
Abstract
Objectives
To study the expression profiles of lncRNA and mRNA in glioblastoma multiforme (GBM) and to find potential core genes in the pathogenesis of this high malignant disease.
Methods
Agilent Microarray (Arrystar v2.0) was used to detect the expressions of 33,045 lncRNAs and 30,215 coding transcripts in 5 GBM and 5 normal brain samples. Differentially expressed lncRNAs and mRNAs were identified through Volcano Plot filtering. The expressions of six lncRNAs were further detected by qPCR to validate the results of microarray. The function of differential mRNA was determined by pathway and GO analysis, and the function of lncRNAs was studied by subgroup analysis and by their physical or functional relationships with corresponding mRNAs.
Results
A total of 815 lncRNAs and 738 mRNAs are found to be differentially expressed between the GBM and normal brain groups. With the expression of these differentially expressed genes, the two group samples could be clearly differentiated. The result of qPCR has showed a good consistency with the microarray, thus proving the accuracy of the microarray data. GO and Pathway analyses have proved that the functions of differentially expressed mRNAs in GBM related closely with many processes that important in the cancer pathogenesis. Core lncRNAs and mRNAs that may play important roles in the pathogenesis of GBM are revealed and listed according to various methods.
Conclusion
The GBM shows an aberrant expression profile of lncRNA and mRNA. Potential core genes are revealed by the lncRNA and mRNA interaction study based on transcriptome profiles in GBM.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glioblastoma multiforme (GBM) is the most common and biologically aggressive subtype of glioma. Even with modern standards of treatment, which includes surgery, followed by radiotherapy and Temozolomide-based chemotherapy, the prognosis of GBM is rather dismal and the average survival time is only 14.6 months (Van Meir et al. 2010). To better understand and to find more effective treatments for the disease, more studies have focused on the head of the cancer pathogenesis, that is, the genome mutations and transcriptome instability.
Once thought to be the dark matter of the human genome, noncoding RNA has proven to be an important component of the transcriptome. Long noncoding RNA (lncRNA) is defined as transcript noncoding RNA with more than 200 neucleotides. The number of it was counted by tens or even hundreds of thousands. It is used to be considered as “junk” transcripts of the genome, but now more and more discoveries have proven that it plays an important role in the gene expression and function regulations, and actively participates in many pathological processes in human malignancies. For example, the well-studied HOX antisense intergenic RNA (HOTAIR) is high expressed in breast cancer and participates in the chromatin remodeling process (Gupta et al. 2010). Another example is the Maternally expressed gene 3 (MEG3), which encodes a lncRNA and is expressed in many normal tissues, but lost in an expanding list of primary human tumors and tumor cell lines (Zhou et al. 2012; Balik et al. 2013; Ying et al. 2013). These examples suggest that lncRNA may play an important role in the genesis and development of cancers.
Efforts have been made to study the relationship between the lncRNA expression and the GBM pathogenesis. Gibb et al. (2011) compiled 272 human serial analyses of gene expression (SAGE) libraries to study the lncRNA transcription patterns across a broad spectrum of human tissues and cancers. They found an extensive, tissue-specific lncRNA expression in normal brain, and a highly aberrant lncRNA expression in GBM. By performing a comparing of the lncRNA expression profile in GBM and normal brain tissues, Han et al. (2012) have found a significant change in Peroxisome proliferator-activated receptor (PPAR) signaling pathway in the GBM and found that ASLNC22381 and ASLNC2081 might play important roles in the recurrence and malignant progression of GBM. Whereas the comprehensive studies for lncRNA in GBM was still very lacking, and our understanding of the roles of lncRNA in this high malignant disease was very limited.
To systematically study the role of lncRNAs in glioblastoma, we built gene expression profiles of GBM and normal brains using lncRNA and mRNA gene expression microarrays. The differentially expressed genes in GBM were then filtered out. The functions of mRNAs were identified by pathway and GO analysis, and the functions of lncRNAs were studied by their physical or co-expressing relationships with the corresponding mRNAs. Genes that may play potential core roles in the pathogenesis of glioblastoma multiforme were screened out based on multiple research directions.
Materials and methods
Acquisition of clinical specimens and ethical standards
Glioblastoma specimens were obtained from archived tissue samples derived from patients with GBM who underwent surgical treatment at Changzheng Hospital, China, from January 2012 to December 2012. Glioma was diagnosed according to the 2007 WHO Classification of Tumors of the Central Nervous System. Normal brain tissues were obtained from severe head trauma patients for whom partial resection of normal brain was required for decompression during surgery. Written informed consent of the patients was provided by their legal surrogates to permit surgical procedures and use of resected tissues. This study was undertaken with the understanding and written consent of each subject, and that the study conforms with World Medical Association Declaration of Helsinki published on the website of the Journal of American Medical Association in 2013. The study was approved by the Specialty Committee on Ethics of Biomedicine Research, Second Military Medical University of China.
Gene microarray
Arraystar Human lncRNA Microarray version 2.0 was designed for the global profiling of human lncRNAs and protein-coding transcripts. 33,045 lncRNAs and 30,215 coding transcripts were detected by the microarray. The lncRNAs were collected from authoritative databases including RefSeq, UCSC Knowngenes, Ensembl, and related literatures. Each transcript was represented by a specific exon or splice junction probe which can identify individual transcript accurately. Positive probes for housekeeping genes and negative probes were also printed onto the array for hybridization quality control.
