
Abstract
Purpose
Glioblastoma (GBM) is one of the most aggressive and incurable primary brain tumors. Identification of novel therapeutic targets is an urgent priority. Programmed cell death 10 (PDCD10), a ubiquitously expressed apoptotic protein, has shown a dual function in different types of cancers and in chemo-resistance. Recently, we reported that PDCD10 was downregulated in human GBM. The aim of this study was to explore the function of PDCD10 in GBM cells.
Methods
PDCD10 was knocked down in three GBM cell lines (U87, T98g and LN229) by lentiviral-mediated shRNA transduction. U87 and T98g transduced cells were used for phenotype study and LN229 and T98g cells were used for apoptosis study. The role of PDCD10 in apoptosis and chemo-resistance was investigated after treatment with staurosporine and temozolomide. A GBM xenograft mouse model was used to confirm the function of PDCD10 in vivo. A protein array was performed in PDCD10-knockdown and control GBM cells.
Results
Knockdown of PDCD10 in GBM cells promoted cell proliferation, adhesion, migration, invasion, and inhibited apoptosis and caspase-3 activation. PDCD10-knockdown accelerated tumor growth and increased tumor mass by 2.1-fold and led to a chemo-resistance of mice treated with temozolomide. Immunostaining revealed extensive Ki67-positive cells and less activation of caspase-3 in PDCD10-knockdown tumors. The protein array demonstrated an increased release of multiple growth factors from PDCD10-knockdown GBM cells.
Conclusions
Loss of programmed cell death 10 activates tumor cells and leads to temozolomide-resistance in GBM, suggesting PDCD10 as a potential target for GBM therapy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Glioblastoma (GBM) is one of the most malignant primary brain tumors in adults. The etiology and pathogenesis of GBM remains unclear. GBM shows remarkable variability of histological features. Vascular hyper-proliferation (neo-angiogenesis) and necrosis are essential diagnostic characteristics that distinguish GBM from lower grade gliomas. Neoplastic cells are highly migratory and can rapidly invade to adjacent brain parenchyma, even into the contralateral hemisphere. GBM is also a genetically heterogeneous tumor [1], suggesting the importance of personalized therapy for GBM patients. The current standard treatment is surgical resection followed by radiotherapy with concomitant and adjuvant temozolomide (TMZ) treatment. Despite this aggressive treatment, the prognosis following diagnosis is still very poor with an overall survival of less than 2 years and a 5-year survival rate of less than 5% [2, 3]. Thus, characterization of new therapeutic targets of GBM is important.
Programmed cell death 10 (PDCD10) originally termed as TFAR15 (TF-1 cell apoptosis related gene) [4] encodes an evolutionarily conserved protein, that is widely expressed in nearly all human tissues and various types of cells including neuronal, glial and endothelial cells (ECs) [5]. PDCD10 is also called CCM3 because it is one of three identified genes causing family cerebral cavernous malformation (CCM), a common brain vascular malformation involving aberrant angiogenesis and chronic hemorrhage [6]. Loss-of-function mutations of PDCD10/CCM3 lead to an earlier onset of CCM and to the most aggressive hemorrhage in CCM among the three identified CCM genes [7]. Hence, the vascular function of PDCD10 has been intensively studied in the past decade. PDCD10 is involved in vasculogenesis, angiogenesis, vessel maturation, apoptosis and senescence, oxidative metabolism and Golgi complex polarization [8] and is also essential for the neuron-glial unit [9]. A variety of signaling pathways underlying the diverse functions of PDCD10 are identified such as p-Akt, p38, p-Erk, VEGFR2, STK24/25, MST4, RhoA, Dll4-Notch and SMAD [10]. More recently, loss of endothelial PDCD10 has been shown to activate MEKK3-KLF2/4 and mTOR signaling pathways resulting in defects of the vascular development [11] and defective autophagy [12], respectively.
Despite its crucial roles in apoptosis, autophagy, angiogenesis, and in regulating several tumor-related signaling pathways, little is known about PDCD10 in cancers. The limited publications showed a dual function of PDCD10 in different cancers, suggesting that its oncological function may be cell type-specific [13,14,15]. Of note, PDCD10 deficiency can result in a resistance to chemotherapy in colorectal cancer [15] and in a higher likeliness to develop brain meningioma [16,17,18]. We recently reported a significant downregulation of PDCD10 in 85% of investigated human primary glioblastoma, and PDCD10 expression was absent in the majority of tumor and endothelial cells in human GBM [19]. Along with these findings, we have identified the crucial role of endothelial PDCD10 in promoting angiogenesis which in turn stimulated tumor growth in a GBM mouse model [20]. However, the direct function of tumor cell originated PDCD10 in GBM remains unclear. The present study attempts to address this issue after genomic modification of PDCD10 expression in GBM tumor cells.
Materials and methods
Cell culture
U87 and LN229 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% of fetal bovine serum (FBS) and 1% of sodium pyruvate. T98g cells were cultured in minimum essential medium Eagle (MEME) supplemented with glutamine (2 mM), non-essential amino acids (1%), sodium pyruvate (1%) and FBS (10%).
Small hairpin RNA (shRNA) knockdown of PDCD10
Knockdown of PDCD10 was achieved by using two different lentiviral shRNA vector systems. An inducible TRIPZ lentiviral shRNA vector for human PDCD10 (shPDCD10:RFP, Clone ID: V2THS_217165) and the empty vector (EV:RFP, catalog# RHS4750) (Thermo Scientific, Schwerte, Germany) were used in U87 and LN229 cells. Another lentiviral shRNA vector for a constitutive knockdown of human PDCD10 (shPDCD10:GFP, catalog# TL302576) and the empty vector (EV:GFP, Catalog# TR30021) (OriGene, Herford, Germany) were applied to T98g cells. Lentiviruses were produced by transfecting shPDCD10 or EV with trans-lentiviral packaging system (Thermo Scientific, Schwerte, Germany) in HEK293 cells according to our previously established protocol [20]. The generated lentiviral-shPDCD10 or -EV supernatants were used to perform transduction in GBM cells. In the case of inducible TRIPZ lentiviral shRNA vector system, knockdown of PDCD10 was induced by the culture of the transduced U87 and LN229 cells with the media containing doxycycline (dox, 1 µg/ml) (Sigma-Aldrich, Munich, Germany). For both vector systems, the non-transduced cells were killed by treatment with puromycin (1 µg/ml) (Sigma-Aldrich, Munich, Germany) 3 days after transduction.
