiation of oligodendrocytes during neural stem cell (NSC) differentiation [29,30,31]. However, although thousands of lncRNAs have been annotated in recent years, only a fraction of them have been examined for participation in any biological function.
Circular RNAs were first identified approximately 20 years ago. The testis-determining gene Sry was found to be circular in mouse testes [32]. In the following two decades circRNAs were thought to be functionless byproducts, until recent studies of circRNA expression profiles showed their tissue specificity, which implied potential regulatory functions for circRNAs [33]. circRNAs have been shown to harbor multiple binding sites for microRNAs. One of the best-studied and most dramatic examples of this is Cdr1as/ciRS-7, which can bind miR-7 as a miRNA sponge, affecting miR-7 target gene activity [34]. In our study, we predicted the co-expression of altered circRNAs and their downstream target genes, and our results showed that these genes are highly enriched in many cancer-related pathways and neurobiological functions, revealing the potential biofunctional roles for circRNAs as drivers of brain cancers or neurological diseases.
Recently, competing endogenous RNA networks have been reported as a novel mechanism to explain a post-transcriptional layer of gene translation regulation. Non-coding RNAs have been shown to play a critical role in tumorigenesis though “miRNA-bridges” [35, 36]. Based on the ceRNA hypothesis, we collected the RNA sequencing profiles of mRNAs, lncRNAs, miRNAs and circRNAs from GBM and matched normal brain tissues and constructed a GBM ceRNA network with experimentally validated miRNA target interactions. This ceRNA network offers considerable clues for understanding the key roles of ceRNA-mediated gene regulatory networks in GBM genesis and regulation. Moreover, it accounts for the possible functions of a substantial proportion of the thousands of uncharacterized lncRNAs and circRNAs.
From the ceRNA network, we identified a substantial amount of cross-talk within the non-coding RNAs, and they subsequently formed a complex network. For example, mRNAs such as GABRD, GRIK3, GRM5, GRIN2C and GRIA2 were co-expressed with multiple lncRNAs or circRNAs, and this co-expression was mediated by miRNAs. This result is consistent with GO or KEGG pathway analyses suggesting that glutamate metabolism is involved in glioma biological functions. It is remarkable that many lncRNAs and circRNAs in the ceRNA network are not annotated. These non-coding RNAs are worth future study, in part to reveal the underlying mechanisms of gene regulation. As shown in Fig. 5b, miRNAs could be placed in the center of the co-expression network because of their unique regulation mechanisms. Previous studies have reported that miR-124 and miR-214 can suppresses the migration and invasion of glioma cells and that miR-132 can cause apoptosis in glioma cells. From the ceRNA network, we first predicted that miR-638 may affect glioma cells via VEGFA or FDZ1 signaling.
The roles of non-coding RNAs in GBM, the most common and malignant primary brain tumor, are generally understudied. Pioneering studies completed the expression profiles of lncRNAs and miRNAs of gliomas and matched normal brain tissue [37, 38]. To the best of our knowledge, this is the first report examining the changing expression of lncRNAs, mRNAs, circRNAs and miRNAs in GBM. GO analysis was performed to further annotate the biological functions of differentially expressed lncRNAs and their target genes. A very interesting phenomena should be noted, as GO analysis showed that lncRNAs were not only involved in cancer genesis, but they were also implicated in a number of GO terms related to glutamate biosynthesis. This result further emphasizes the importance of glutamate in GBM genesis, development and infiltration. KEGG pathway analysis confirmed the results of GO analysis, as d-glutamine and d-glutamate metabolic pathways (ko00471) were among the top differentially regulated pathways. Previous studies have demonstrated that glutamate helps glioma cells kill surrounding neurons and is responsible for glioma-associated seizures. Our results suggest that lncRNAs may be potential therapeutic targets for controlling tumors and seizures.