RNA labeling and array hybridization
Sample labeling and array hybridization were performed according to the Agilent One-Color Microarray-Based Gene Expression Analysis protocol (Agilent Technology) with minor modifications. Briefly, mRNA was purified from total RNA after removal of rRNA (mRNA-ONLY™ Eukaryotic mRNA Isolation Kit, Epicentre). Then, each sample was amplified and transcribed into fluorescent cRNA along the entire length of the transcripts without 3′ bias utilizing a random priming method. The labeled cRNAs were purified by RNeasy Mini Kit (Qiagen). The concentration and specific activity of the labeled cRNAs (pmol Cy3/μg cRNA) were measured by NanoDrop ND-1000. One microgram of each labeled cRNA was fragmented by adding 11 μl 10× Blocking Agent and 2.2 μl of 25× Fragmentation Buffer and then heated the mixture at 60 °C for 30 min, finally, 55 μl 2× GE Hybridization buffer was added to dilute the labeled cRNA. One hundred microliters of hybridization solution was dispensed into the gasket slide and assembled to the lncRNA expression microarray slide. The slides were incubated for 17 h at 65 °C in an Agilent Hybridization Oven. The hybridized arrays were washed, fixed, and scanned with using the Agilent DNA Microarray Scanner (part number G2505B).
Data acquisition and processing
Agilent Feature Extraction software (version 10.7.3.1) was used to analyze acquired array images. Quantile normalization and subsequent data processing were performed with the GeneSpring GX v12.0 software package (Agilent Technologies). After quantile normalization of the raw data, lncRNAs and mRNAs that at least two out of ten samples have flags in Present or Marginal (“All Targets Value”) were chosen for further data analysis. Differentially expressed lncRNAs and mRNAs with statistical significance between the two groups were identified through Volcano Plot filtering. Hierarchical Clustering was performed using the Agilent GeneSpring GX software (version 12.0).
qPCR
Total RNA was extracted from 5 frozen GBM and 5 normal brain specimens using TRIzol reagent (Invitrogen Life Technologies), and then reverse-transcribed using Fermentas RT reagent Kit (Perfect Real Time) according to the manufacturer’s instructions. Two microgram of total RNA was converted to cDNA according to the manufacturer’s protocol. LncRNAs expression was measured by qPCR using SYBR Premix Ex Taq on MX3000 instrument. The primers used in this study were shown in Table S1. PCR was performed in a total reaction volume of 8 μl, including 5 μl 2× PCR master mix(Superarray), 0.5 μl of PCR Forward Primer (10 uM), 0.5 μl of PCR Reverse Primer (10 uM), 2 μl of cDNA, and diluted to 8 μl with double-distilled water. The quantitative real-time PCR reaction was set at an initial denaturation step of 10 min at 95 °C; and 95 °C (10 s), 60 °C (60 s), 95 °C (10 s) in a total 40 cycles, with a final step heating slowly from 60 to 99 °C. All samples normalized to GAPDH to calculate relative lncRNA concentrations.
GO and pathway analysis for the differentially expressed mRNA
Differentially expressed mRNAs screened by Volcano Plot filtering were further studied with Gene Ontology (http://www.geneontology.org), and KEGG (http://www.genome.jp/kegg) database to study the functions of the genes and to find the pathways they participated. The p value cut-off was defined as 0.05.
LncRNA classification and subgroup analysis
Enhancer LncRNAs profiling
LncRNAs with enhancer-like function are identified using GENCODE annotation of the human genes (Harrow et al. 2006; Orom et al. 2010). All these lncRNAs with enhancer-like function were analyzed in the microarray dataset, and the lncRNAs with large fold changes (≥2) and small p values (≤0.05) between the GBM and normal brain groups were filtered out.
Rinn lincRNAs profiling
Rinn et al. developed an approach for identifying lncRNAs based on a distinctive chromatin signature that marks actively transcribed genes (Guttman et al. 2009; Khalil et al. 2009). With the method, they have found 3,289 lincRNAs which could bind to the chromatin proteins and show clear evolutionary conservation. All probes for lincRNAs based on John Rinn’s papers were included in our study and further analysis was made to filter out differentially expressed lincRNAs in the scope.
HOX cluster profiling
Rinn et al. (2007) characterized the transcriptional landscape of the four human Hox loci and identified a total of 407 discrete transcribed regions in the four Hox loci. Analysis was made to study the profiling data of all probes targeting these 407 discrete transcribed regions in our microarry dataset. The lncRNAs in the scope with large fold changes (≥2), and the small p values (≤0.05) between the GBM and normal brain groups were filtered out.
LincRNAs nearby coding gene co-expression analysis
The differentially expressed lncRNAs and mRNAs were screened, respectively, then the positions of these genes in the chromatin were signed, and the physical distances between any nearby pairs of lncRNA and mRNA were calculated. When the expressions of the lincRNA and its nearby coding genes were both changed (either up or down) more than twofold, and the distance between them was <200 kb, they were characterized.
Enhancer LncRNAs nearby coding gene co-expression analysis
Similarly, this analysis was based on the co-expression relationship between the differentially expressed enhancer-like LncRNAs and their nearby (distance <300 kb) coding genes. When the expressions of the enhancer-like lincRNA and its nearby coding gene were both changed (either up or down) more than twofold, they were characterized.