Real-time RT-PCR (RT2-PCR)
Total RNA was extracted using the innuPREP RNA mini kit (Analytik Jena, Jena, Germany) and the cDNA was synthesized using the iScript cDNA kit (Bio-Rad, Munich, Germany) according to the manufacturer’s instructions. Real-time PCR was performed according to our established protocol [21]. Primer sequences are: forward 5′-TGG CAG CTG ATG ATG TAG AAG-3′ and reverse 5′-TCG TGC CTT TTC GTT TAG GT-3′ for PDCD10; forward 5′-AGC CAC ATC GCT CAG ACA-3′ and reverse 5′-GCC CAA TAC GAC CAA ATC C-3′ for reference gene GAPDH. The relative expression of target gene was calculated by \({{\text{2}}^ - }^{{\Delta \Delta {C_{\text{t}}}}}\) method.
Cell phenotype study
U87 and T98g cell proliferation and adhesion were measured respectively by WST-1 assay and by crystal violet staining according to our previous protocols [20]. For cell invasion and migration assays, cells were first cultured in serum-free media for 24 h in order to synchronize cell cycle thereby minimizing the influence of proliferation on cell invasion and migration. Thereafter, cell invasion, scratch migration assay, and spheroid migration assay were performed as described previously [20].
Apoptosis induction and detection
Because apoptotic stimuli caused detachment of U87 cells that led to an inaccurate quantification of apoptotic cells, we replaced U87 with LN229 for apoptotic study. LN229 and T98g cells (3 × 105) were seeded in 6-well plate. After overnight incubation, apoptosis was induced by treatment with 100 nM of staurosporine (STS, Sigma-Aldrich), a well-accepted apoptosis inducer and used as positive control in the present study, or with 150 µM of TMZ (Sigma-Aldrich), the first line drug for chemotherapy of GBM in clinical practice, for different time periods as indicated in individual experiments followed by the nuclear staining with Hoechst-33258 dye. Pictures were acquired from 4 to 6 randomized fields of each well. Cells with condensed chromatin and apoptotic bodies were counted as apoptotic cells. Data were presented as mean of percentage of apoptotic cells from 4 to 6 wells per group. Additionally, activation of caspase-3 after induction of apoptosis was detected by Western blot and immunofluorescent staining using an antibody specifically against cleaved caspase-3 (1:400, Cell Signaling Technology, Frankfurt, Germany) and the activity of caspase-3 was evaluated by using caspase-3 colorimetric protease assay kit (Life Technology, Darmstadt, Germany) according to the manufacturer’s instruction.
MTT assay
After the treatment with STS and TMZ, the viability of LN229 and T98g cells was evaluated by MTT assay. MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide) (5 mg/ml) (Life technology, Darmstadt, Germany) was added to the culture and incubated for 3 h. To dissolve the MTT formazan, 150 µl of MTT solvent (4 mM HCl, 0.1% NP40 in isopropanol) was added to each well and the plate was incubated on an orbital shaker for 15 min at room temperature. The absorbance was detected at 590 nm in a plate reader.
Western blot
Total protein extraction and western blot were performed as described previously [22]. The following antibodies were used: PDCD10 (1:500; Abcam, Cambridge, UK); cleaved caspase-3 (1:400) and GAPDH (1:1000; both Cell Signaling Technology, Frankfurt, Germany). The semi-quantification of the blots were done by using software image J software 1.50b.
Glioblastoma xenograft model and treatment
The animal experiments were performed strictly according to the ethics contract (No. 84-02.04.2012.A348). A GBM xenograft model was established using female nude mice (4–6 weeks old) based on a previous protocol with modifications [20]. Briefly, 100 µl of cell suspension was prepared with 1 million of shPDCD10-U87 or control-U87 cells. Then the individual cell suspension was mixed with VEGF (1000 ng/ml) (R&D Systems, Wiesbaden-Nordenstadt, Germany), bFGF (1000 ng/ml) (R&D Systems) and 100 µl of Matrigel (BD, Heidelberg, Germany). The mixture was subcutaneously implanted into the flank of the nude mice (n = 8, each for control and for shPDCD10). To maintain the stable knockdown of PDCD10 in vivo, 2 mg/ml dox was added to the drinking water. In addition, TMZ (p.o., 50 mg/kg, × 5 days) or vehicle was administered daily to the control (dox-) and shPDCD10 mice 7 days after implantation (n = 5 for each group). Tumor size was evaluated using a caliper measuring tumor length and width twice a week. The volume of the tumor (VT) was calculated by the following formula: VT = π/6 × length × width2. The xenograft tumors were removed 3 weeks after implantation and weighted before sectioning for further histological studies and immunostaining.
Immunohistochemistry and immunofluorescent staining
Immunohistochemistry and immunofluorescent staining were performed according to the protocol described previously [23]. The following antibodies were used: proliferation marker mouse anti-Ki67 (1:200, Zytomed, Berlin, Germany); mouse anti-CD31 (1:40, Dako, Hamburg, Germany) and rabbit anti-cleaved caspase-3 (1:400, Cell Signaling Technology). The images were acquired using a fluorescence microscope (Axio Imager M2, Zeiss, Oberkochen, Germany).