Conclusion
In our study, we acquired the expression profiles of lncRNAs, mRNAs, circRNAs and miRNAs detected by microarray in GBM and matched normal brain tissues. GO and KEGG pathway analyses allowed us to annotate the potential functions of differentially expressed lncRNAs and circRNAs. Co-expression networks were constructed for both lncRNA–mRNA and circRNA–miRNA–mRNA interactions, which contributes to our understanding of tumorigenesis, cancer progression and even therapeutic targeting. We hope to inspire researchers to study the role of non-coding RNAs in GBM.
Declaration
Ethics approval and consent to participate: the study including all experimental protocols was approved by the Ethics Committee of the west china hospital of Sichuan university and all patients gave pre-operative written informed consent.
References
Venkatesh T, Suresh PS, Tsutsumi R (2015) Non-coding RNAs: functions and applications in endocrine-related cancer. Mol Cell Endocrinol 416(1):1–21
Sumazin P et al (2011) An extensive microRNA-mediated network of RNA-RNA interactions regulates established oncogenic pathways in glioblastoma. Can Res 147(2):370–381
Chen Y, Li C, Tan C, Liu X (2016) Circular RNAs: a new frontier in the study of human diseases. J Med Genet 53(6):359–365
Li JH, Liu S, Zhou H, Qu LH, Yang JH (2013) Starbase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale clip-seq data. Nucleic Acids Res 42(D1): 92–97
Ju YP, Lee JE, Park et al (2014) Roles of long non-coding RNAs on tumorigenesis and glioma development. Brain Tumor Res Treat (1):1–6
Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP (2011) CeRNA hypothesis: the rosetta stone of a hidden RNA language? Cell 146(3):353–358
Riquelme I, Ili C, Roa JC, Brebi P (2016) Long non-coding RNAs in gastric cancer: mechanisms and potential applications. Oncotarget. https://doi.org/10.18632/oncotarget.9396
Tay Y, Rinn J, Pandolfi PP (2014) The multilayered complexity of ceRNA crosstalk and competition. Nature 505(7483):344–352
Thomson DW, Dinger ME (2016) Endogenous microRNA sponges: evidence and controversy. Nat Rev Genet 17(5):272 –283
Li Y, Zheng Q, Bao C, Li S, Guo W, Zhao J et al (2015) Circular RNA is enriched and stable in exosomes: a promising biomarker for cancer diagnosis. Cell Res 25(8):981
Shao T, Wu A, Chen J, Chen H, Lu J, Bai J et al (2015) Identification of module biomarkers from the dysregulated ceRNA-ceRNA interaction network in lung adenocarcinoma. Mol Biosyst 11(11):3048–3058
Dou C, Cao Z, Yang B, Ding N, Hou T, Luo F et al (2016) Changing expression profiles of lncRNAs, mRNAs, circRNAs and miRNAs during osteoclastogenesis. Sci Rep 6:21499
Wang PF, Liu N, Song HW, Yao K, Jiang T, Li SW et al (2016) Idh-1r132h mutation status in diffuse glioma patients: implications for classification. Oncotarget 7(21):31393–31400
Zou P, Xu H, Chen P, Yan Q, Zhao L, Zhao P et al (2013) Idh1/idh2 mutations define the prognosis and molecular profiles of patients with gliomas: a meta-analysis. PLoS ONE 8(7):e68782–e68782
Li Y, Wang Z, Wang Y, Zhao Z, Zhang J, Lu J et al (2016) Identification and characterization of lncRNA mediated transcriptional dysregulation dictates lncRNA roles in glioblastoma. Oncotarget 19(29):45027–45041
Han L, Zhang K, Shi Z, Zhang J et al (2012) LncRNA profile of glioblastoma reveals the potential role of lncRNAs, in contributing to glioblastoma pathogenesis. Int J Oncol 40(6):2004–2012
Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A et al (2013) String v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res 41(Database issue):808–815
Kanehisa M, Araki M, Goto S et al (2008) Kegg for linking genomes to life and the environment. Nucleic Acids Res 36(1):d480–d484