LncRNA and mRNA interaction network analysis
Random variance model (RVM) t test to filtering the differentially expressed genes
The RVM t test was applied to filter the differentially expressed genes for the control and experiment group because the RVM t test can raise degrees of freedom effectively in the cases of small samples. After the significant analysis and False discovery rate (FDR) analysis, the differentially expressed genes were selected according to the p value threshold (p < 0.05) (Wright and Simon 2003; Yang et al. 2005; Clarke et al. 2008).
Co-expression network
The co-expression Networks were built according to the normalized signal intensity of specific expression genes. For each pair of genes, the Pearson correlation was calculated, and the significant correlation pairs were chosen to construct the network (Prieto et al. 2008). The gene co-expression network was constructed in GBM and normal brain group, respectively. Within the network analysis, a degree is the simplest and most important measure of the centrality of a gene within a network and determines the relative importance. A degree is defined as the number of directly linked neighbors. Degree in GBM group was recorded as exp_degree, which in normal brain group was recorded as con_degree. A clustering coefficient is a measure of the degree to which nodes in a graph tend to cluster together. It was calculated by the local measure (Watts and Strogatz 1998). To exclude other genes’ impact in each co-expression network, we further performed normalization of the degree, i.e., divided by the maximum value of the gene degree in each network [Normalized degree(i) = Degree(i)/Degree(Max)]. Then, the difference value of a gene’s normalized degree (delta normalized degree, represented as |diffK|) was calculated between the two co-expression networks.
Results
Glioblastoma demonstrates significantly altered lncRNA and mRNA expression patterns comparing with that of the normal brain
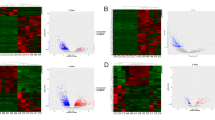
The lncRNA and mRNA expression profiles were detected in 5 GBMs and 5 normal brain tissues (GEO dataset: GSE51146). Volcano Plot filtering has found 526 lncRNAs and 395 mRNAs to be up-regulated in the GBM samples comparing with the normal brain samples (fold change >2, and p < 0.05). On the other hand, 289 lncRNAs and 343 mRNAs were found to be down-regulated in the GBM group comparing with the normal brain group (fold change >2, and p < 0.05) (Fig. 1, table S2 and S3). The Hierarchical Clustering analysis found that with the expression of these genes, the samples could be clearly classified into two groups, i.e., the GBM and Normal brain group (Fig. 2).

Box plot of mRNA (a) and lncRNA (d). They are used to look at and compare the distributions of gene expression values in the samples after normalization. Scatter plot of mRNA (b) and lncRNA (e). The variation of gene expressions could be visualized in these plots. Volcano plot of mRNA (c) and lncRNA (f). They are constructed using fold-change values and p values, and thus the relationship between fold change (magnitude of change) and statistical significance (which takes both magnitude of change and variability into consideration) could be visualized. They also allow subsets of genes to be isolated, based on those values. The vertical lines correspond to 2.0-fold up and down, and the horizontal line represents a p value of 0.05. So, the red point in the plot represents the differentially expressed genes with statistical significance

Hierarchical clustering of mRNA (a) and lncRNA (b) by Z-score method. Based on the expression levels of mRNAs or LncRNAs, the ten samples could be classified into two groups (GBM and normal brain). The dendrogram shows the relationships among the expression levels of samples
Real-time quantitative PCR showed a good consistency of the result with the microarray data
The detection of the expression level of selected six lncRNAs (ENST00000411660, NR_026664, HIT000337391, ENST00000451515,NR_015392, and uc002cds.2) by qPCR showed a good consistency with the microarray results (Fig. 3).

Comparing of the microarray and qPCR results. The qPCR results of selected six lncRNAs (ENST00000411660, NR_026664, HIT000337391, ENST00000451515,NR_015392, and uc002cds.2) showed a good consistency with the microarray data
Many key genes and pathways are involved in the pathogenesis of glioblastoma revealed by GO and pathway analysis
GO analysis reveals the functions of differential expressed (both up-regulated and down-regulated) mRNA in GBMs (Figure S3). It could be found that the functions of these mRNAs are related with many processes that important in the cancer pathogenesis, such as homophilic cell adhesion, cell–cell adhesion, calcium ion binding, p53 binding, 1-phosphatidylinositol binding, transcription regulator activity. Pathway analysis shows that the up-regulated mRNAs are participated in DNA replication, cell signaling (Focal adhesion, Bacterial invasion of epithelial cells, T cell receptor signaling pathway, etc.), and processes in many malignant cancerous disease (human renal cell carcinoma, chronic myeloid leukemia, etc.). On the other hand, pathways that the down-regulated mRNAs involved in refer to RNA degradation, primary immunodeficiency, long-term depression, etc. (Table 1).
Subgroup analysis revealed lists of lncRNA and mRNA that may play important roles in GBM
Enhancer LncRNAs profiling
A total of 48 differentially expressed lncRNAs with enhancer-like function between the GBM and normal brain groups are filtered (fold change >2 and p < 0.05). The top 10 up- and down-regulated enhancer lncRNAs were listed in Table 2.
Rinn lincRNAs profiling
A total of 84 lincRNAs which show a distinctive chromatin signature and a clear evolutionary conservation were filtered out based on John Rinn’s paper (fold change >2 and p < 0.05). The top 10 up- and down-regulated lncRNAs were listed in Table 3.