Protein array
To study the underlying mechanism of PDCD10 in GBM cells, protein array was performed using a protein array kit (cataloge# ARY007, R&D Systems). The nitrocellulose membrane was precoated with antibodies against 55 different growth factors and angiogenic proteins. Two membranes were incubated with the media collected from EV-U87 and shPDCD10-U87 cells followed by the detection according to manufacturer’s protocol. To optimally visualize the individual dots, the chemiluminescence detection was done after 1 min, 5 min and 10 min exposure. The semi-quantification of the blot dots was performed by using image J software 1.50b. The data were presented as the mean of a pair of duplicated dots for proteins upregulated more than 1.5-fold in the PDCD10-knockdown group (shPDCD10) compared to the control (EV).
Statistical analysis
Data are presented as mean ± SD. Statistical analysis was performed using the WinSTAT software. Results between two groups were analyzed by the student t test. A P value less than 0.05 was considered statistically significant.
Results
Confirmation of PDCD10 knockdown in the GBM cells
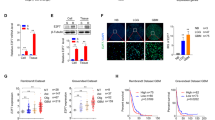
The succesful transduction was evidenced by the expression of red fluorescence protein in U87 (U87:RFP) and LN229 (LN229:RFP) cells or of green fluorescence protein in T98g cells (T98g:GFP) (Fig. 1a). The knockdown of PDCD10 in these transduced cells was confirmed at mRNA and protein levels by RT2-PCR (P < .001) (Fig. 1b) and Western blotting (Fig. 1c), respectively. Semi-quantification of the blots indicated a reduction of PDCD10 protein expression by 63% in U87 cells (P < .01), 57% in T98g cells (P < .01) and 62% in LN229 cells (P < .05) in comparison to corresponding controls (Fig. 1c).

Stable knockdown of PDCD10 was achieved using two different PDCD10-specific lentiviral shRNA vector systems and two different empty vectors (EV). a The representative photos showed successfully transduced cells expressing red fluorescence protein (shPDCD10:RFP, for U87 and LN229 cells) or green fluorescent protein (shPDCD10:GFP, for T98g cells). b and c Confirmation of PDCD10 knockdown by RT2-PCR (b) and Western blot (c). *P < .05, **P < .01 and ***P < .001, compared with empty vector (EV)
Knockdown of PDCD10 activated GBM cells in vitro
To investigate the consequence of PDCD10 knockdown on GBM cell behavior, the proliferation, adhesion, invasion and migration assays were performed in two different GBM cell lines (U87 and T98g) in which PDCD10 was knocked down respectively by using two different lentiviral vectors. The WST-1 assay demonstrated a proliferation increase of 59% (P < .01) in shPDCD10-U87 and 53% (P < .001) in shPDCD10-T98g cells after knockdown of PDCD10 (Fig. 2a). The adhesion assay showed a significantly higher adhesion ability in both shPDCD10-U87 (P < .01) and shPDCD10-T98g cells (P < .001) compared to the corresponding controls (Fig. 2b). An enhanced invasion was also observed in PDCD10-knockdown cells. The invaded cells increased to 120% (P < .01) in shPDCD10-U87 and 127% (P < .05) in shPDCD10-T98g cells (Fig. 2c). Furthermore, a remarkable enhanced mobility was detected in both PDCD10 knockdown-GBM cells by scratch assay (Fig. 2d). The increased migration activity of shPDCD10-U87 cells (P < .01) and shPDCD10-T98g cells (P < .001) was confirmed by spheroid migration assay (Fig. 2e).

Knockdown of PDCD10 in GBM cells promotes proliferation (a), adhesion (b) invasion (c) and migration (d and e). a Proliferation increase of PDCD10 knockdown GBM cells. Cell proliferation was detected 24 h after seeding cells in 96-well plate by WST-1 assay at 450 nm. b Adhesion increase of PDCD10 knockdown GBM cells. Cells were seeded in a 96-well plate precoated with matrigel. After 90 min of incubation, cells were stained with crystal violet followed by the absorbance detection at 550 nm. c Increased invasion of PDCD10 knockdown GBM cells. U87 and T98g cells were seeded in the transwell insert precoated with matrigel followed by the culture in serum-free medium for 24 h. Invasive cells were stained by crystal violet after 24 h of incubation and the absorbance was measured in a plate reader at 550 nm. Scale bar: 50 µm. d and e Migration increase of PDCD10 knockdown GBM cells. Before migration assay, cells were cultured in serum-free medium for 24 h. For the scratch assay (d), the cells were reseed onto the 35 mm petri dishes followed by 16 h of starvation. The photos were recorded at 10 h after scratching. The migrated cells were counted in six fields each petri dish. Scale bar: 200 µm. For the spheroid migration assay (e), the spheroids were photographed after 48 h of incubation. The data were presented as the mean of the longest and shortest spheroid diameters of each spheroid. Scale bar: 100 µm. All data presented in this figure are representative of at least three independent experiments. *P < .05, **P < .01 and ***P < .001, compared to EV
Knockdown of PDCD10 stimulated tumor growth in glioblastoma xenograft model
shPDCD10-U87 cells were applied in the animal study. RT2-PCR proved a stable knockdown of PDCD10 in the shPDCD10-U87 xenograft tumors (Fig. 3a). Under this knockdown condition, the tumor growth rate was significantly faster in shPDCD10-mice than in the control mice as inspected from day 10 to 21 after implantation (Fig. 3b). The weight of the tumor mass at day 21 after implantation was 2.1-fold heavier in the shPDCD10 group than that in the control group (P < .01) (Fig. 3c). H&E staining revealed dense and orientated tumor cells on xenograft tumor sections, similar to that seen in human GBM (Fig. 3d). Interestingly, microvessel-like structures that mostly contained blood cells were frequently observed on shPDCD10 sections (arrows, H&E staining in Fig. 3d). The immunohistochemistry photos and the quantitative analysis showed a considerable more Ki67-positive cells (arrowheads) on the sections from shPDCD10-tumors than that from the control tumors (Fig. 3d) (P < .05). Furthermore, many tumor cells (arrowheads, Fig. 3d) as well as some vessel-like structures (arrows, Fig. 3d) appeared CD31-positive exclusively in shPDCD10-sections. Quantitative analysis indicated a significantly higher number of CD31-positive vessel-like structures in shPDCD10-sections (P < .05) (Fig. 3d).