Wagle N, Berger MF, Davis MJ, Blumenstiel B, Defelice M, Pochanard P et al (2012) High-throughput detection of actionable genomic alterations in clinical tumor samples by targeted, massively parallel sequencing. Cancer Discov 2(1):82–93
Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10(3):1–10
Langmead B, Salzberg SL (2012) Fast gapped-read alignment with bowtie 2. Nat Methods 9(4):357–359
Mao BX, Cai T, Olyarchuk J, Wei (2005) L: Automated genome annotation and pathway identification using the KEGG orthology (KO) as a controlled vocabulary. Bioinformatics 21(19):3787–3793
Mortazavi A, Williams BA, Mccue K, Schaeffer L, Wold B (2008) Mapping and quantifying mammalian transcriptomes by RNA-seq. Nat Methods 5(7):621–628
Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L (2010) Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28(5):511–515
Trapnell C, Pachter L, Salzberg SI (2009) Discovering splice junctions with RNA-seq. Bioinformatics 25(9):1105–1111
Jin M, Zhang T, Liu C, Badeaux M et al (2014) MiRNA-128 suppresses prostate cancer by inhibiting BMI-1 to inhibit tumor-initiating cells. Cancer Res 74(15):4183–4195
Cheng CJ, Bahal R, Babar IA et al (2015) MicroRNA silencing for cancer therapy targeted to the tumour microenvironment. Nature 518(7537):107–110
Tay Y, Kats L, Salmena L, Weiss D et al (2011) Coding-independent regulation of the tumor suppressor pten by competing endogenous mRNAs. Cell 147(2):344–357
Yan Y, Zhang L, Jiang Y, Xu T, Mei Q, Wang H et al (2015) LncRNA and mRNA interaction study based on transcriptome profiles reveals potential core genes in the pathogenesis of human glioblastoma multiforme. J Cancer Res Clin Oncol 141(5):827–838
Johnstone KA, Dubose AJ, Futtner CR, Elmore MD, Brannan CI, Resnick JL (2006) A human imprinting centre demonstrates conserved acquisition but diverged maintenance of imprinting in a mouse model for angelman syndrome imprinting defects. Hum Mol Genet 15(3):393–404
Sutherland HF, Wadey R, Mckie JM, Taylor C, Atif U, Johnstone KA et al (1996) Identification of a novel transcript disrupted by a balanced translocation associated with digeorge syndrome. Am J Hum Genet 59(1):23–31
Willingham AT, Schultz PG (2005) A strategy for probing the function of noncoding RNAs finds a repressor of NFAT. Science 309(5740):1570–1573
Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK et al (2013) Natural RNA circles function as efficient microRNA sponges. Nature 495(7441):384–388
Zhao ZJ, Shen J (2015) Circular RNA participates in the carcinogenesis and the malignant behavior of cancer. RNA Biol 4(3):501–589
Xu H, Guo S, Li W, Yu P (2015) The circular RNA Cdr1as, via miR-7 and its targets, regulates insulin transcription and secretion in islet cells. Sci Rep 5(1):385–393
Le TD, Zhang J, Liu L, Li J (2016) Computational methods for identifying miRNA sponge interactions. Brief Bioinform 18(4):577–590
Cao Y, Wang P, Ning S, Xiao W, Xiao B, Li X (2016) Identification of prognostic biomarkers in glioblastoma using a long non-coding RNA-mediated, competitive endogenous RNA network. Oncotarget 5(27):41737–41747
Kiang MY, Zhang XQ, Leung KK (2015) Long non-coding RNAs: the key players in glioma pathogenesis. Cancers 7(3):1406–1424
Funding
This study was funded by Sichuan province science and technology support Plan Nos. 2012S20152 and 0040205301911.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
We declare that we have no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Yuan, Y., Jiaoming, L., Xiang, W. et al. Analyzing the interactions of mRNAs, miRNAs, lncRNAs and circRNAs to predict competing endogenous RNA networks in glioblastoma. J Neurooncol 137, 493–502 (2018). https://doi.org/10.1007/s11060-018-2757-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-018-2757-0