HOX cluster profiling
A total of 11 differentially expressed lincRNAs with discrete transcribed regions in the four Hox loci were characterized (fold change ≥2.0, p value ≤0.05), listed in Table 4.
LincRNAs nearby coding gene data analysis
Five pairs of lincRNA and its nearby coding gene were found to be changed more than twofold simultaneously (Table 5).
Enhancer LncRNAs nearby coding gene data analysis
Five pairs of enhancer lncRNA and its nearby coding gene were found to be changed more than twofold at the same time (Table 6).
Core lncRNAs in glioblastoma were revealed by analyzing their co-expression relationship with mRNAs
The co-expression network of mRNA and lncRNA was constructed in glioblastoma and normal brain group, respectively, according to the similarity of gene expression patterns in each group. (Figure S2: mRNA-lncRNA co-expression network in glioblastoma; Figure S3: mRNA-lncRNA co-expression network in normal brain tissues) The nodes in the figures stand for the genes, and the lines between nodes stand for the relationship between the genes. The node size is on behalf of the gene’s degree. The greater the degree is, the more interactions between the target gene and other genes. The clustering coefficient is on behalf of the density of an area of the network. Genes with greater clustering coefficient indicates the region of this gene is more important for the regulation of whole network. Genes with a large degree value and clustering coefficient in normal brain and GBM samples (clustering coefficient >0.6, Degree ≥40) were listed in table S4 and table S5, respectively.
The critical change of co-expression status of a gene between the co-expression network of GBM and normal brain suggests that the expression of the gene very possibly undertakes a pivotal change in the process of GBM pathogenesis (Carlson et al. 2006). Gene like this may play an important role in the GBM’s genesis and development, so a way to find these genes is to select the genes that have the most pivotal changes in the co-expression network of the GBM compared with that of the normal brain. Through analyzing the changes of co-expression status of genes between the GBM and Normal brain samples, we have got a list of genes (|diffK| > 0.6) that may participate in the pathogenesis of GBM and deserves for further study (Table 7).
Discussion
With the completion of the human genome project, it has been found that the number of total protein-encoding genes in human is <20,000, nearly equals to that of the lower organisms like Caenorhabditis elegans (Birney et al. 2007). Coding genes accounts for <2 % of the entire human genome sequence and the function of the remaining 98 % was largely unknown. In fact, it has been found that 90 % of these noncoding sequences are transcribed, producing a huge number of noncoding RNA, in which 80 % are lncRNA.
The abundance of lncRNA has effectively doubled the number of potential gene targets in cancer gene expression networks. Large scale, high-throughput studies are crucial to understanding the regulation of lncRNA expression in cancer. Glioma is the most common cancer in central neurological system, and GBM is the most common and aggressive form of that. A systematic study on lncRNA in GBM is not only beneficial to find more valuable targets for prognosis predicting and therapeutic intervention for this disease, but also help to develop new types of anticancer drugs, for example, using large antisense RNA to silent the expression of oncogenes (Morris 2009), or targeting lncRNA protein interactions to interfere the expression of certain molecules (Tsai et al. 2011). In this study, by lncRNA and mRNA gene expression microarray, we have built the complete lncRNA and mRNA expression profiles of 5 GBMs and 5 Normal brains. Then, the differentially expressed lncRNAs and mRNAs were filtered by Volcano Plot method. A total of 815 lncRNAs (526 up-regulated and 289 down-regulated) and 738 mRNAs (395 up-regulated and 343 down-regulated) are found to be differentially expressed between the GBM group and normal brain group. With the expression of these genes, the two group samples could be clearly differentiated by Hierarchical Clustering. We selected six lncRNAs and detected their expressions with qPCR. The result of qPCR has showed a good consistency with the microarray, thus proving the accuracy and reliability of the microarray data. In the next step, the main problem is how to filter out key targets possibly associated with tumor pathological process from these differentially expressed genes. We have used a series of methods to identify the possible key targets of potential research value.
With Gencode annotation, Orom et al. (2010) has identified more than 3,000 lncRNAs to be expressed in various human cell lines and tissues. By depletion of a number of these lncRNAs and by reporter assays, they have proved that these lncRNAs exhibit functions similar to classically defined enhancers. Enhancers are important elements regulating the gene expression, many of which may participate in the pathological mechanisms of cancers. Therefore, we analyzed our microarray data to screen the significantly differentially expressed lncRNAs with enhancer-like function in our GBM and normal brain samples. The targets with the biggest fold changes and the smallest p values were listed, which could be worthy for further study.
By a new approach using chromatin-state maps, Guttman et al. (2009) has identified nearly 1,600 large intervening noncoding RNAs (lincRNAs) which show strong purifying selection in their genomic loci, exonic sequences, and promoter regions, with greater than 95 % showing clear evolutionary conservation. Although the absence of evolutionary conservation cannot prove the absence of function, the lncRNAs with markedly high rate of conservation are most likely to be functional. Rinn et al. had proved that some of these lincRNAs are transcriptionally regulated by key transcription factors and defined a unique collection of functional lincRNAs that are highly conserved and implicated in diverse biological process. We detected the probes listed in Rinn’s article and screened a list of lincRNA differentially expressed in our GBM samples.