Increased tumor growth in a shPDCD10-transduced human glioblastoma xenograft. shPDCD10-transduced U87 cells were subcutaneously implanted into the flank of nude mice. To maintain the stable knockdown of PDCD10 in vivo, 2 mg/ml doxycycline (dox) and 1% sucrose were added to the drinking water. For the control group (Con), shPDCD10-transduced U87 cells were cultured in dox-free medium in vitro before implantation. The control mice received drinking water containing no dox but 1% sucrose. The xenograft tumors were removed from the mice 21 days after implantation. a PDCD10 downregulation in xenograft tumors as detected by RT2-PCR (n = 3 for each group). b and c PDCD10 knockdown promotes tumor growth. The tumor growth curve (b) and the tumor mass weighed at the end of the experiments (c) indicates a significantly quicker tumor growth rate and bigger tumors in shPDCD10 mice than in controls (n = 8 for each group). d PDCD10 knockdown stimulates tumor cell proliferation and the formation of vessel-like structures. H&E staining shows the histological morphology and vessel-like structure (arrows). Ki67 immunostaining reveals proliferating cells (arrowheads) in the tumor sections. Many tumor cells (arrowheads) and some vessel-like structures (arrows) appeared CD31-positive exclusively on shPDCD10-tumor sections. The red blood cells in the vessels appeared in dark brown, indicative of the formation of functional microvessels inside the tumor. Scale bar: 50 µm. The quantitative analysis revealed a significantly higher number of Ki67-positive cells and CD31-positive vessel-like structures in shPDCD10-tumor sections compared to the controls (Con) (n = 4 for each group). *P < .05, **P < .01 and ***P < .001, compared to Con
Knockdown of PDCD10 induced apoptosis-resistance in vitro and chemo-resistance in vivo involving inhibition of caspase-3 activation
To study the role of PDCD10 in apoptosis, shPDCD10- and EV-transduced LN229 and T98g cells were treated with STS and TMZ. Hoechst 33258 staining showed a 29% (P < .01) reduction of apoptotic cells with STS and a 47% (P < .01) reduction with TMZ for the shPDCD10-LN229 group compared with the corresponding controls (Fig. 4a). In T98g cells, knockdown of PDCD10 (shPDCD10 group) reduced the percentage of apoptotic cells by 52% (P < .01) of the corresponding controls (EV) with STS and by 34% (P < .01) with TMZ (Fig. 4a). MTT assay confirmed a significantly higher viability of shPDCD10-LN229 and shPDCD10-T98g cells in response to STS and TMZ treatment (Fig. 4b). Caspase-3 detected by immunostaining (Fig. 4c), enzyme activity assay (Fig. 4d) and western blot (Fig. 4e) demonstrated less caspase-3 activation in shPDCD10-LN229 and shPDCD10-T98g cells than in cells without knockdown.

PDCD10 knockdown in GBM cells reduces apoptosis in vitro and induces a chemo-resistance in vivo involving inhibition of caspase-3 activation. a PDCD10 knockdown in LN229 and T98g cells induces apoptosis resistance in vitro. EV- and shPDCD10-transduced GBM cells were treated with staurosporine (STS, 100 nM, 16 h) or with temozolomide (TMZ, 150 µM, 24 h) followed by Hoechst 33258 nuclear staining. Cells with condensed chromatin and apoptotic bodies were counted as apoptotic cells. The counting was done in six randomized fields from four wells per group. Scar bar: 20 µm. b Reduced cytotoxicity of STS and TMZ in PDCD10 knockdown LN229 and T98g cells. LN229 and T98g cells were treated with STS (100 nM, 72 h) and TMZ (150 µM, 72 h). Cell viability was detected by MTT assay at 590 nm. C-E: Inhibition of caspase-3 activation in PDCD10 knockdown GBM cells. Cells were treated with STS (100 nM, 16 h) and TMZ (150 µM, 24 h) and active (cleaved) caspase-3 (Cas-3) was detected by immunofluorescent staining (c), enzyme activity assay (d) and western blot (e). sh shPDCD10. *P < .05, **P < .01 and ***P < .001, compared to EV; Scale bar: 20 µm. F and G: PDCD10 knockdown inhibits caspase-3 activation (f) and leads to a chemo-resistant to TMZ treatment in vivo (g). Human glioblastoma shPDCD10-xenograft mice and control xenograft mice (as described in Fig. 2 legend) were treated daily with TMZ (p.o., 50 mg/kg for 5 days) or with vehicle 7 days after implantation. The tumors were removed from the mice 21 d after implantation and the tumor mass was weighted before sectioning. The representative photos show the activation of caspase-3 (orange color) in the control (Con) and in shPDCD10 tumor sections detected by immunofluorescence staining. Scale bar: 20 µm. Quantitative analysis indicated a significant reduction of the Cas-3-positive cells in shPDCD10-tumor than that in control tumor (f). As a consequence, PDCD10 knockdown significantly reduced the efficiency of chemo-therapy induced by TMZ treatment in comparison to the control (g). n = 5 for each group. * P < .05 compared to control (Con)
Next, we examined whether PDCD10 knockdown affected the efficiency of chemotherapy by TMZ in vivo. Immunofluorescent staining revealed a significant decrease in the number of active caspase-3-positive cells in shPDCD10-sections (P < .05) (Fig. 4f). As a consequence, TMZ treatment resulted in only 44.8% reduction of tumor mass in shPDCD10-mice compared to 52.1% in control mice, indicating that knockdown of PDCD10 led to a significant resistance to TMZ treatment (P < .05) (Fig. 4g).
Knockdown of PDCD10 stimulated the release of multiple soluble factors from GBM cells
To explore the mechanism of the tumor suppressor function of PDCD10, a protein array of 55 growth factors and antigenic proteins was performed with the media collected from EV-U87 and shPDCD10-U87 cultures. Each protein was detected in duplicate. Figure 5 presents the dot-blot images after a 5 min exposure. Semi-quantification revealed that 19 of 55 target proteins were upregulated more than 1.5-fold in the shPDCD10 group in comparison to the EV control (Fig. 5b). Eight of these showed more than twofold upregulation in the shPDCD10-blot (Fig. 5c). The most significantly increased soluble factor released from PDCD10-knockdown GBM cells was angiogenin (14-fold of EV), a crucial molecule involved in cancer and angiogenesis.

PDCD10 knockdown in GBM cells stimulates the release of multiple soluble factors. A protein array was carried out with the media from shPDCD10-U87 and EV-U87 cells. a Dot-blots on X-ray film (5 min exposure). b Semi-quantification of the blots showed 19 upregulated proteins (> 1.5-fold of EV) among 55 detected proteins. c Summary of eight proteins which were upregulated more than twofold in shPDCD10 cells in comparison with EV control
Discussion
PDCD10, a ubiquitously expressed adaptor protein, plays a pivotal role in vasculogenesis and angiogenesis [8], cell apoptosis, autophagy, senescence [10, 12] and chemotherapy-resistance [15]. So far, the understanding of the function and the underlying mechanism of PDCD10 is mainly derived from the studies carried out in the endothelial vascular context [11, 22, 24] because loss-of-function mutation in PDCD10/CCM3 causes familial CCM, one of the most common vascular malformations in the central nervous system (CNS). However, only a few publications deal with PDCD10 in the oncological field despite its crucial role in apoptosis and the regulation of multiple survival signaling pathways. The co-incidence of CCM and brain tumors is very rare. Nevertheless, the overlap of CCM with low grade astrocytoma [25] or with GBM [26, 27] have been shown, but these reports did not mention the genetic alteration of CCM causal genes including PDCD10/CCM3 in those patients. Interestingly, a direct association of PDCD10/CCM3 mutation with multiple meningiomas has recently been reported [16,17,18]. Our group has demonstrated the absence of PDCD10 in the majority of endothelial and tumor cells of human GBM [19], which is associated with the proliferation of both cell types. Moreover, loss of endothelial PDCD10 activated GBM tumor cells in vitro and stimulated tumor growth in vivo [20]. Along with these findings, the present study further explored the role of GBM cell-originated PDCD10. Here, we report that stable knockdown of PDCD10 activated GBM cells in vitro (Fig. 2) and stimulated xenograft tumor growth in vivo (Fig. 3). The present new data together with our previously identified tumor-promoter effect via pro-angiogenesis mediated by the loss of endothelial PDCD10 [20] suggest that absence of PDCD10 in tumor cells and in endothelial cells may synergistically contribute to the tumor growth and tumor infiltration in human GBM. In addition, we noted CD31-positive cells and vessel like structures exclusively on the sections from PDCD10-knockdown xenograft tumors. Some of these microvessels contained blood cells suggesting the formation of functional vessels after knockdown of PDCD10 in tumor (arrows in CD31 staining; Fig. 3d). Given the fact that the xenograft tumor is formed by the implanted U87 cells, the presence of CD31-positive cells in tumors raises high interest to further study whether loss of tumor-originated PDCD10 is involved in the transition of tumor cells to endothelial cells, thereby inducing tumor-angiogenesis. This may serve as an additional and important mechanism for the neo-angiogenesis in human GBM besides the angiogenesis induced by loss of endothelial PDCD10.
The apoptotic function of PDCD10 appears context-dependent [24, 28,29,30]. Moreover, the controversial data concerning PDCD10 in different types of cancer cells has been reported [13,14,15, 31]. Furthermore, increasing evidence shows an altered expression of PDCD10 in response to different chemotherapies in cancers [31,32,33]. MicroRNA-425-5p regulates the chemo-resistance in colorectal cancer cells through downregulation of PDCD10 [15]. Downregulation of PDCD10 was associated with the resistance to chemotherapy-triggered apoptosis by a gene chip analysis [31]. We were thus interested in exploring whether PDCD10 is involved in chemo-resistance in GBM. TMZ is the first line drug for chemotherapy of GBM in clinical practice. It impairs DNA repair, thereby blocking cell cycle and promoting apoptosis, as well as to lesser extent suppressing autophagy and senescence [34, 35]. Regardless of its certain benefices for the O6-methylguanine-DNA-methyltransferase (MGMT) hypermethylated cases, GBM tumors often are resistant to the TMZ treatment [35]. Here, we evaluate apoptosis in vitro and the tumor growth after TMZ treatment in vivo after knockdown of PDCD10 in GBM cells. In this part of the study, apoptosis was induced by TMZ and by STS in two different GBM cells (LN229 and T98g). Knockdown of PDCD10 led to a significant reduction of apoptosis in both TMZ and STS models (Fig. 4a), to an increase in cell viability (Fig. 4b) and to an inhibition of caspase-3 activation (Fig. 4c–e). Moreover, TMZ-treatment reduced the tumor mass by 44.8% in PDCD10-knockdown group (shPDCD10), and 52.1% in control group, indicating a chemo-resistance effect induced by PDCD10 knockdown (Fig. 4g). This chemo-resistant effect was associated with lesser activation of caspase 3 detected in shPDCD10-tumor sections (Fig. 4f). These new findings are in agreement with our previous observation that selective expression of PDCD10 in the pseudopalisading area, a hypoxia region in human GBM tumor, is associated with the activation of caspase-3 and apoptosis in human GBM, whereas absence of PDCD10 in the infiltration area of the tumor is associated with tumor cell proliferation [19]. Taken together, our data strongly suggest that loss of tumor-originated PDCD10 results in apoptosis-resistance which may potentially affect the efficiency of chemotherapy targeting apoptosis in human GBM.