By ideas similar to the above, we examined the expressions of lncRNA associated with the Hox loci in this data set. The classic case in the study of Hox-loci-associated lncRNAs is the finding of HOTAIR. It has been found in a recent study that in the progression of breast cancer, systemic dysfunctions were found in the lncRNAs associated with the Hox loci. One of these lncRNAs, named as HOTAIR, is abnormally over-expressed in the primary tumors and metastases and can be used as a direct predictor for the risk of breast cancer metastasis and poor survival of the patients. Up-regulating the HOTAIR expression causes a pan-genome rearrangement of PRC2 binding mode, leads to the opening of a series of genes, and thus increases the invasion and metastasis of cancer cells. Instead, knocking down its expression inhibits the tumor cell invasion, especially for those cell lines with an increased activity of PRC2 (Gupta et al. 2010). In Glioblastoma, we have also found an aberrant expression profile of the Hox-loci-associated lncRNAs compared with that of the normal brain. Apparent up-regulation was found in genes like nc-HOXC11-109, and noticeable down-regulation was found in genes like nc-HOXC5-254. It is possible to find in glioma some key genes with a similar action of HOTAIR in further in-depth study.
Not like mRNA, a difficulty in lncRNA study is that the functions of most of the lncRNAs are not determined, and there is no existing database could be used to find their functional annotations. To solve this problem, we have tried to construct a correlation between mRNA and lncRNA, so the functions of lncRNA could be indirectly determined by the study of its corresponding mRNA. First, by GO annotation and Pathway analysis, we made a systemic analysis for the functions of the differentially expressed mRNAs. The results showed that the functions of these targets related closely with many aspects of tumor progression and involved in many cancer-associated pathways. This shows that the differential mRNAs screened by the gene microarray analysis are closely related with the cancer pathogenesis, which may very possibly contain a lot of key genes in GBM. Then, we have established the contact of lncRNA and mRNA in two ways: One way is to use the physical adjacency of the mRNA and lncRNA, and the other is to use the relationship of co-expression pattern among genes.
Acting to the adjacent genes is at least one mechanism for the lncRNA to play its regulatory role, although distant targets could also be regulated by some lncRNAs in a lot of circumstances. We made a co-expression analysis for the mRNA and its adjacent (<300 kb) lincRNA or enhancer-like lncRNA. When the expressions of both are significantly changed in GBM, it very likely indicates there is some interaction between the mRNA and its adjacent lncRNA. In this way, we have determined some lncRNAs like AC133528.2, CR626360, and AC051649.6 as the targets for further study. It is noteworthy that some corresponding mRNA related closely to cancers, such as ITSN1 (Ma et al. 2010, 2011; Russo and O’Bryan 2012), NEU4 (Yamanami et al. 2007; Shiozaki et al. 2011; Tringali et al. 2012), LSP1 (Gorodnova et al. 2010; Vachon et al. 2012), and CAD (Otsuki et al. 1981; Morin et al. 2012). It suggests that the associated lncRNAs of these genes may play important roles in GBM.
The principle of co-expression network study is that by comparing the similarity or difference of the expression pattern of any two genes among samples, whether there is any correlations between them could be determined (Pujana et al. 2007). If the expression pattern of a gene shows an identical or opposite curve with one another, there is likely to be some interaction between the two genes. By bioinformatics analysis, the co-expression network in cancer and control groups were, respectively, constructed, and by comparing the differences between them, potential core genes could possibly be identified (Carlson et al. 2006). With this method, Yang et al. (2011) has successfully found a key lncRNA called HEIH in Hepatocellular Carcinoma. In the present study, we used the same method to find a list of lncRNAs and mRNAs, in which very possibly contains some key transcripts closely related to the pathogenesis of GBM.
The study on lncRNA has greatly expanded the field of gene research in GBM. We look forward to more targets to be found to give more hopes for the ultimate cure of this common and high malignant disease in the central nervous system.