To uncover the underlying mechanism leading to the activation of GBM cells, the stimulation of tumor growth and the reduction of apoptosis, we performed an array of 55 proteins with the media collected from shPDCD10- and EV-U87 cells (Fig. 5a). Semi-quantification of the dot blots demonstrated that eight proteins were increased by more than twofold (Fig. 5c). These soluble factors all play crucial roles in regulating tumor cell proliferation, migration and invasion as well as angiogenesis. For example, angiogenin (ANG), the most significantly increased soluble factor, is a secreted growth factor and is potently angiogenic protein in comparison with most other angiogenic factors, as it can induce blood vessel formation in different tissues at femtomolar doses [36]. ANG is also the first known human tumor-derived protein including GBM. ANG can directly promote GBM tumor cell proliferation but can also constantly translocate into the nuclei of tumor cells and promote cell proliferation by activating NF-κB signaling pathway [37]. The upregulation of ANG is strongly correlated with an invasive cancer phenotype involving the stimulation of matrix metallopeptidase-2 (MMP2) expression through activation of Erk1/2 [38]. The second highest upregulated factor hepatocyte growth factor (HGF) is a ligand of receptor tyrosine kinases mesenchymal epithelial transition (MET) which is dysregulated in approximately 90% of GBM. Thus, the HGF/MET pathway is a vital regulator of multiple pro-survival pathways in GBM, thereby contributing to oncogenesis and tumor. HGF autocrine activation predicts sensitivity to MET inhibition in glioblastoma progression [39,40,41]. The intrinsic feedback loop activated by HGF-autocrine leads to amplify or promote the drivers of GBM through various mechanisms including inducing cell cycle progression, cell migration, invasion and angiogenesis and inhibition of apoptosis. Endoglin, another upregulated factor, is a homodimeric cell membrane glycoprotein receptor for transforming growth factor β and bone morphogenetic proteins. Its expression is implicated in the development of resistance to vascular endothelial growth factor (VEGF)-targeted tumor therapy [42] and is associated with high grade of glioma patients. Thus, endoglin is suggested as a specific and sensitive marker for evaluation of angiogenesis in GBM as well as a target for GBM therapy [43]. Taken together, we believe that all these upregulated soluble factors resulting from PDCD10 knockdown could synergistically act on the GBM cells themselves through an autocrine manner as well as on non-tumor cells such as endothelial cells through a paracrine mechanism to trigger GBM cells, inhibit apoptosis and eventually stimulate tumor growth and tumor invasion.
In conclusion, the present study demonstrated a tumor suppressor-like function of tumor-originated PDCD10 in vitro and in vivo in GBM. Given that loss of endothelial PDCD10 can also promote GBM tumor growth through stimulating angiogenesis, we suggest that the absence of PDCD10 in the majority of infiltrating tumor cells and endothelial cells in human GBM may potentially contribute to GBM progression and chemotherapy-resistance, mostly like through increase in secreting multiple growth factors. Thus targeting PDCD10 may be a novel strategy to potentiate other therapeutic options for GBM in the future.
References
Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, Alexe G, Lawrence M, O’Kelly M, Tamayo P, Weir BA, Gabriel S, Winckler W, Gupta S, Jakkula L, Feiler HS, Hodgson JG, James CD, Sarkaria JN, Brennan C, Kahn A, Spellman PT, Wilson RK, Speed TP, Gray JW, Meyerson M, Getz G, Perou CM, Hayes DN, Cancer Genome Atlas Research N (2010) Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17(1):98–110. https://doi.org/10.1016/j.ccr.2009.12.020
Alexander BM, Cloughesy TF (2017) Adult glioblastoma. J Clin Oncol 35(21):2402–2409. https://doi.org/10.1200/JCO.2017.73.0119
Cloughesy TF, Cavenee WK, Mischel PS (2014) Glioblastoma: from molecular pathology to targeted treatment. Annu Rev Pathol 9:1–25. https://doi.org/10.1146/annurev-pathol-011110-130324
Wang Y, Liu H, Zhang Y, Ma D (1999) cDNA cloning and expression of an apoptosis-related gene, humanTFAR15 gene. Sci China Ser C Life Sci 42(3):323–329. https://doi.org/10.1007/BF03183610
Petit N, Blecon A, Denier C, Tournier-Lasserve E (2006) Patterns of expression of the three cerebral cavernous malformation (CCM) genes during embryonic and postnatal brain development. Gene Expr Patterns 6(5):495–503. https://doi.org/10.1016/j.modgep.2005.11.001
Bergametti F, Denier C, Labauge P, Arnoult M, Boetto S, Clanet M, Coubes P, Echenne B, Ibrahim R, Irthum B, Jacquet G, Lonjon M, Moreau JJ, Neau JP, Parker F, Tremoulet M, Tournier-Lasserve E, Societe Francaise de N (2005) Mutations within the programmed cell death 10 gene cause cerebral cavernous malformations. Am J Hum Genet 76(1):42–51. https://doi.org/10.1086/426952
Shenkar R, Shi C, Rebeiz T, Stockton RA, McDonald DA, Mikati AG, Zhang L, Austin C, Akers AL, Gallione CJ, Rorrer A, Gunel M, Min W, De Souza JM, Lee C, Marchuk DA, Awad IA (2015) Exceptional aggressiveness of cerebral cavernous malformation disease associated with PDCD10 mutations. Genet Med 17(3):188–196. https://doi.org/10.1038/gim.2014.97