Abbreviations
- GBM:
-
Glioblastoma multiforme
- LncRNA:
-
Long noncoding RNA
- HOTAIR:
-
HOX antisense intergenic RNA
- MALAT1:
-
Metastasis-associated lung adenocarcinoma transcript 1
- MEG3:
-
Maternally expressed gene 3
- SAGE:
-
Serial analyses of gene expression
- GAPDH:
-
Glyceraldehyde 3-phosphate dehydrogenase
- FDR:
-
False discovery rate
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- RVM t test:
-
Random variance model t test
- lincRNAs:
-
Large intervening noncoding RNAs
- PRC2:
-
The polycomb complex 2
- ITSN1:
-
Intersectin 1
- UCSC:
-
University of California, Santa Cruz
- HEIH:
-
LncRNA high expression in hepatocellular carcinoma
References
Balik V, Srovnal J, Sulla I, Kalita O, Foltanova T, Vaverka M, Hrabalek L, Hajduch M (2013) MEG3: a novel long noncoding potentially tumour-suppressing RNA in meningiomas. J Neurooncol 112:1–8
Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR, Margulies EH, Weng Z, Snyder M, Dermitzakis ET, Thurman RE, Kuehn MS, Taylor CM, Neph S, Koch CM, Asthana S, Malhotra A, Adzhubei I, Greenbaum JA, Andrews RM, Flicek P, Boyle PJ, Cao H, Carter NP, Clelland GK, Davis S, Day N, Dhami P, Dillon SC, Dorschner MO, Fiegler H, Giresi PG, Goldy J, Hawrylycz M, Haydock A, Humbert R, James KD, Johnson BE, Johnson EM, Frum TT, Rosenzweig ER, Karnani N, Lee K, Lefebvre GC, Navas PA, Neri F, Parker SC, Sabo PJ, Sandstrom R, Shafer A, Vetrie D, Weaver M, Wilcox S, Yu M, Collins FS, Dekker J, Lieb JD, Tullius TD, Crawford GE, Sunyaev S, Noble WS, Dunham I, Denoeud F, Reymond A, Kapranov P, Rozowsky J, Zheng D, Castelo R, Frankish A, Harrow J, Ghosh S, Sandelin A, Hofacker IL, Baertsch R, Keefe D, Dike S, Cheng J, Hirsch HA, Sekinger EA, Lagarde J, Abril JF, Shahab A, Flamm C, Fried C, Hackermuller J, Hertel J, Lindemeyer M, Missal K, Tanzer A, Washietl S, Korbel J, Emanuelsson O, Pedersen JS, Holroyd N, Taylor R, Swarbreck D, Matthews N, Dickson MC, Thomas DJ, Weirauch MT, Gilbert J, Drenkow J, Bell I, Zhao X, Srinivasan KG, Sung WK, Ooi HS, Chiu KP, Foissac S, Alioto T, Brent M, Pachter L, Tress ML, Valencia A, Choo SW, Choo CY, Ucla C, Manzano C, Wyss C, Cheung E, Clark TG, Brown JB, Ganesh M, Patel S, Tammana H, Chrast J, Henrichsen CN, Kai C, Kawai J, Nagalakshmi U, Wu J, Lian Z, Lian J, Newburger P, Zhang X, Bickel P, Mattick JS, Carninci P, Hayashizaki Y, Weissman S, Hubbard T, Myers RM, Rogers J, Stadler PF, Lowe TM, Wei CL, Ruan Y, Struhl K, Gerstein M, Antonarakis SE, Fu Y, Green ED, Karaoz U, Siepel A, Taylor J, Liefer LA, Wetterstrand KA, Good PJ, Feingold EA, Guyer MS, Cooper GM, Asimenos G, Dewey CN, Hou M, Nikolaev S, Montoya-Burgos JI, Loytynoja A, Whelan S, Pardi F, Massingham T, Huang H, Zhang NR, Holmes I, Mullikin JC, Ureta-Vidal A, Paten B, Seringhaus M, Church D, Rosenbloom K, Kent WJ, Stone EA, Batzoglou S, Goldman N, Hardison RC, Haussler D, Miller W, Sidow A, Trinklein ND, Zhang ZD, Barrera L, Stuart R, King DC, Ameur A, Enroth S, Bieda MC, Kim J, Bhinge AA, Jiang N, Liu J, Yao F, Vega VB, Lee CW, Ng P, Shahab A, Yang A, Moqtaderi Z, Zhu Z, Xu X, Squazzo S, Oberley MJ, Inman D, Singer MA, Richmond TA, Munn KJ, Rada-Iglesias A, Wallerman O, Komorowski J, Fowler JC, Couttet P, Bruce AW, Dovey OM, Ellis PD, Langford CF, Nix DA, Euskirchen G, Hartman S, Urban AE, Kraus P, Van Calcar S, Heintzman N, Kim TH, Wang K, Qu C, Hon G, Luna R, Glass CK, Rosenfeld MG, Aldred SF, Cooper SJ, Halees A, Lin JM, Shulha HP, Zhang X, Xu M, Haidar JN, Yu Y, Ruan Y, Iyer VR, Green RD, Wadelius C, Farnham PJ, Ren B, Harte RA, Hinrichs AS, Trumbower H, Clawson H, Hillman-Jackson J, Zweig AS, Smith K, Thakkapallayil A, Barber G, Kuhn RM, Karolchik D, Armengol L, Bird CP, de Bakker PI, Kern AD, Lopez-Bigas N, Martin JD, Stranger BE, Woodroffe A, Davydov E, Dimas A, Eyras E, Hallgrimsdottir IB, Huppert J, Zody MC, Abecasis GR, Estivill X, Bouffard GG, Guan X, Hansen NF, Idol JR, Maduro VV, Maskeri B, McDowell JC, Park M, Thomas PJ, Young AC, Blakesley RW, Muzny DM, Sodergren E, Wheeler DA, Worley KC, Jiang H, Weinstock GM, Gibbs RA, Graves T, Fulton R, Mardis ER, Wilson RK, Clamp M, Cuff J, Gnerre S, Jaffe DB, Chang JL, Lindblad-Toh K, Lander ES, Koriabine M, Nefedov M, Osoegawa K, Yoshinaga Y, Zhu B, de Jong PJ (2007) Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447:799–816
Carlson MR, Zhang B, Fang Z, Mischel PS, Horvath S, Nelson SF (2006) Gene connectivity, function, and sequence conservation: predictions from modular yeast co-expression networks. BMC Genomics 7:40
Clarke R, Ressom HW, Wang A, Xuan J, Liu MC, Gehan EA, Wang Y (2008) The properties of high-dimensional data spaces: implications for exploring gene and protein expression data. Nat Rev Cancer 8:37–49
Gibb EA, Vucic EA, Enfield KS, Stewart GL, Lonergan KM, Kennett JY, Becker-Santos DD, MacAulay CE, Lam S, Brown CJ, Lam WL (2011) Human cancer long non-coding RNA transcriptomes. PLoS One 6:e25915
Gorodnova TV, ESh K, Yanus GA, Katanugina AS, Abysheva SN, Togo AV, Imyanitov EN (2010) Distribution of FGFR2, TNRC9, MAP3K1, LSP1, and 8q24 alleles in genetically enriched breast cancer patients versus elderly tumor-free women. Cancer Genet Cytogenet 199: 69–72
Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ, Tsai MC, Hung T, Argani P, Rinn JL, Wang Y, Brzoska P, Kong B, Li R, West RB, van de Vijver MJ, Sukumar S, Chang HY (2010) Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 464:1071–1076
Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, Huarte M, Zuk O, Carey BW, Cassady JP, Cabili MN, Jaenisch R, Mikkelsen TS, Jacks T, Hacohen N, Bernstein BE, Kellis M, Regev A, Rinn JL, Lander ES (2009) Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 458:223–227
Han L, Zhang K, Shi Z, Zhang J, Zhu J, Zhu S, Zhang A, Jia Z, Wang G, Yu S, Pu P, Dong L, Kang C (2012) LncRNA profile of glioblastoma reveals the potential role of lncRNAs in contributing to glioblastoma pathogenesis. Int J Oncol 40:2004–2012
Harrow J, Denoeud F, Frankish A, Reymond A, Chen CK, Chrast J, Lagarde J, Gilbert JG, Storey R, Swarbreck D, Rossier C, Ucla C, Hubbard T, Antonarakis SE, Guigo R (2006) GENCODE: producing a reference annotation for ENCODE. Genome Biol 7(Suppl 1): S4.1–S9
Khalil AM, Guttman M, Huarte M, Garber M, Raj A, Rivea MD, Thomas K, Presser A, Bernstein BE, van Oudenaarden A, Regev A, Lander ES, Rinn JL (2009) Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci USA 106:11667–11672
Ma Y, Wang B, Li W, Ying G, Fu L, Niu R, Gu F (2010) Reduction of intersectin1-s induced apoptosis of human glioblastoma cells. Brain Res 1351:222–228
Ma Y, Wang B, Li W, Liu X, Wang J, Ding T, Zhang J, Ying G, Fu L, Gu F (2011) Intersectin1-s is involved in migration and invasion of human glioma cells. J Neurosci Res 89:1079–1090
Morin A, Fritsch L, Mathieu JR, Gilbert C, Guarmit B, Firlej V, Gallou-Kabani C, Vieillefond A, Delongchamps NB, Cabon F (2012) Identification of CAD as an androgen receptor interactant and an early marker of prostate tumor recurrence. FASEB J 26:460–467
Morris KV (2009) RNA-directed transcriptional gene silencing and activation in human cells. Oligonucleotides 19:299–306
Orom UA, Derrien T, Guigo R, Shiekhattar R (2010) Long noncoding RNAs as enhancers of gene expression. Cold Spring Harb Symp Quant Biol 75:325–331
Otsuki T, Mori M, Tatibana M (1981) Phosphorylation and dephosphorylation of carbamoyl-phosphate synthetase II complex of rat ascites hepatoma cells. J Biochem 89:1367–1374
Prieto C, Risueno A, Fontanillo C, De las Rivas J (2008) Human gene coexpression landscape: confident network derived from tissue transcriptomic profiles. PLoS One 3: e3911
Pujana MA, Han JD, Starita LM, Stevens KN, Tewari M, Ahn JS, Rennert G, Moreno V, Kirchhoff T, Gold B, Assmann V, Elshamy WM, Rual JF, Levine D, Rozek LS, Gelman RS, Gunsalus KC, Greenberg RA, Sobhian B, Bertin N, Venkatesan K, Ayivi-Guedehoussou N, Sole X, Hernandez P, Lazaro C, Nathanson KL, Weber BL, Cusick ME, Hill DE, Offit K, Livingston DM, Gruber SB, Parvin JD, Vidal M (2007) Network modeling links breast cancer susceptibility and centrosome dysfunction. Nat Genet 39:1338–1349
Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, Goodnough LH, Helms JA, Farnham PJ, Segal E, Chang HY (2007) Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 129:1311–1323
Russo A, O’Bryan JP (2012) Intersectin 1 is required for neuroblastoma tumorigenesis. Oncogene 31:4828–4834
Shiozaki K, Yamaguchi K, Takahashi K, Moriya S, Miyagi T (2011) Regulation of sialyl Lewis antigen expression in colon cancer cells by sialidase NEU4. J Biol Chem 286:21052–21061
Tringali C, Cirillo F, Lamorte G, Papini N, Anastasia L, Lupo B, Silvestri I, Tettamanti G, Venerando B (2012) NEU4L sialidase overexpression promotes beta-catenin signaling in neuroblastoma cells, enhancing stem-like malignant cell growth. Int J Cancer 131:1768–1778
Tsai MC, Spitale RC, Chang HY (2011) Long intergenic noncoding RNAs: new links in cancer progression. Cancer Res 71:3–7