Fischer A, Zalvide J, Faurobert E, Albiges-Rizo C, Tournier-Lasserve E (2013) Cerebral cavernous malformations: from CCM genes to endothelial cell homeostasis. Trends Mol Med 19(5):302–308. https://doi.org/10.1016/j.molmed.2013.02.004
Louvi A, Nishimura S, Gunel M (2014) Ccm3, a gene associated with cerebral cavernous malformations, is required for neuronal migration. Development 141(6):1404–1415. https://doi.org/10.1242/dev.093526
Draheim KM, Fisher OS, Boggon TJ, Calderwood DA (2014) Cerebral cavernous malformation proteins at a glance. J Cell Sci 127(Pt 4):701–707. https://doi.org/10.1242/jcs.138388
Zhou Z, Tang AT, Wong WY, Bamezai S, Goddard LM, Shenkar R, Zhou S, Yang J, Wright AC, Foley M, Arthur JS, Whitehead KJ, Awad IA, Li DY, Zheng X, Kahn ML (2016) Cerebral cavernous malformations arise from endothelial gain of MEKK3-KLF2/4 signalling. Nature 532(7597):122–126. https://doi.org/10.1038/nature17178
Marchi S, Corricelli M, Trapani E, Bravi L, Pittaro A, Delle Monache S, Ferroni L, Patergnani S, Missiroli S, Goitre L, Trabalzini L, Rimessi A, Giorgi C, Zavan B, Cassoni P, Dejana E, Retta SF, Pinton P (2015) Defective autophagy is a key feature of cerebral cavernous malformations. EMBO Mol Med 7(11):1403–1417. https://doi.org/10.15252/emmm.201505316
Barrier A, Lemoine A, Boelle PY, Tse C, Brault D, Chiappini F, Breittschneider J, Lacaine F, Houry S, Huguier M, Van der Laan MJ, Speed T, Debuire B, Flahault A, Dudoit S (2005) Colon cancer prognosis prediction by gene expression profiling. Oncogene 24(40):6155–6164. https://doi.org/10.1038/sj.onc.1208984
Fu X, Zhang W, Su Y, Lu L, Wang D, Wang H (2016) MicroRNA-103 suppresses tumor cell proliferation by targeting PDCD10 in prostate cancer. Prostate 76(6):543–551. https://doi.org/10.1002/pros.23143
Zhang Y, Hu X, Miao X, Zhu K, Cui S, Meng Q, Sun J, Wang T (2016) MicroRNA-425-5p regulates chemoresistance in colorectal cancer cells via regulation of Programmed Cell Death 10. J Cell Mol Med 20(2):360–369. https://doi.org/10.1111/jcmm.12742
Riant F, Bergametti F, Fournier HD, Chapon F, Michalak-Provost S, Cecillon M, Lejeune P, Hosseini H, Choe C, Orth M, Bernreuther C, Boulday G, Denier C, Labauge P, Tournier-Lasserve E (2013) CCM3 mutations are associated with early-onset cerebral hemorrhage and multiple meningiomas. Mol Syndromol 4(4):165–172. https://doi.org/10.1159/000350042
Fauth C, Rostasy K, Rath M, Gizewski E, Lederer AG, Sure U, Zschocke J, Felbor U (2015) Highly variable intrafamilial manifestations of a CCM3 mutation ranging from acute childhood cerebral haemorrhage to late-onset meningiomas. Clin Neurol Neurosurg 128:41–43. https://doi.org/10.1016/j.clineuro.2014.10.023
Labauge P, Fontaine B, Neau JP, Bergametti F, Riant F, Blecon A, Marchelli F, Arnoult M, Lannuzel A, Clanet M, Olschwang S, Denier C, Tournier-Lasserve E (2009) Multiple dural lesions mimicking meningiomas in patients with CCM3/PDCD10 mutations. Neurology 72(23):2044–2046. https://doi.org/10.1212/WNL.0b013e3181a92b13
Lambertz N, El Hindy N, Kreitschmann-Andermahr I, Stein KP, Dammann P, Oezkan N, Mueller O, Sure U, Zhu Y (2015) Downregulation of programmed cell death 10 is associated with tumor cell proliferation, hyperangiogenesis and peritumoral edema in human glioblastoma. BMC Cancer 15:759. https://doi.org/10.1186/s12885-015-1709-8
Zhu Y, Zhao K, Prinz A, Keyvani K, Lambertz N, Kreitschmann-Andermahr I, Lei T, Sure U (2016) Loss of endothelial programmed cell death 10 activates glioblastoma cells and promotes tumor growth. Neuro-oncology 18(4):538–548. https://doi.org/10.1093/neuonc/nov155
You C, Zhao K, Dammann P, Keyvani K, Kreitschmann-Andermahr I, Sure U, Zhu Y (2017) EphB4 forward signalling mediates angiogenesis caused by CCM3/PDCD10-ablation. J Cell Mol Med 21(9):1848–1858. https://doi.org/10.1111/jcmm.13105
Zhu Y, Wu Q, Xu JF, Miller D, Sandalcioglu IE, Zhang JM, Sure U (2010) Differential angiogenesis function of CCM2 and CCM3 in cerebral cavernous malformations. Neurosurg Focus 29(3):E1. https://doi.org/10.3171/2010.5.FOCUS1090
El Hindy N, Keyvani K, Pagenstecher A, Dammann P, Sandalcioglu IE, Sure U, Zhu Y (2013) Implications of Dll4-Notch signaling activation in primary glioblastoma multiforme. Neuro-oncology 15(10):1366–1378. https://doi.org/10.1093/neuonc/not071
You C, Sandalcioglu IE, Dammann P, Felbor U, Sure U, Zhu Y (2013) Loss of CCM3 impairs DLL4-Notch signalling: implication in endothelial angiogenesis and in inherited cerebral cavernous malformations. J Cell Mol Med 17(3):407–418. https://doi.org/10.1111/jcmm.12022
Zhang JY, Ming ZY, Wu AH (2012) Is cerebral cavernous malformation a pre-glioma lesion? Chin Med J 125(24):4511–4513
Mian MK, Nahed BV, Walcott BP, Ogilvy CS, Curry WT (2012) Glioblastoma multiforme and cerebral cavernous malformations: intersection of pathophysiologic pathways. J Clin Neurosci 19(6):884–886. https://doi.org/10.1016/j.jocn.2011.07.017
Wilson DM, Cohen B, Keshari K, Vogel H, Steinberg G, Dillon W (2014) Case report: glioblastoma multiforme complicating familial cavernous malformations. Clin Neuroradiol 24(3):293–296. https://doi.org/10.1007/s00062-013-0249-3
Schleider E, Stahl S, Wustehube J, Walter U, Fischer A, Felbor U (2011) Evidence for anti-angiogenic and pro-survival functions of the cerebral cavernous malformation protein 3. Neurogenetics 12(1):83–86. https://doi.org/10.1007/s10048-010-0261-6
Lauenborg B, Kopp K, Krejsgaard T, Eriksen KW, Geisler C, Dabelsteen S, Gniadecki R, Zhang Q, Wasik MA, Woetmann A, Odum N (2010) Programmed cell death-10 enhances proliferation and protects malignant T cells from apoptosis. APMIS 118(10):719–728. https://doi.org/10.1111/j.1600-0463.2010.02669.x
Chen L, Tanriover G, Yano H, Friedlander R, Louvi A, Gunel M (2009) Apoptotic functions of PDCD10/CCM3, the gene mutated in cerebral cavernous malformation 3. Stroke 40(4):1474–1481. https://doi.org/10.1161/STROKEAHA.108.527135
Huerta S, Harris DM, Jazirehi A, Bonavida B, Elashoff D, Livingston EH, Heber D (2003) Gene expression profile of metastatic colon cancer cells resistant to cisplatin-induced apoptosis. Int J Oncol 22(3):663–670
Gonzalez-Fernandez R, Morales M, Avila J, Martin-Vasallo P (2012) Changes in leukocyte gene expression profiles induced by antineoplastic chemotherapy. Oncol Lett 3(6):1341–1349. https://doi.org/10.3892/ol.2012.669
Urfali-Mamatoglu C, Kazan HH, Gunduz U (2018) Dual function of programmed cell death 10 (PDCD10) in drug resistance. Biomed Pharmacother 101:129–136. https://doi.org/10.1016/j.biopha.2018.02.020
Friedman HS, Kerby T, Calvert H (2000) Temozolomide and treatment of malignant glioma. Clin Cancer Res 6(7):2585–2597
Messaoudi K, Clavreul A, Lagarce F (2015) Toward an effective strategy in glioblastoma treatment. Part I: resistance mechanisms and strategies to overcome resistance of glioblastoma to temozolomide. Drug Discov Today 20(7):899–905. https://doi.org/10.1016/j.drudis.2015.02.011
Sheng J, Xu Z (2016) Three decades of research on angiogenin: a review and perspective. Acta Biochim Biophys Sinica 48(5):399–410. https://doi.org/10.1093/abbs/gmv131
Xia W, Fu W, Cai X, Wang M, Chen H, Xing W, Wang Y, Zou M, Xu T, Xu D (2015) Angiogenin promotes U87MG cell proliferation by activating NF-kappaB signaling pathway and downregulating its binding partner FHL3. PLoS ONE 10(2):e0116983. https://doi.org/10.1371/journal.pone.0116983
Miyake M, Goodison S, Lawton A, Gomes-Giacoia E, Rosser CJ (2015) Angiogenin promotes tumoral growth and angiogenesis by regulating matrix metallopeptidase-2 expression via the ERK1/2 pathway. Oncogene 34(7):890–901. https://doi.org/10.1038/onc.2014.2
Cecchi F, Rabe DC, Bottaro DP (2012) Targeting the HGF/Met signaling pathway in cancer therapy. Expert Opin Ther Targets 16(6):553–572. https://doi.org/10.1517/14728222.2012.680957
Xie Q, Bradley R, Kang L, Koeman J, Ascierto ML, Worschech A, De Giorgi V, Wang E, Kefene L, Su Y, Essenburg C, Kaufman DW, DeKoning T, Enter MA, O’Rourke TJ, Marincola FM, Vande Woude GF (2012) Hepatocyte growth factor (HGF) autocrine activation predicts sensitivity to MET inhibition in glioblastoma. Proc Natl Acad Sci USA 109(2):570–575. https://doi.org/10.1073/pnas.1119059109
Cruickshanks N, Zhang Y, Yuan F, Pahuski M, Gibert M, Abounader R (2017) Role and therapeutic targeting of the HGF/MET pathway in glioblastoma. Cancers. https://doi.org/10.3390/cancers9070087
Rosen LS, Gordon MS, Robert F, Matei DE (2014) Endoglin for targeted cancer treatment. Curr Oncol Rep 16(2):365. https://doi.org/10.1007/s11912-013-0365-x
Seon BK, Haba A, Matsuno F, Takahashi N, Tsujie M, She X, Harada N, Uneda S, Tsujie T, Toi H, Tsai H, Haruta Y (2011) Endoglin-targeted cancer therapy. Curr Drug Deliv 8(1):135–143
Acknowledgements
The authors thank Dr. Anja Prinz and Dr. Kai Zhao for their contributions to establishing knockdown cell lines. We also thank Ms. Rita Haase for her technical assistance. X.Y.W. and Y.L.W. received a scholarship from the Medical Faculty, University of Duisburg-Essen. This study was supported financially by the IFORES-program at the Medical Faculty, University of Duisburg-Essen to Y.Z.
Funding
This study was supported financially by the IFORES-program at the Medical Faculty, University of Duisburg-Essen to Y.Z.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All applicable international, national, and /or institutional guidelines for the care and use of animals were followed. All procedures performed in studies involving animals were in accordance with the ethical standards of the institution or practice at which the studies were conducted. The University of Duisburg-Essen approved all animal experiments (No. 84-02.04.2012.A348).
Informed consent
For this type of study formal consent is not required.
Research involving human and animal participants
This article does not contain any studies with human participants performed by any of the authors.
Rights and permissions
About this article

Cite this article
Nickel, AC., Wan, XY., Saban, DV. et al. Loss of programmed cell death 10 activates tumor cells and leads to temozolomide-resistance in glioblastoma. J Neurooncol 141, 31–41 (2019). https://doi.org/10.1007/s11060-018-03017-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-018-03017-7