Vachon CM, Scott CG, Fasching PA, Hall P, Tamimi RM, Li J, Stone J, Apicella C, Odefrey F, Gierach GL, Jud SM, Heusinger K, Beckmann MW, Pollan M, Fernandez-Navarro P, Gonzalez-Neira A, Benitez J, van Gils CH, Lokate M, Onland-Moret NC, Peeters PH, Brown J, Leyland J, Varghese JS, Easton DF, Thompson DJ, Luben RN, Warren RM, Wareham NJ, Loos RJ, Khaw KT, Ursin G, Lee E, Gayther SA, Ramus SJ, Eeles RA, Leach MO, Kwan-Lim G, Couch FJ, Giles GG, Baglietto L, Krishnan K, Southey MC, Le ML, Kolonel LN, Woolcott C, Maskarinec G, Haiman CA, Walker K, Johnson N, McCormack VA, Biong M, Alnaes GI, Gram IT, Kristensen VN, Borresen-Dale AL, Lindstrom S, Hankinson SE, Hunter DJ, Andrulis IL, Knight JA, Boyd NF, Figuero JD, Lissowska J, Wesolowska E, Peplonska B, Bukowska A, Reszka E, Liu J, Eriksson L, Czene K, Audley T, Wu AH, Pankratz VS, Hopper JL, dos-Santos-Silva I (2012) Common breast cancer susceptibility variants in LSP1 and RAD51L1 are associated with mammographic density measures that predict breast cancer risk. Cancer Epidemiol Biomark Prev 21:1156–1166
Van Meir EG, Hadjipanayis CG, Norden AD, Shu HK, Wen PY, Olson JJ (2010) Exciting new advances in neuro-oncology: the avenue to a cure for malignant glioma.CA. Cancer J Clin 60:166–193
Watts DJ, Strogatz SH (1998) Collective dynamics of ‘small-world’ networks. Nature 393:440–442
Wright GW, Simon RM (2003) A random variance model for detection of differential gene expression in small microarray experiments. Bioinformatics 19:2448–2455
Yamanami H, Shiozaki K, Wada T, Yamaguchi K, Uemura T, Kakugawa Y, Hujiya T, Miyagi T (2007) Down-regulation of sialidase NEU4 may contribute to invasive properties of human colon cancers. Cancer Sci 98:299–307
Yang H, Crawford N, Lukes L, Finney R, Lancaster M, Hunter KW (2005) Metastasis predictive signature profiles pre-exist in normal tissues. Clin Exp Metastasis 22:593–603
Yang F, Zhang L, Huo XS, Yuan JH, Xu D, Yuan SX, Zhu N, Zhou WP, Yang GS, Wang YZ, Shang JL, Gao CF, Zhang FR, Wang F, Sun SH (2011) Long noncoding RNA high expression in hepatocellular carcinoma facilitates tumor growth through enhancer of zeste homolog 2 in humans. Hepatology 54:1679–1689
Ying L, Huang Y, Chen H, Wang Y, Xia L, Chen Y, Liu Y, Qiu F (2013) Downregulated MEG3 activates autophagy and increases cell proliferation in bladder cancer. Mol BioSyst 9:407–411
Zhou Y, Zhang X, Klibanski A (2012) MEG3 noncoding RNA: a tumor suppressor. J Mol Endocrinol 48:R45–R53
Acknowledgments
This study was supported by the National “863” High Technique Project (2007AA02Z483), National Natural Science Foundation of China (30930094, 81101908, 81272781, 30973076), Program for academic leaders in health sciences (No. XBR2011030) and “Shu Guang” project (No. 11SG37) in Shanghai. We want to thank Yanfen Ge and her colleagues at KangCheng Bio-tech Inc., and Qi Li and his colleagues at Gminix Company, for the assistance in bioinformatic analysis and many other help during the course of this study.
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Yong Yan and Lei Zhang have contributed equally to the article.
Electronic supplementary material
Below is the link to the electronic supplementary material.
432_2014_1861_MOESM1_ESM.tif
Supplementary material 1 (TIFF 1201 kb) Figure S1: GO analysis of differential expressed (A, C, and E, up-regulated; B, D, and F, down-regulated) mRNA in GBMs. A and B, Biological process; C and D, Cellular component; E and F, Molecular function
432_2014_1861_MOESM2_ESM.tif
Supplementary material 2 (TIFF 9904 kb) Figure S2: Gene co-expression network of the normal brain. The solid round spots stand for the mRNAs, the Hexagon with a circle spots stand for the lncRNAs. The lines between spots stand for the relationship between the genes. The spot size is on behalf of the capability of a gene to act with its adjacent genes
432_2014_1861_MOESM3_ESM.tif
Supplementary material 3 (TIFF 10098 kb) Figure S3: Gene co-expression network of the GBM. The solid round spots stand for the mRNAs, the Hexagon with a circle spots stand for the lncRNAs. The lines between spots stand for the relationship between the genes. The spot size is on behalf of the capability of a gene to act with its adjacent genes
Rights and permissions
About this article

Cite this article
Yan, Y., Zhang, L., Jiang, Y. et al. LncRNA and mRNA interaction study based on transcriptome profiles reveals potential core genes in the pathogenesis of human glioblastoma multiforme. J Cancer Res Clin Oncol 141, 827–838 (2015). https://doi.org/10.1007/s00432-014-1861-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00432-014-1861-6